

Thermal Polymorphism in CsCB11H12
 , , , , , ,
, , , , , ,
Abstract
:
1. Introduction
2. Results
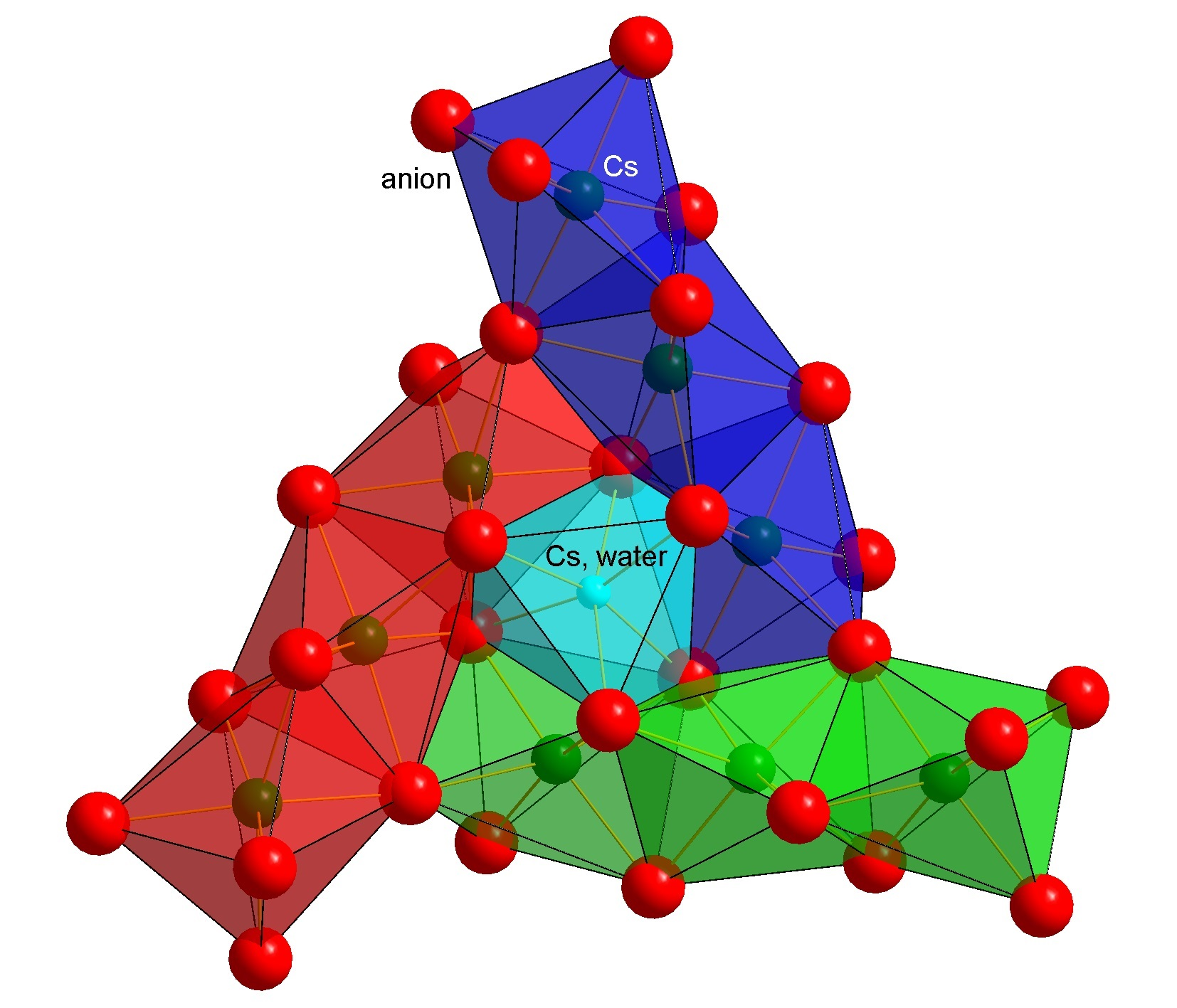
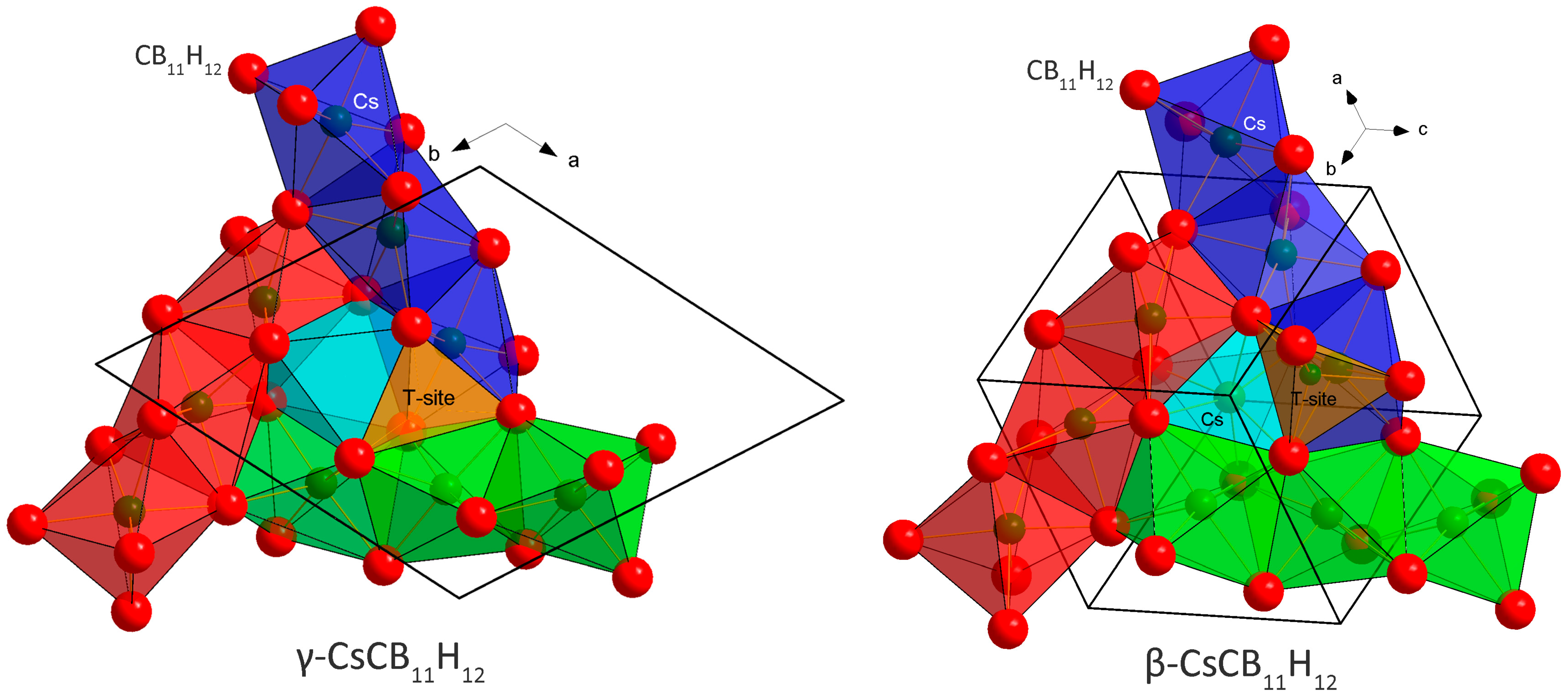
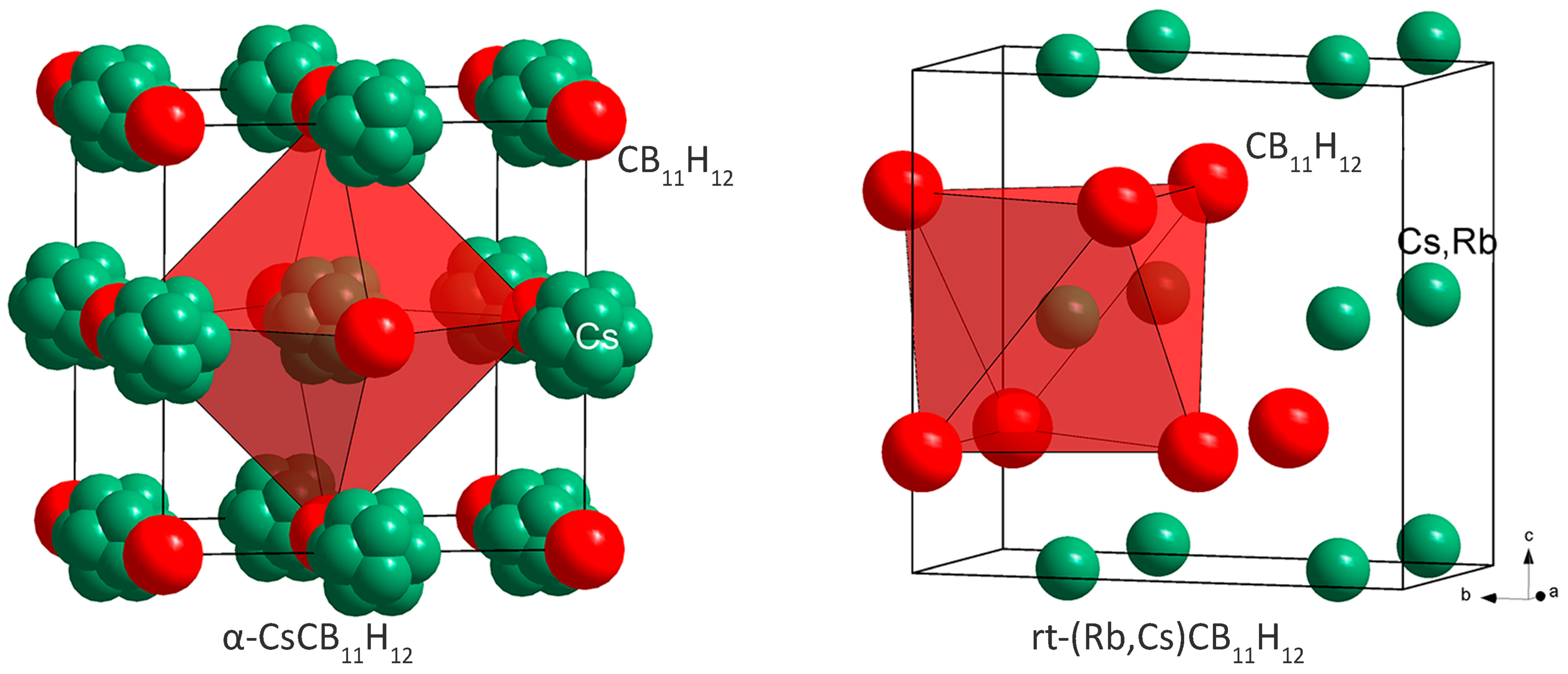
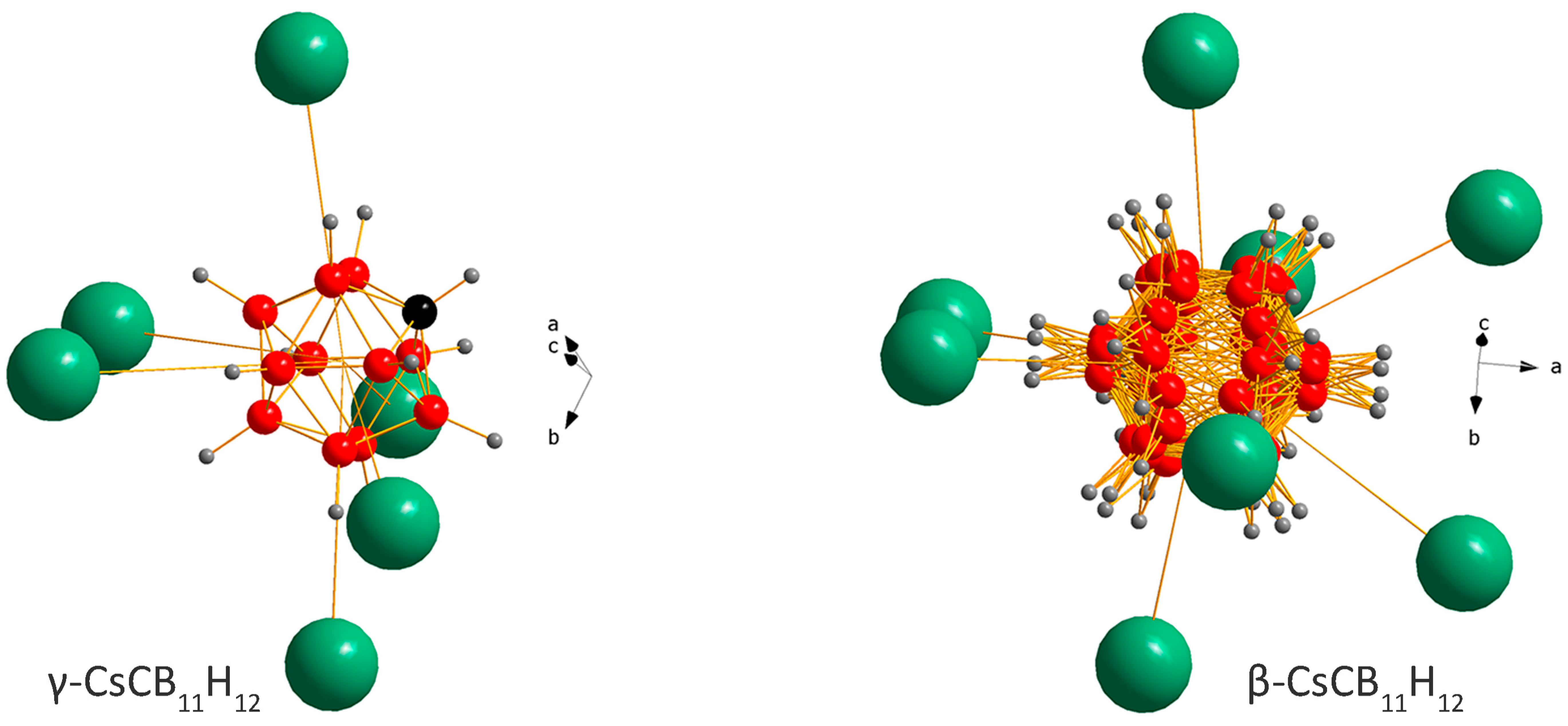
2.1. Crystal Structures and Thermal Polymorphism
2.1.1. CsCB11H12
2.1.2. Cs0.93Rb0.07CB11H12
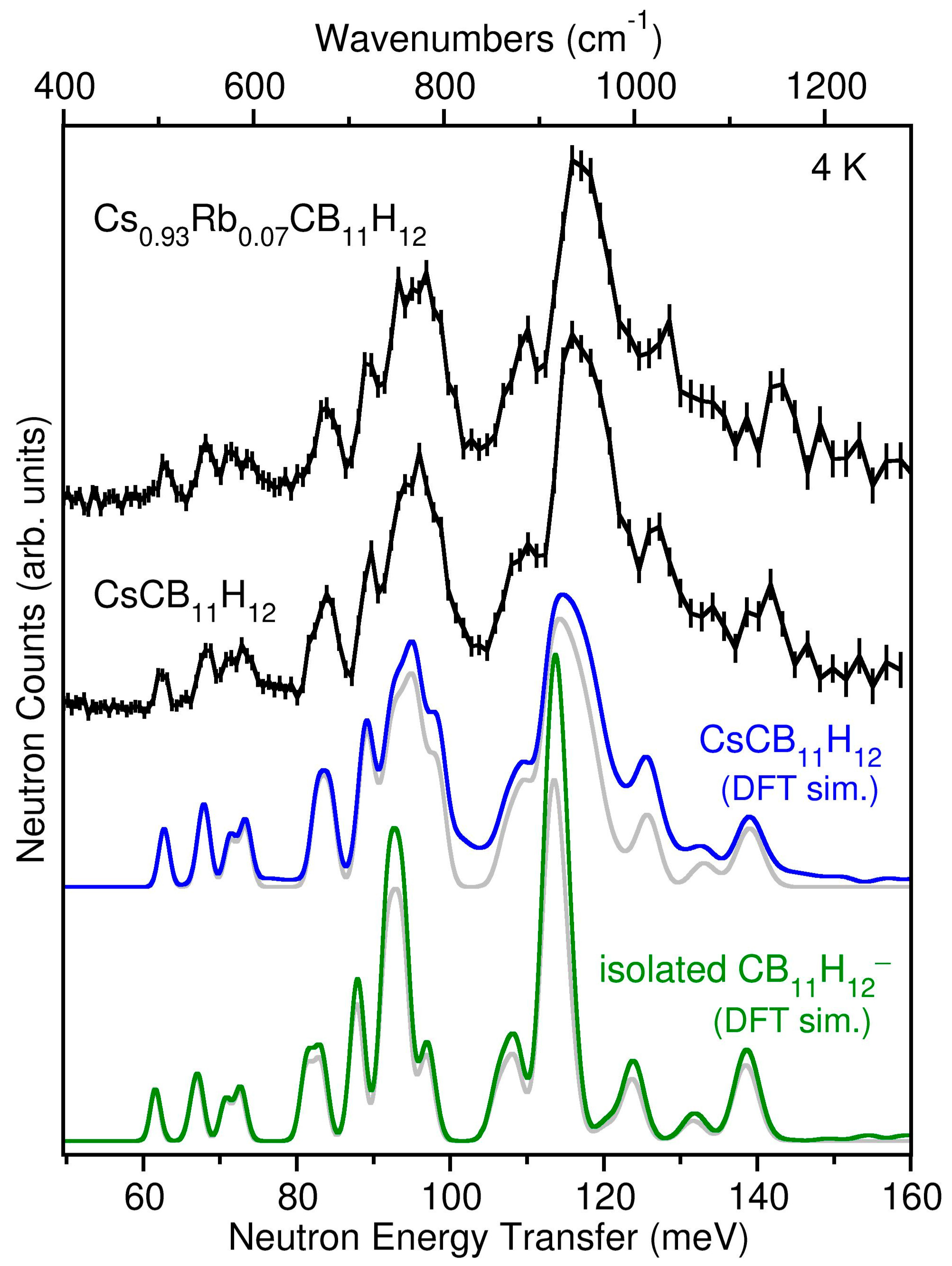
2.2. Anion Dynamics
3. Discussion
3.1. Metastable and Stable Polymorphs
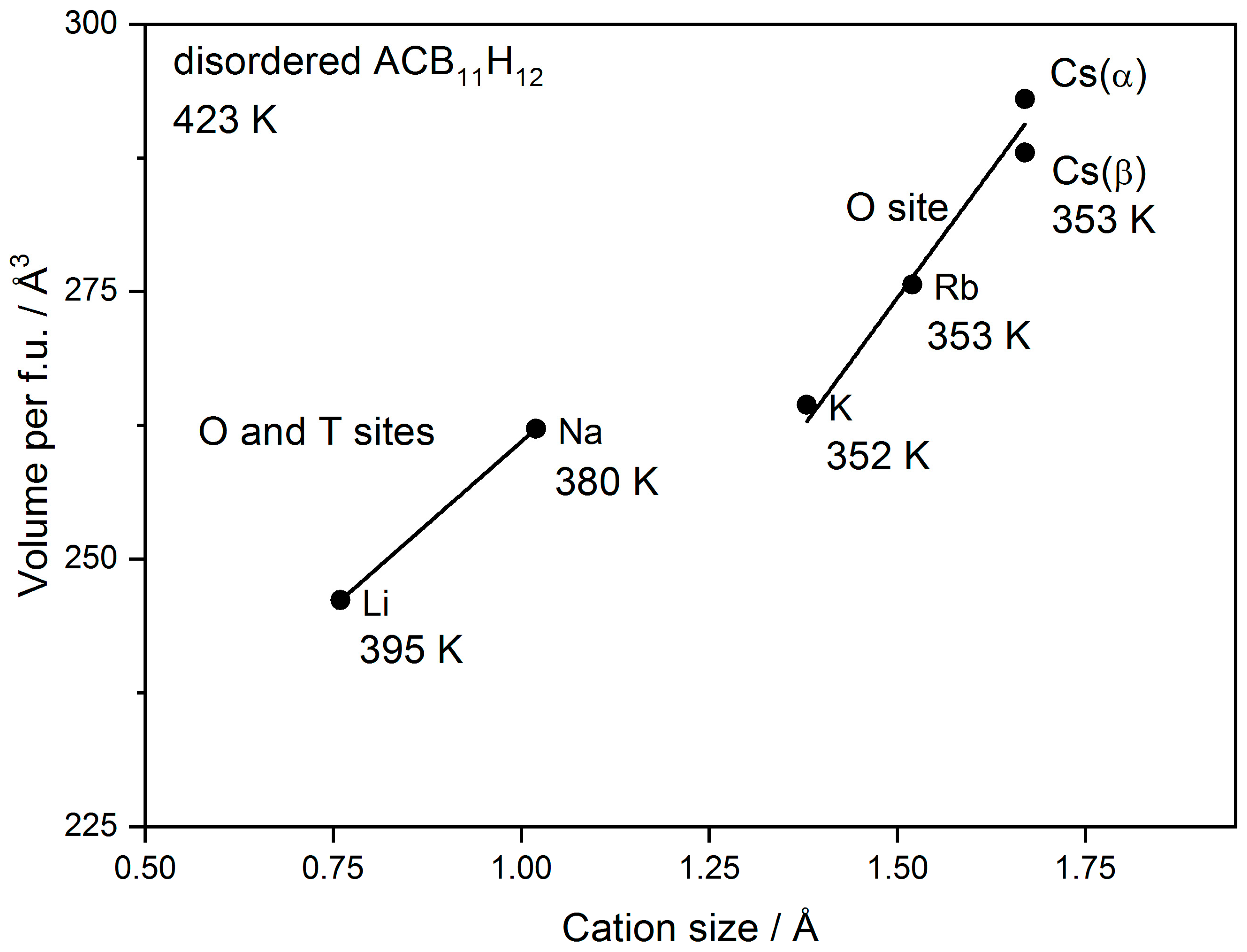
3.2. Cation Coordination in Alkali-Metal Carba-Hydridoborates
4. Materials and Methods
4.1. Samples Preparation
4.2. Synchrotron Radiation X-ray Powder Diffraction (SR-XPD)
4.3. Differential Scanning Calorimetry (DSC)
4.4. Raman Spectroscopy
4.5. Fourier Transformation Infrared Spectroscopy (FTIR)
4.6. Neutron Scattering Measurements
4.7. Density Functional Theory (DFT) Calculations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Grimes, R.N. Carboranes; Elsevier: Burlington, VT, USA, 2011; pp. 1–1107. [Google Scholar]
