
Isolation and Characterization of an Unknown Process-Related Impurity in Furosemide and Validation of a New HPLC Method
 ,
,
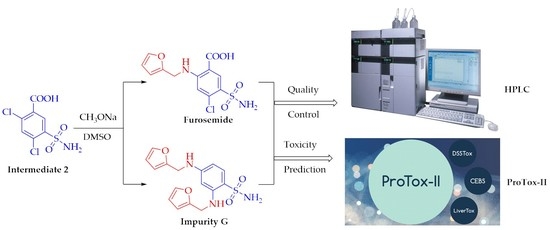
Abstract
:
1. Introduction
2. Results
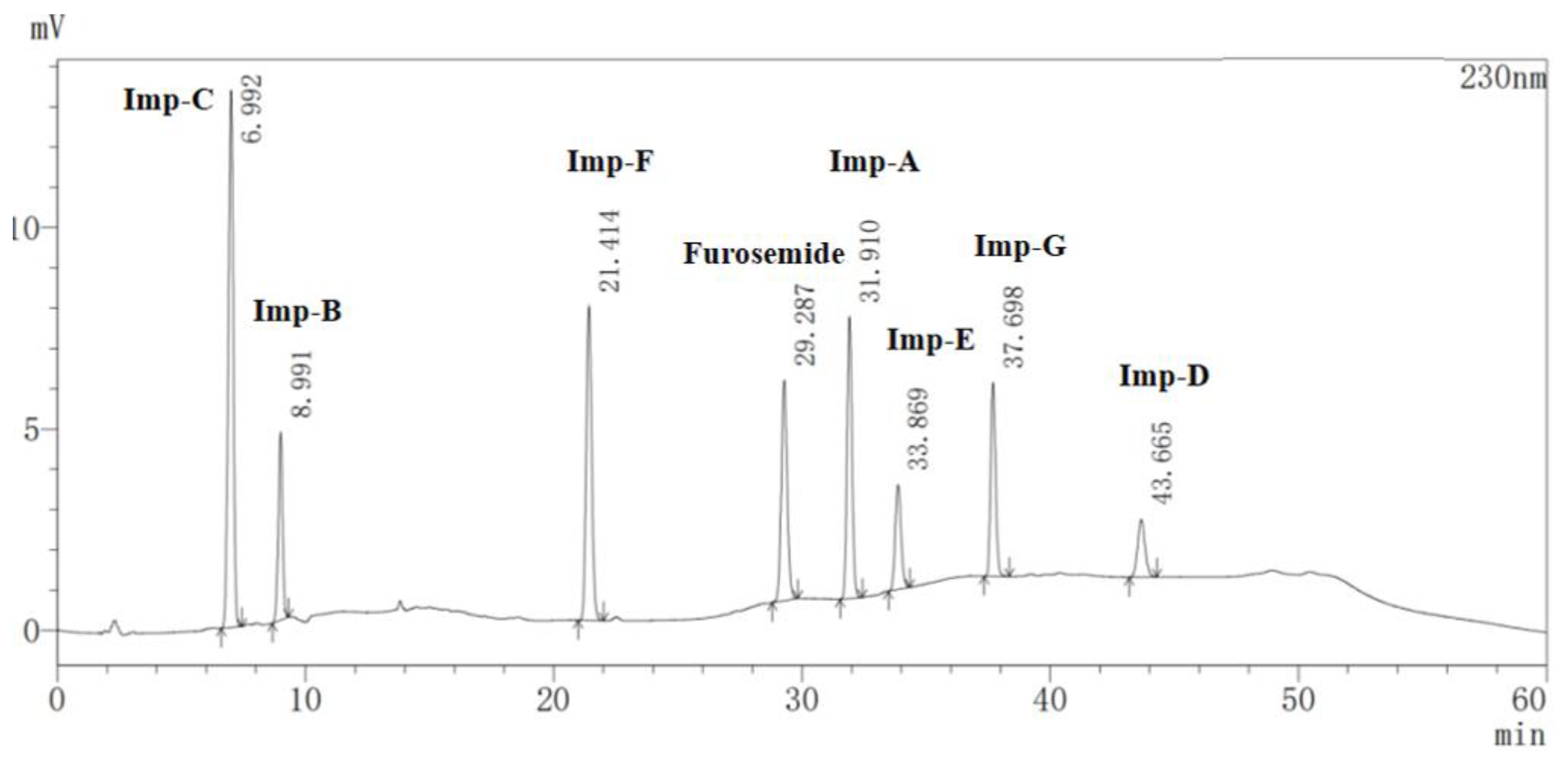
2.1. Detection and Separation of Impurity G
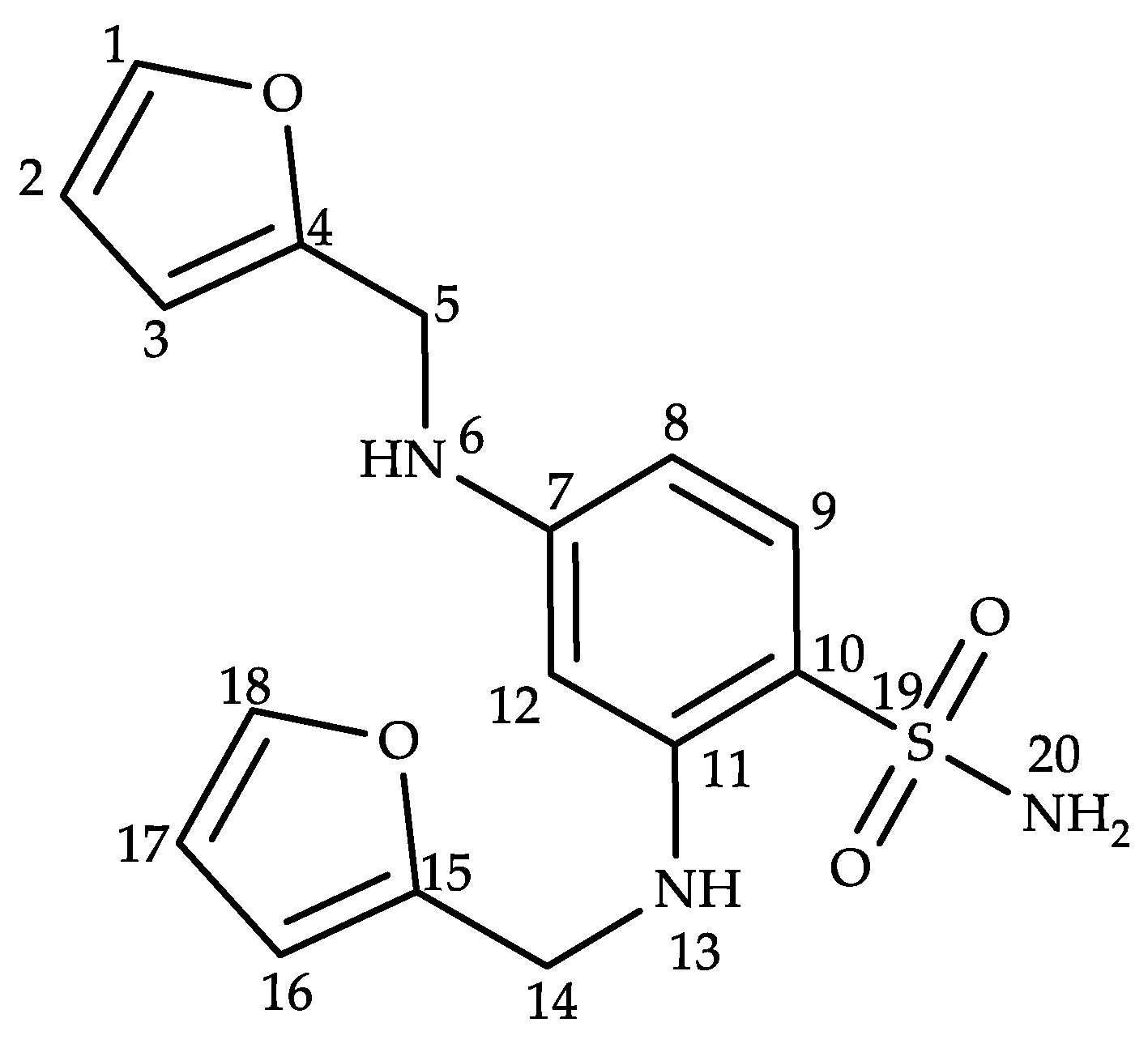
2.2. Structural Characterization of Impurity G
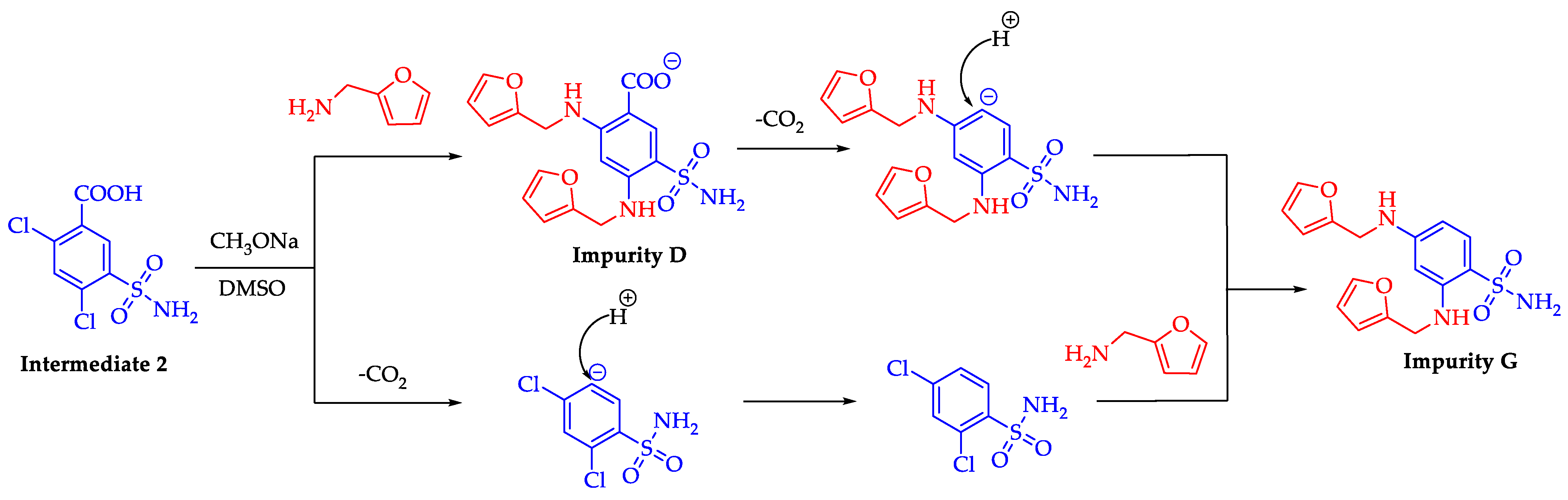
2.3. Formation Pathway and Controlling of Impurity G
2.4. Optimization of the HPLC-UV Method
2.5. HPLC Method Validation
2.5.1. Specificity
2.5.2. Limits of Detection (LODs) and Limits of Quantitation (LOQs)
2.5.3. Linearity
2.5.4. Accuracy
2.5.5. Repeatability and Intermediate Precision
2.5.6. Robustness
2.6. Prediction of Toxicity of Impurity G by ProTox-II Platform
3. Materials and Methods
3.1. Materials and Chemicals
3.2. Analytical HPLC
3.3. HPLC-MS Analysis
3.4. Nuclear Magnetic Resonance Spectroscopy (NMR)
3.5. Fourier Transform Infrared Spectroscopy (FT-IR)
3.6. Isolation of Impurity G
3.7. Preparation of Solutions
3.7.1. Preparation of Specificity Solution and Standard Solution
