3.2.1. Synthesis
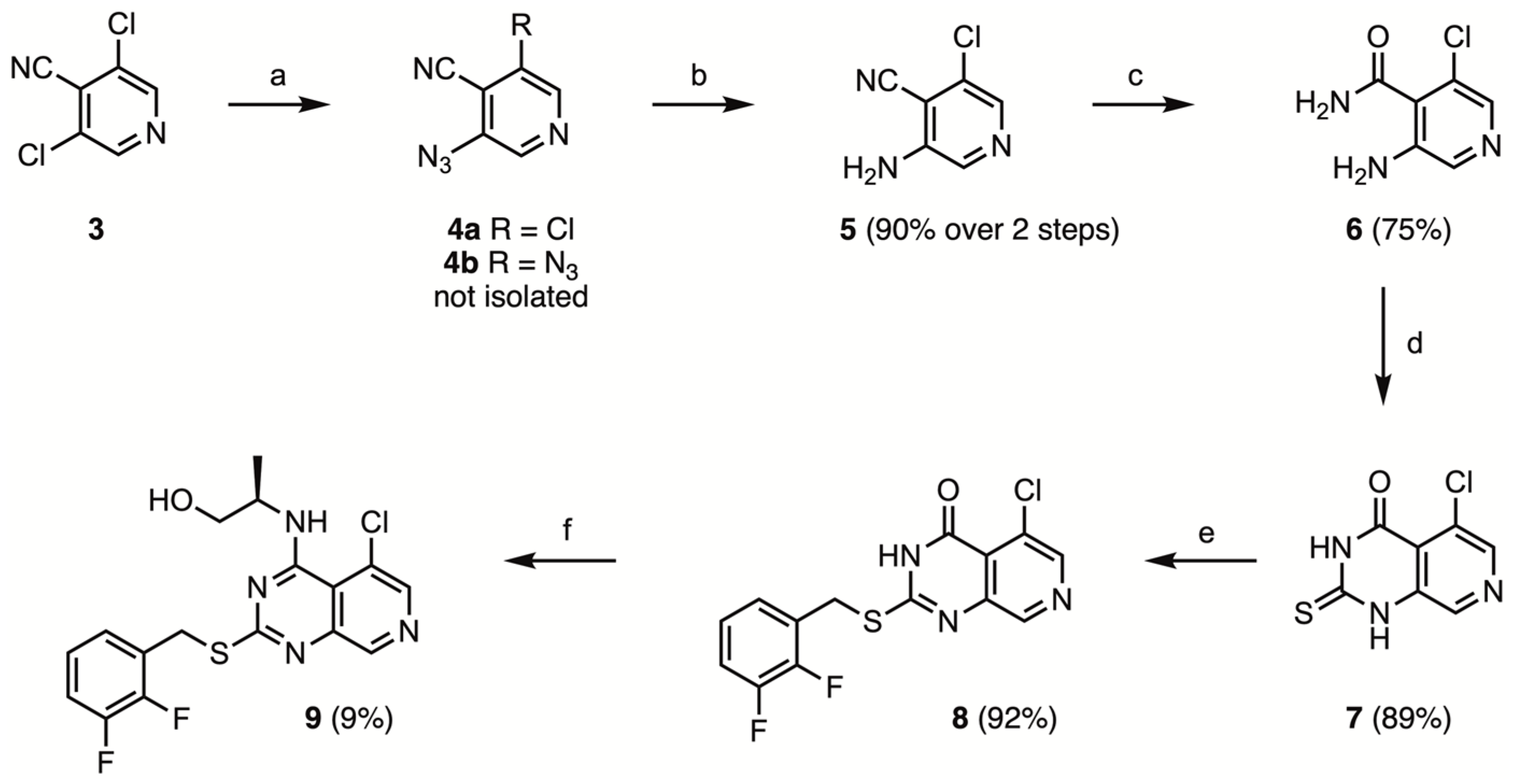
3-Amino-5-chloropyridine-4-carbonitrile (5)
A mixture of 3,5-dichloropyridine-4-carbonitrile 3 (13.76 g, 80.0 mmol, 1.00 eq.) and NaN3 (5.72 g, 8.80 mmol, 1.10 eq.) in DMF (100 mL) was heated to 80 °C for 19 h. After cooling to room temperature, the reaction mixture was transferred to a separatory funnel together with H2O (200 mL) and EtOAc (200 mL). After phase separation the aqueous phase was extracted with EtOAc (2 × 200 mL). The combined organic phases were washed with water (2 × 200 mL) and brine (2 × 200 mL), dried over Na2SO4 and coated on Celite. Filtration over silica using 30% EtOAc/PE gave the crude intermediated product 4a, which was used without further purification in the next step. The obtained solid was dissolved ACN (600 mL) and NaI (107 g, 720 mmol, 9.00 eq.) was added. The suspension was cooled in an ice bath and FeCl3 (19.4 g, 120 mmol, 1.50 eq.) was added. After stirring for 10 min at 0 °C, the mixture was stirred for 30 min at room temperature, when full conversion was observed by TLC. The reaction mixture was transferred to a separatory funnel along with EtOAc (200 mL) and water (100 mL) and quenched with Na2S2O3. After phase separation, the aqueous phase was extracted with EtOAc (2 × 200 mL). The combined organic phases were washed with H2O (2 × 100 mL) and brine (2 × 100 mL), dried over Na2SO4 and filtered over silica. The obtained brown solid was recrystallized twice from methanol to afford the title compound (11.041 g, 71.89 mmol, 90%) as an off-white solid. mp 170—172 °C.
1H NMR (400 MHz, d6-DMSO): δ (ppm) 8.14 (s, 1H), 7.85 (s, 1H), 6.85 (s, 2H); 13C NMR (101 MHz, d6-DMSO): δ (ppm) 147.9, 137.5, 133.8, 130.8, 113.5, 98.7.
3-Amino-5-chloropyridine-4-carboxamide (6)