- Tang, W.S.; Unemoto, A.; Zhou, W.; Stavila, V.; Matsuo, M.; Wu, H.; Orimo, S.-I.; Udovic, T.J. Unparalleled lithium and sodium superionic conduction in solid electrolytes with large monovalent cage-like anions. Energy Environ. Sci. 2015, 8, 3637–3645. [Google Scholar] [CrossRef] [Green Version]
- Brighi, M.; Murgia, F.; Černý, R. Closo-hydroborate sodium salts: An emerging class of room-temperature solid electrolytes. Cell Rep. Phys. Sci. 2020, 1, 100217d. [Google Scholar] [CrossRef]
- Timmermans, J. Plastic Crystals: A Historical Overview. J. Phys. Chem. Solids 1961, 18, 1–8. [Google Scholar] [CrossRef]
- Skripov, A.V.; Soloninin, A.V.; Babanova, O.A.; Skoryunov, R.V. Anion and Cation Dynamics in Polyhydroborate Salts: NMR Studies. Molecules 2020, 25, 2940. [Google Scholar] [CrossRef] [PubMed]
- Murgia, F.; Brighi, M.; Piveteau, L.; Avalos, C.E.; Gulino, V.; Nierstenhöfer, M.C.; Ngene, P.; de Jongh, P.; Černý, R. Enhanced Room-Temperature Ionic Conductivity of NaCB11H12 via High-Energy Mechanical Milling. Appl. Mater. Interfaces 2021, 13, 61346–61356. [Google Scholar] [CrossRef]
- Dimitrievska, M.; Wu, H.; Stavila, V.; Babanova, O.A.; Skoryunov, R.V.; Soloninin, A.V.; Zhou, W.; Trump, B.A.; Andersson, M.S.; Skripov, A.V.; et al. Structural and Dynamical Properties of Potassium Dodecahydro-monocarba-closo-dodecaborate: KCB11H12. J. Phys. Chem. C 2020, 124, 17992–18002. [Google Scholar] [CrossRef]
- Bareiss, K.U.; Friedly, A.; Schleid, T. The unexpected crystal structure of cesium dodecahydro-monocarba-closo-dodecaborate Cs[CB11H12]. Z. Naturforsch. 2020, 75, 1049–1059. [Google Scholar] [CrossRef]
- Romerosa, A.M. Thermal, structural and possible ionic-conductor behaviour of CsB10CH13 and Cs B11CH12. Thermochim. Acta 1993, 217, 123–128. [Google Scholar] [CrossRef]