3.7.2. Preparation of Sample Solution
3.7.3. Preparation of Spiked Solution for Method Validation
3.8. Toxicity Prediction of Furosemide and Impurity G
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cannon, P.J.; Kilcoyne, M.M. Ethacrynic acid and furosemide: Renal pharmacology and clinical use. Prog. Cardiovasc. Dis. 1969, 12, 99–118. [Google Scholar] [CrossRef] [PubMed]
- Carone, L.; Oxberry, S.G.; Twycross, R.; Charlesworth, S.; Mihalyo, M.; Wilcock, A. Furosemide. J. Pain Symptom Manag. 2016, 52, 144–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duffy, M.; Jain, S.; Harrell, N.; Kothari, N.; Reddi, A.S. Albumin and Furosemide Combination for Management of Edema in Nephrotic Syndrome: A Review of Clinical Studies. Cells 2015, 4, 622–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blázquez-Bermejo, Z.; Farré, N.; Caravaca Perez, P.; Llagostera, M.; Morán-Fernández, L.; Fort, A.; de Juan Bagudá, J.; García-Cosio, M.D.; Ruiz-Bustillo, S.; Delgado, J.F. Dose of furosemide before admission predicts diuretic efficiency and long-term prognosis in acute heart failure. ESC Heart Fail. 2022, 9, 656–666. [Google Scholar] [CrossRef]