3-Amino-5-chloropyridine-4-carbonitrile (9.55 g, 62.2 mmol, 1.00 eq.), which had been finely ground with a pestle and mortar, was added to solution of 50% aqueous CsOH (1.4 mL, 7.8 mmol, 13 mol%) in 20% aqueous ammonia (80 mL) in a 100 mL screw-capped reaction tube equipped with a stir bar. After heating to 100 °C for 1 h, all material had dissolved, and the mixture was cooled to 0 °C in an ice bath. The resulting white crystalline precipitate was filtered and washed with ice water and dried under vacuum (40 °C) to afford the title compound as a white solid (7.957 g, 46.37 mmol, 75%).
1H NMR (400 MHz, d6-DMSO): δ (ppm) 7.99 (s, 1H), 7.82 (br. s, 2H), 7.77 (s, 1H), 5.50 (br. s, 2H); 13C NMR (101 MHz, d6-DMSO): δ (ppm) 165.5, 142.3, 136.2, 135.3, 127.1, 126.7.
2-Thioxo-5-chloropyrido[3,4-d]pyrimidine-4-one (7)
DBU (5.0 mL, 33.5 mmol, 2.3 eq.) was added dropwise to a suspension of 2-chloro-5-aminopyridine-4-carboxamide (2.50 g, 14.6 mmol, 1.00 eq.) in dry DMF (25 mL) and CS2 (5.0 mL, 83 mmol, 5.7 eq.), under N2-atmosphere, while cooling in an ice bath. Following complete addition, the reaction mixture was heated to 60 °C for 3 h and then cooled in an ice bath, and poured over 100 mL ice. Next, the reaction mixture was acidified (pH = 2–3) with 1M HCl under continuous stirring. The obtained precipitate was washed with H2O, MeOH, and EtOAc, and dried under vacuum to afford the title compound (2.783 g, 13.0 mmol, 89%) as a green solid that was used without further purification. mp > 300 °C.
2-(2,3-Difluorobenzylmercapto)-5-chloropyrido[3,4-d]pyrimidine-4-one (8)
2-Thioxo-5-chloropyrido[3,4-d]pyrimidine-4-one (427 mg, 2.00 mmol, 1.00 eq.) and dry DMF (20 mL) were added to a flame dried, N2-flushed 100 mL round-bottom flask under N2-atmosphere. The suspension was sonicated for 15 min, after which triethylamine (0.3 mL, 2.2 mmol, 1.1 eq.) was added, resulting in dissolution of the starting material. Next, a solution of 2,3-difluorobenzyl bromide (414 mg, 2.00 mmol, 1.00 eq.) in dry DMF (5 mL) was added dropwise. The reaction mixture was stirred at room temperature for 1 h, concentrated in vacuo and 20 mL H2O was added. The formed precipitate was filtered, washed with water, MeOH and Et2O, and dried under vacuum to afford the title compound (621 mg, 1.83 mmol, 92%) as a beige solid. mp > 300 °C.