- Romerosa, A.M. Thermal, Structural and Possible Ionic-Conductor Behaviour of CsB10CH13 and Cs B11CH12. Ph.D. Thesis, University of Barcelona, Barcelona, Spain, 1992. [Google Scholar]
- Meisel, K. Kristallstrukturen von Thoriumphosphiden. Z. Anorg. Allg. Chem. 1939, 240, 300–312. [Google Scholar] [CrossRef]
- Jorgensen, M.; Zhou, W.; Wu, H.; Udovic, T.J.; Paskevicius, M.; Černý, R.; Jensen, T.R.J. Polymorphism of Calcium Decahydrido-closo-decaborate and Characterisation of its Hydrates. Inorg. Chem. 2021, 60, 10943–10957. [Google Scholar] [CrossRef]
- Her, J.H.; Wu, H.; Verdal, N.; Zhou, W.; Stavila, V.; Udovic, T.J. Structures of the Strontium and Barium Dodecahydro-closo-Dodecaborates. J. Alloys Compd. 2012, 514, 71–75. [Google Scholar] [CrossRef]
- Verdal, N.; Zhou, W.; Stavila, V.; Her, J.-H.; Yousufuddin, M.; Yildirim, T.; Udovic, T.J. Alkali and Alkaline-Earth Metal Dodecahydro-closo-Dodecaborates: Probing Structural Variations via Neutron Vibrational Spectroscopy. J. Alloys Compds. 2011, 509S, S694–S697. [Google Scholar] [CrossRef]