- Tuttolomondo, A.; Pinto, A.; Parrinello, G.; Licata, G. Intravenous high-dose furosemide and hypertonic saline solutions for refractory heart failure and ascites. Semin. Nephrol. 2011, 31, 513–522. [Google Scholar] [CrossRef]
- Li, Y.; Tang, X.; Zhang, J.; Wu, T. Nutritional support for acute kidney injury. Cochrane Db Syst. Rev. 2010, 1, Cd005426. [Google Scholar]
- Walter, S.; Karl, S.; Wilhelm, S.; Rudi, W. Process for the Preparation of Sulfamylanthranilic Acids. DE 1277860B, 19 September 1968. [Google Scholar]
- Walter, S.; Karl, S. Process for the Preparation of Sulfamylanthranilic Acids. DE 1220436B, 7 July 1966. [Google Scholar]
- Walter, S.; Karl, S.; Rudi, W. Process for the Preparation of Sulfamylanthranilic Acids. DE 1213846B, 4 April 1966. [Google Scholar]
- Karl, S.; Helmut, N.; Walter, S. Diuretic Sulphamylantranilic Acids. DE 1806581 A1, 3 June 1971. [Google Scholar]
- Yellin, H.; Konfino, E. Process for the Preparation of 4-Chloro-n-furfuryl-5-sulfamoyl-anthranilic Acid. US 3780067A, 18 December 1973. [Google Scholar]