1H NMR (400 MHz, d6-DMSO): δ (ppm) 13.04 (br. s, 1H), 8.85 (s, 1H), 8.50 (s, 1H), 7.51–7.42 (app. t, J = 7.0 Hz, 1H), 7.39–7.28 (m, 1H), 7.22–7.11 (m, 1H), 4.55 (s, 2H); HRMS (ESI-Q-TOF): m/z [M+H]+ calcd. for C14H8Cl1F2N3O1S1: 340.01173; found: 340.0114.
(2R)-2-[(5-Chloro-2-(2,3-difluorobenzylmercapto)-pyrido[3,4-d]pyrimidine-4-yl)amino]propanol (9)
In a flame dried, N2-flushed screw capped reaction tube 2-(2,3-difluorobenzylmercapto)-5-chloropyrido[3,4-d]pyrimidine-4-one (600 mg, 1.76 mmoL, 1.00 eq.) and POCl3 (10 mL) were added. After heating to 120 °C for 2 h, the mixture was cooled down to room temperature, and concentrated to dryness in vacuo. The obtained material was resuspended in ethyl acetate, poured over ice and neutralized with saturated aqueous NaHCO3. The obtained aqueous solution was extracted with ethyl acetate, and the combined organic phases were dried over MgSO4 and concentrated in vacuo. The crude intermediate was then dissolved in dry DMF (6 mL), and Et3N (0.50 mL, 3.6 mmol, 2.0 eq.) and (R)-alaninol (0.50 mL, 6.40 mmol, 3.6 eq.) were added dropwise while cooling in an ice bath. After stirring overnight at room temperature, the reaction mixture was concentrated in vacuo. Water (20 mL) and EtOAc (20 mL) were added. Following phase separation, the aqueous phase was extracted with EtOAc (2 × 20 mL). The combined organic phases were washed with water (2 × 20 mL) and brine (2 × 20 mL), dried over Na2SO4 and coated on Celite. Filtration over silica using 1:1 EtOAc/Et2O followed by column chromatography using 40–100% Et2O/PE afforded the title compound (61 mg, 0.15 mmol, 9%) as a white solid. mp 146–148 °C.
1H NMR (400 MHz, d6-DMSO): δ (ppm) 8.83 (s, 1H), 8.43 (s, 1H), 8.04 (d, J = 7.4 Hz, 1H), 7.45–7.37 (app t., J = 7.1 Hz, 1H), 7.34–7.23 (m, 1H), 7.19–7.09 (m, 1H), 5.15 (t, J = 4.5 Hz, 1H), 4.47 (s, 2H), 4.37–4.24 (m, 1H), 3.64–3.48 (m, 2H), 1.21 (d, J = 6.6 Hz, 3H); 13C NMR (101 MHz, d6-DMSO): δ (ppm) 168.0, 156.2, 149.6 (dd, J = 12.6, 245.4 Hz), 149.5, 148.1 (12.9, 246.8 Hz), 146.3, 142.9, 127.9 (d, J = 11.1 Hz), 126.2 (t, J = 2.6 Hz), 124 (t, J = 2.6 Hz), 124.6 (d, J = 4.7, 7.0 Hz), 124.5, 116.2 (d, J = 11.1 Hz), 114.3, 63.0, 48.9, 27.3, 16.3; 19F NMR (377 MHz, d6-DMSO): δ (ppm) −139.30 (m, 1F), −142.74 (m, 1F); HRMS (ESI-Q-TOF): m/z [M+H]+ calcd. for C17H15Cl1F2N4O1S1: 397.06958; found: 397.0697.