- V_Sim, Open-Source Software. Available online: https://gitlab.com/l_sim/v_sim-website/-/tree/master/download (accessed on 5 January 2023).
- Dimitrievska, M.; Shea, P.; Kweon, K.E.; Bercx, M.; Varley, J.B.; Tang, W.S.; Skripov, A.V.; Stavila, V.; Udovic, T.J.; Wood, B.C. Carbon Incorporation and Anion Dynamics as Synergistic Drivers for Ultrafast Diffusion in Superionic LiCB11H12 and NaCB11H12. Adv. Energy Mater. 2018, 8, 1703422. [Google Scholar] [CrossRef]
- Verdal, N.; Wu, H.; Udovic, T.J.; Stavila, V.; Zhou, W.; Rush, J.J. Evidence of a Transition to Reorientational Disorder in the Cubic Alkali-Metal Dodecahydro-closo-Dodecaborates. J. Solid State Chem. 2011, 184, 3110–3116. [Google Scholar] [CrossRef]
- Verdal, N.; Udovic, T.J.; Rush, J.J.; Cappelletti, R.; Zhou, W. Reorientational Dynamics of the Dodecahydro-Closo-Dodecaborate Anion in Cs2B12H12. J. Phys. Chem. A 2011, 115, 2933–2938. [Google Scholar] [CrossRef]