- Wang, Z.; Yang, Y.S.; Liu, J.Z.; Shen, S.S.; Zhang, X.Y. A Kind of Preparation Method of Furosemide. CN 106117168A, 16 November 2016. [Google Scholar]
- Chen, B.; Gao, Z.Q.; Liu, Y.; Zheng, Y.M.; Han, Y.; Zhang, J.P.; Hu, C.Q. Embryo and Developmental Toxicity of Cefazolin Sodium Impurities in Zebrafish. Front. Pharmacol. 2017, 8, 403. [Google Scholar] [CrossRef] [Green Version]
- Sigvardson, K.; Adams, S.; Barnes, T.; Blom, K.; Fortunak, J.; Haas, M.; Reilly, K.; Repta, A.; Nemeth, G. The isolation and identification of a toxic impurity in XP315 drug substance. J. Pharm. Biomed. Anal. 2002, 27, 327–334. [Google Scholar] [CrossRef]
- International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Harmonised Tripartite Guideline: Impurities in New Drug Substances Q3A (R2). Current Step 4 Version. 24 October 2006. Available online: https://www.ema.europa.eu/en/ich-q3a-r2-impurities-new-drug-substances-scientific-guideline (accessed on 10 February 2023).
- EP Monograph on Furosemide, EP 10.0. 2020, pp. 2718–2719. Available online: https://www.drugfuture.com/standard/search.aspx/ (accessed on 10 February 2023).