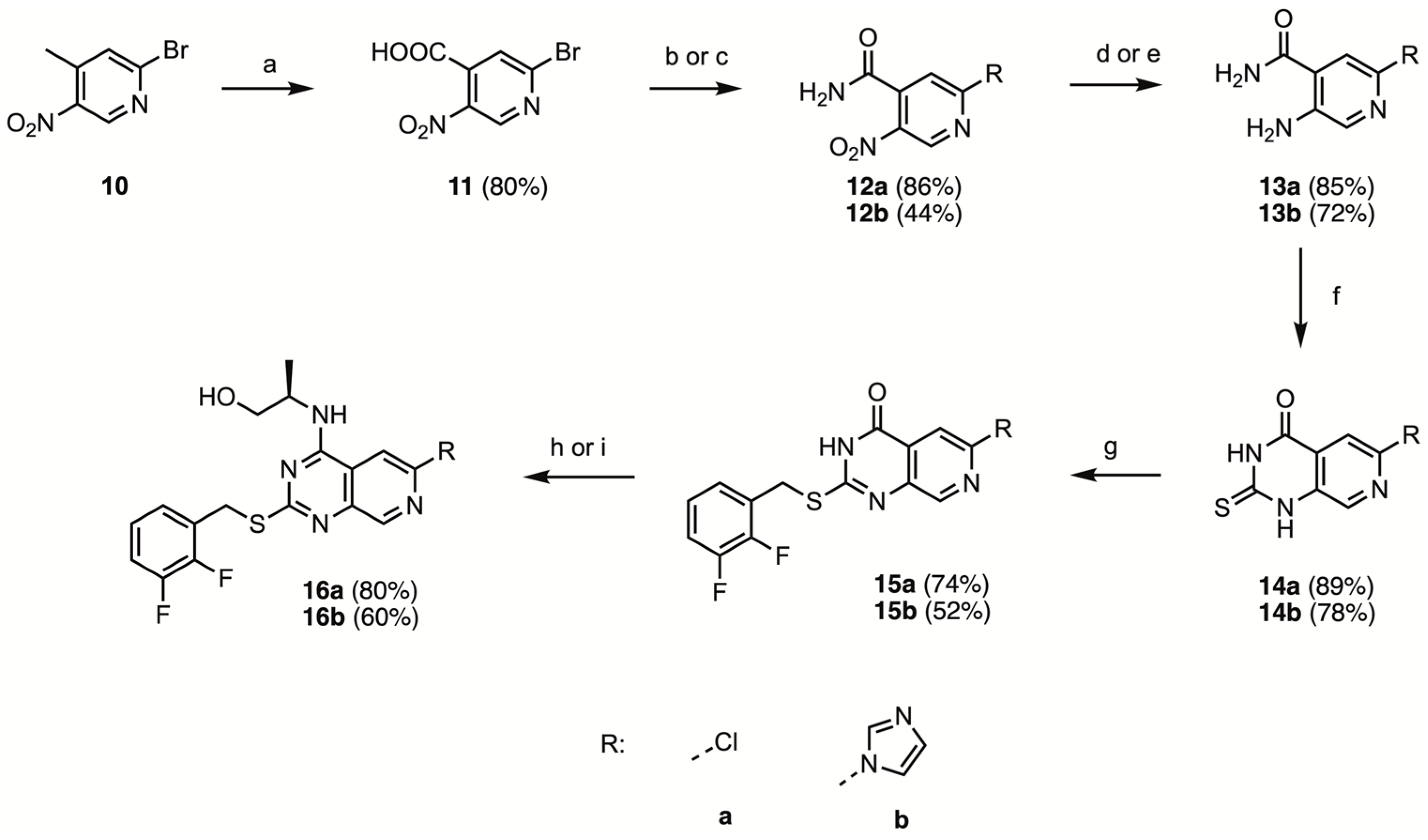
2-Bromo-5-nitropyridine-4-carboxylic acid (11)
2-Bromo-4-methyl-5-nitropyridine 10 (25.0 g, 115 mmol, 1.00 eq.) was dissolved in concentrated H2SO4 (100 mL). The mixture was cooled in an ice bath, and K2Cr2O7 (50.83 g, 173 mmol, 1.5 eq.) was added portion-wise. Following complete addition, the mixture was stirred in an ice bath for 1 h and at room temperature for 24 h. The resulting green solution was slowly poured onto 1L of ice. The obtained precipitate was filtered, washed with water until the eluent was no longer green, and dried under vacuum to afford the title compound as white solid (22.780 g, 92.23 mmol, 80%).