- Rius, J.; Romerosa, A.; Teixidor, F.; Casabó, J.; Miravitllest, C. Phase Transitions in Cesium 7,8-Dicarbaundecaborate(12): A New One-Dimensional Cesium Solid Electrolyte at 210 °C. Inorg. Chem. 1991, 30, 1376–1379. [Google Scholar] [CrossRef]
- Tiritiris, I.; Schleid, T. Die Kristallstrukturen der Dicaesium-Dodekahalogeno-closo-Dodekaborate Cs2[B12X12] (X = Cl, Br, I) und ihrer Hydrate. Z. Anorg. Allg. Chem. 2004, 630, 1555–1563. [Google Scholar] [CrossRef]
- Peryshkov, D.V.; Bukovsky, E.V.; Lacroix, M.R.; Wu, H.; Zhou, W.; Jones, W.M.; Lozinšek, M.; Folsom, T.C.; Heyliger, D.L.; Udovic, T.J.; et al. Latent Porosity in Alkali Metal M2B12F12 Salts: Structures and Rapid Room-Temperature Hydration/Dehydration Cycles. Inorg. Chem. 2017, 56, 12023–12041. [Google Scholar] [CrossRef]
- Ostwald, W. Studien über die Bildung und Umwandlung fester Körper. 1. Abhandlung: Übersättigung und Überkaltung. Z. Phys. Chem. 1879, 22, 289–330. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. A 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Toby, B.H.; Von Dreele, R.B. GSAS-II: The Genesis of a Modern Open-Source All Purpose Crystallography Software Package. J. Appl. Crystallogr. 2013, 46, 544–549. [Google Scholar] [CrossRef]
- Favre-Nicolin, V.; Černý, R. FOX, “Free Objects for Crystallography”: A modular approach to ab initio structure determination from powder diffraction. J Appl. Crystallogr. 2002, 35, 734–743. [Google Scholar] [CrossRef] [Green Version]
- Coelho, A.A. Whole-profile structure solution from powder diffraction data using simulated annealing. J. Appl. Crystallogr. 2000, 33, 899–908. [Google Scholar] [CrossRef]
- Larson, A.C.; Von Dreele, R.B. General Structure Analysis System; Report LAUR 86–748; Los Alamos National Laboratory: Los Alamos, NM, USA, 1994. [Google Scholar]
- Putz, H.; Brandenburg, K. (Eds.) Diamond-Crystal and Molecular Structure Visualization; Crystal Impact: Bonn, Germany.