- Miller, J.M.; Robert, J.; Sørensen, A. Reversed-phase ion-pair liquid chromatographic method for determining the impurities of furosemide. J. Pharm. Biomed. Anal. 1993, 11, 257–261. [Google Scholar] [CrossRef]
- Santos, C.A.D.; Mazzola, P.G.; Polacwiecz, B.; Knirsch, M.C.; Cholewa, O.; Penna, T.C.V. Stability of furosemide and aminophylline in parenteral solutions. Brazilian J. Pharm. Sci. 2011, 47, 89–96. [Google Scholar]
- Chen, L.-J.; Burka, L.T. Chemical and enzymatic oxidation of furosemide: Formation of pyridinium salts. Chem. Res. Toxicol. 2007, 20, 1741–1744. [Google Scholar] [CrossRef] [PubMed]
- Ghanekar, A.; Gibbs, C.W., Jr. Stability of furosemide in aqueous systems. J. Pharm Sci 1978, 67, 808–811. [Google Scholar] [CrossRef]
- Bundgaard, H.; Nørgaard, T.; Nielsen, N.M. Photodegradation and hydrolysis of furosemide and furosemide esters in aqueous solutions. Int. J. Pharm. 1988, 42, 217–224. [Google Scholar] [CrossRef]
- Kurmi, M.; Kumar, S.; Singh, B.; Singh, S. Implementation of design of experiments for optimization of forced degradation conditions and development of a stability-indicating method for furosemide. J. Pharm. Biomed. Anal. 2014, 96, 135–143. [Google Scholar] [CrossRef] [PubMed]
- USP Monograph on Furosemide, USP43-NF38. 2020, p. 2053. Available online: https://www.uspnf.com/errata/furosemide-tablets-2020-08-01 (accessed on 10 February 2023).
- Mandrioli, M.; Tura, M.; Scotti, S.; Gallina Toschi, T. Fast Detection of 10 Cannabinoids by RP-HPLC-UV Method in Cannabis sativa L. Molecules 2019, 24, 2113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuang, T.; Chen, Y.; Xu, J.; Qi, Z.; Ye, J.; Xu, C.; Du, W.; Liu, B.; Zhang, G. Isolation, structural characterization and quality control strategy of an unknown process-related impurity in sugammadex sodium. J. Pharm. Biomed. Anal. 2021, 200, 114072. [Google Scholar] [CrossRef]
- Zhuang, T.; Zhang, W.; Cao, L.; He, K.; Wang, Y.; Li, J.; Chen, L.; Liu, B.; Zhang, G. Isolation, identification and characterization of two novel process-related impurities in olanzapine. J. Pharm. Biomed. Anal. 2018, 152, 188–196. [Google Scholar] [CrossRef]
- Zhuang, T.; Jin, J.; Ma, Y.; Ren, X.; Liang, L.; Du, W.; Liu, B.; Liu, X.; Zhang, G. Structural elucidation and synthesis of a dimeric degradation impurity during long-term stability studies of oxycodone hydrochloride injection. New J. Chem. 2021, 45, 18651–18658. [Google Scholar] [CrossRef]
- Häussermann, A.; Rominger, F.; Straub, B.F. CO2 on a tightrope: Stabilization, room-temperature decarboxylation, and sodium-induced carboxylate migration. Chem. Eur. J. 2012, 18, 14174–14185. [Google Scholar] [CrossRef]
- Lee, D.; Chang, S. Direct C-H amidation of benzoic acids to introduce meta- and para-amino groups by tandem decarboxylation. Chem. Eur. J. 2015, 21, 5364–5368. [Google Scholar] [CrossRef]
- International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Harmonised Tripartite Guideline: Validation of Analytical Procedures: Text and Methodology Q2 (R1). Current Step 4 Version. November 2005. Available online: https://www.ich.org/page/quality-guidelines (accessed on 10 February 2023).
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018, 46, 257–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouback, T.A.; Pokhrel, S.; Albeshri, A.; Aljohani, A.M.; Samad, A.; Alam, R.; Hossen, M.S.; Al-Ghamdi, K.; Talukder, M.E.K.; Ahammad, F.; et al. Pharmacophore-Based Virtual Screening, Quantum Mechanics Calculations, and Molecular Dynamics Simulation Approaches Identified Potential Natural Antiviral Drug Candidates against MERS-CoV S1-NTD. Molecules 2021, 26, 4961. [Google Scholar] [CrossRef] [PubMed]
- Ślusarczyk, S.; Senol Deniz, F.S.; Abel, R.; Pecio, Ł.; Pérez-Sánchez, H.; Cerón-Carrasco, J.P.; den-Haan, H.; Banerjee, P.; Preissner, R.; Krzyżak, E.; et al. Norditerpenoids with Selective Anti-Cholinesterase Activity from the Roots of Perovskia atriplicifolia Benth. Int. J. Mol. Sci. 2020, 21, 4475. [Google Scholar] [CrossRef] [PubMed]
- Rolta, R.; Yadav, R.; Salaria, D.; Trivedi, S.; Imran, M.; Sourirajan, A.; Baumler, D.J.; Dev, K. In silico screening of hundred phytocompounds of ten medicinal plants as potential inhibitors of nucleocapsid phosphoprotein of COVID-19: An approach to prevent virus assembly. J. Biomol. Struct. Dyn. 2021, 39, 7017–7034. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | δH (ppm) | δC (ppm) | DEPT | 1H-1H COSY | HMBC |
|---|---|---|---|---|---|
| 1 | 7.58 (d, J = 1.0 Hz, 1H) | 142.5 | CH | H2(3JHH) | C3(3JCH), C4(3JCH) |
| 2 | 6.38 (dd, J = 3.0, 1.9 Hz, 1H) | 110.8 | CH | H1(3JHH), H3(3JHH) | C4(3JCH), C3(2JCH) |
| 3 | 6.28 (d, J = 3.1 Hz, 1H) | 107.6 | CH | H2(3JHH) | C1(3JCH), C2(2JCH), C4(2JCH) |
| 4 | – | 153.2 | – | – | – |
| 5 | 4.27 (d, J = 4.6 Hz, 2H) | 39.8 | CH2 | – | C3(3JCH), C4(2JCH) |
| 6 | 6.22 (t, J = 5.4 Hz, 1H) | – | – | – | C12(3JCH), C8(3JCH), C4(3JCH), C5(2JCH) |
| 7 | – | 113.8 | – | – | – |
| 8 | 6.02 (d, J = 3.0 Hz, 1H) | 101.2 | CH | H9(3JHH) | C4(5JCH), C12(3JCH), C7(2JCH), C9(2JCH) |
| 9 | 7.35 (d, J = 9.2 Hz, 1H) | 130.4 | CH | H8(3JHH) | C15(6JCH), C12(4JCH), C11(3JCH), C10(2JCH) |
| 10 | – | 146.4 | – | – | – |
| 11 | – | 113.9 | – | – | – |
| 12 | 6.01 (d, J = 2.2 Hz, 1H) | 94.0 | – | – | C15(5JCH), C9(4JCH), C8(3JCH), C7(2JCH) |
| 13 | 6.62 (t, J = 5.2 Hz, 1H) | – | CH | – | C11(3JCH), C15(3JCH), C14(2JCH), C12(2JCH) |
| 14 | 4.34 (d, J = 4.6 Hz, 2H) | 40.2 | CH2 | – | C10(4JCH), C16(3JCH), C15(2JCH) |
| 15 | – | 152.9 | – | – | – |
| 16 | 6.34 (d, J = 3.1 Hz, 1H) | 107.5 | CH | H17(3JHH) | C18(3JCH), C15(2JCH), C17(2JCH) |
| 17 | 6.43 (dd, J = 3.0, 1.9 Hz, 1H) | 110.9 | CH | H18(3JHH), H16(3JHH) | C15(3JCH), C16(2JCH), C18(2JCH) |
| 18 | 7.60 (d, J = 0.9 Hz, 1H) | 142.7 | CH | H17(3JHH) | C15(3JCH), C16(3JCH) |
| 20 | 6.92 (s, 2H) | – | – | – | – |
| Compound | Relative Retention Time (RRT) | Resolution | Theoretical Plates | Symmetry Factor |
|---|---|---|---|---|
| Furosemide | 1.00 | 6.517 | 71,898 | 1.087 |
| Imp-A | 1.09 | 4.890 | 120,398 | 1.097 |