- Udovic, T.J.; Brown, C.M.; Leão, J.B.; Brand, P.C.; Jiggetts, R.D.; Zeitoun, R.; Pierce, T.A.; Peral, I.; Copley, J.R.D.; Huang, Q.; et al. The Design of a Bismuth-based Auxiliary Filter for the Removal of Spurious Background Scattering Associated with Filter-Analyzer Neutron Spectrometers. Nucl. Instr. Meth. A 2008, 588, 406–413. [Google Scholar] [CrossRef]
- Copley, J.R.D.; Cook, J.C. The Disk Chopper Spectrometer at NIST: A New Instrument for Quasielastic Neutron Scattering Studies. Chem. Phys. 2003, 292, 477–485. [Google Scholar] [CrossRef]
- Azuah, R.T.; Kneller, L.R.; Qiu, Y.; Tregenna-Piggott, P.L.W.; Brown, C.M.; Copley, J.R.D.; Dimeo, R.M. DAVE: A Comprehensive Software Suite for the Reduction, Visualization, and Analysis of Low Energy Neutron Spectroscopic Data. J. Res. Natl. Inst. Stan. 2009, 114, 341–358. [Google Scholar] [CrossRef]
- Paul, R.L.; Sahin, D.; Cook, J.C.; Brocker, C.; Lindstrom, R.M.; O’Kelly, D.J. NGD Cold-Neutron Prompt Gamma-Ray Activation Analysis Spectrometer at NIST. J. Radioanal. Nucl. Chem. 2015, 304, 189–193. [Google Scholar] [CrossRef]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A Modular and Open-Source Software Project for Quantum Simulations of Materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmuller, J.; Hafner, J. Ab initio Force Constant Approach to Phonon Dispersion Relations of Diamond and Graphite. Europhys. Lett. 1995, 32, 729–734. [Google Scholar] [CrossRef]
- Yildirim, T. Structure and Dynamics from Combined Neutron Scattering and First-Principles Studies. Chem. Phys. 2000, 261, 205–216. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymorph | s.g. | a [Å] | c [Å] | V [Å3] | Z | Texp [K] |
|---|---|---|---|---|---|---|
| γ | R3 | 20.9533 (1) | 13.2400 (1) | 5034.1 (1) | 18 | 303 |
| 20.7818 (2) | 13.0935 (2) | 4897.3 (1) | 156 | |||
| β | I43d | 15.1493 (6) | 3476.8 (4) | 12 | 423 | |
| α | Fm3 | 10.6491 (9) | 1207.7 (3) | 4 | 523 | |
| α′ | P63mc | 7.3987 (5) | 12.5639 (3) | 595.5 (2) | 2 | 539 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Černý, R.; Brighi, M.; Wu, H.; Zhou, W.; Dimitrievska, M.; Murgia, F.; Gulino, V.; de Jongh, P.E.; Trump, B.A.; Udovic, T.J. Thermal Polymorphism in CsCB11H12. Molecules 2023, 28, 2296. https://doi.org/10.3390/molecules28052296
Černý R, Brighi M, Wu H, Zhou W, Dimitrievska M, Murgia F, Gulino V, de Jongh PE, Trump BA, Udovic TJ. Thermal Polymorphism in CsCB11H12. Molecules. 2023; 28(5):2296. https://doi.org/10.3390/molecules28052296
Chicago/Turabian StyleČerný, Radovan, Matteo Brighi, Hui Wu, Wei Zhou, Mirjana Dimitrievska, Fabrizio Murgia, Valerio Gulino, Petra E. de Jongh, Benjamin A. Trump, and Terrence J. Udovic. 2023. "Thermal Polymorphism in CsCB11H12" Molecules 28, no. 5: 2296. https://doi.org/10.3390/molecules28052296