| Imp-B | 0.31 | 34.377 | 12,953 | 0.952 |
| Imp-C | 0.24 | – | 5670 | 0.931 |
| Imp-D | 1.49 | 5.815 | 100,376 | 1.089 |
| Imp-E | 1.16 | 9.637 | 97,672 | 1.129 |
| Imp-F | 0.73 | 18.664 | 44,339 | 1.097 |
| Imp-G | 1.29 | 13.078 | 174,284 | 1.119 |
| Compound | LOD | LOQ | |||||
|---|---|---|---|---|---|---|---|
| µg/mL | % | S/N | µg/mL | % | S/N | RSD (%) | |
| Furosemide | 0.012 | 0.0012 | 4.11 | 0.061 | 0.0061 | 14.27 | 0.76 |
| Imp-A | 0.020 | 0.0020 | 3.59 | 0.099 | 0.0099 | 17.82 | 0.78 |
| Imp-B | 0.019 | 0.0019 | 2.70 | 0.097 | 0.0097 | 13.02 | 1.48 |
| Imp-C | 0.005 | 0.0005 | 2.63 | 0.024 | 0.0024 | 11.82 | 0.35 |
| Imp-D | 0.098 | 0.0098 | 3.07 | 0.488 | 0.0488 | 16.18 | 1.04 |
| Imp-E | 0.096 | 0.0044 | 3.27 | 0.222 | 0.0222 | 16.37 | 0.65 |
| Imp-F | 0.019 | 0.0019 | 4.14 | 0.097 | 0.0097 | 19.46 | 0.58 |
| Imp-G | 0.020 | 0.0020 | 4.33 | 0.059 | 0.0059 | 13.04 | 1.61 |
| Compound | Concentration (µg/mL) | Correlation Coefficient (r) | Regression Equation |
|---|---|---|---|
| Furosemide | 0.100–1.998 | 0.9999 | y = 59,180 x + 6139 |
| Imp-A | 0.146–2.917 | 1.0000 | y = 66,308 x − 59 |
| Imp-B | 0.150–2.993 | 1.0000 | y = 39,245 x + 47 |
| Imp-C | 0.146–2.911 | 1.0000 | y = 130,336 x + 325 |
| Imp-D | 0.148–2.960 | 1.0000 | y = 20,195 x − 124 |
| Imp-E | 0.149–2.985 | 1.0000 | y = 30,029 x + 1 |
| Imp-F | 0.145–2.904 | 1.0000 | y = 82,955 x + 332 |
| Imp-G | 0.153–3.057 | 1.0000 | y = 56,489 x + 236 |
| Compound | Accuracy | Repeatability | Intermediate Precision | |||
|---|---|---|---|---|---|---|
| 50% | 100% | 150% | RSD% (n = 9) | RSD% (n = 6) | RSD% (n = 6) | |
| Imp-A | 101.4 | 101.2 | 101.7 | 0.30 | 2.82 | 2.59 |
| Imp-B | 102.2 | 102.1 | 102.0 | 0.47 | 2.88 | 2.70 |
| Imp-C | 102.1 | 102.0 | 102.0 | 0.33 | 2.88 | 1.42 |
| Imp-D | 105.5 | 102.8 | 103.6 | 1.50 | 2.86 | 3.94 |
| Imp-E | 101.6 | 101.3 | 102.1 | 0.52 | 2.82 | 1.93 |
| Imp-F | 102.3 | 102.0 | 102.0 | 0.33 | 2.90 | 4.26 |
| Imp-G | 103.5 | 104.8 | 104.1 | 2.27 | 2.48 | 2.43 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, A.; Xue, Y.; Zeng, Y.; Li, J.; Zhou, H.; Wang, Z.; Chen, Y.; Chen, H.; Jin, J.; Zhuang, T. Isolation and Characterization of an Unknown Process-Related Impurity in Furosemide and Validation of a New HPLC Method. Molecules 2023, 28, 2415. https://doi.org/10.3390/molecules28052415
Xu A, Xue Y, Zeng Y, Li J, Zhou H, Wang Z, Chen Y, Chen H, Jin J, Zhuang T. Isolation and Characterization of an Unknown Process-Related Impurity in Furosemide and Validation of a New HPLC Method. Molecules. 2023; 28(5):2415. https://doi.org/10.3390/molecules28052415
Chicago/Turabian StyleXu, Ao, Yunlin Xue, Yuyu Zeng, Jing Li, Huiling Zhou, Zhen Wang, Yin Chen, Hui Chen, Jian Jin, and Tao Zhuang. 2023. "Isolation and Characterization of an Unknown Process-Related Impurity in Furosemide and Validation of a New HPLC Method" Molecules 28, no. 5: 2415. https://doi.org/10.3390/molecules28052415