1H NMR (400 MHz, d6-DMSO): δ (ppm) 13.03 (br. s, 1H), 9.11 (s, 1H), 8.14 (s, 1H); 13C NMR (101 MHz, d6-DMSO): δ (ppm) 163.7, 146.39, 146.31, 142.2, 138.5, 127.1;
2-Chloro-5-nitropyridine-4-carboxamide (12a)
Oxalyl chloride (20 mL) was added to an ice-cold solution of 2-bromo-5-nitropyridine-4-carboxylic acid 11 (10.00 g, 40.49 mmol) in dry DCM (200 mL) at 0 °C and under N2-atmosphere. After stirring for 5 min, dry DMF (0.2 mL) was added. The reaction mixture was stirred at 0 °C for 1 h and at room temperature overnight. The resulting solution was concentrated in vacuo. The obtained residue was dissolved in dry DCM (50 mL) and added dropwise to a heavily stirred, ice cold 25% aqueous ammonia solution (300 mL). After stirring for 15 min, the formed precipitate was filtered, washed with saturated aqueous NaHCO3 and water, and dried under vacuum overnight to afford the title compound (7.498 g, 34.71 mmol, 86%) as a white solid that was used without further purification.
2-Imidazolyl-5-nitropyridine-4-carboxamide (12b)
In a flame dried N2-flushed round-bottom 100 mL two-necked flask, equipped with a N2-balloon, septum and stir bar, 2-chloro-5-nitropyridine-4-carboxylic acid 11 (1 g, 4.049 mmol, 1.00 eq.), 1,1-carbonyldiimidazole (985 mg, 6.07 mmol, 1.50 eq.) and dry THF (20 mL) were combined. The reaction mixture was stirred at room temperature for 30 min, then aqueous ammonia (3.0 mL, 40 mmol, 10 eq.) was added in one portion. The mixture was stirred overnight at room temperature, and then was transferred to a separatory funnel along with EtOAc (50 mL) and water (20 mL). Following phase separation, the aqueous phase was extracted with EtOAc (1 × 20 mL). The combined organic phases were washed with water and brine, dried over Na2SO4 and concentrated to dryness in vacuo. The obtained solid was triturated with ethanol, filtered and washed with ice cold ethanol to afford the title compound as a white solid (416 mg, 1.78 mmol, 44%). mp 259–261 °C.
1H NMR (400 MHz, d6-DMSO): δ (ppm) 9.20 (s, 1H), 8.72 (s, 1H), 8.31 (br. s, 1H), 8.16 (br. s, 1H), 8.11 (s, 1H), 8.13–8.05 (m, 2H), 7.20 (s, 1H); 13C NMR (101 MHz, d6-DMSO): δ (ppm) 164.9, 151.3, 145.9, 144.0, 140.1, 136.0, 131.1, 117.0, 111.4; HRMS (ESI-Q-TOF): m/z [M+H]+ calcd. for C9H7N5O3: 234.06216; found: 234.0625.
2-Chloro-5-aminopyridine-4-carboxamide (13a)
Activated iron powder (8.31g, 149 mol, 5 eq.) was added to a mixture of 2-chloro-5-nitropyridine-4-carboxamide 12a (6.00 g, 29.8 mmol, 1.00 eq.) in water (15 mL), EtOH (30 mL) and AcOH (30 mL). The resulting suspension was sonicated for 1 h and then filtered over 1:1 Celite/MgSO4, which was washed with EtOAc (500 mL). The filtrate was transferred to a separatory funnel along with water (100 mL). After phase separation, the aqueous phase was extracted with EtOAc (2 × 100 mL). The combined organic phases were washed with saturated aqueous NaHCO3 (3 × 100 mL), water (3 × 100 mL) and brine (2 × 100 mL), dried over Na2SO4 and coated on Celite. Flash chromatography using 50–100% EtOAc/PE afforded the title compound (4.340 g, 25.29 mmol, 85%) as an off-white solid.
1H NMR (400 MHz, d6-DMSO): δ (ppm) 8.12 (br. s, 1H), 7.93 (s, 1H), 7.57 (br. s, 1H), 7.54 (s, 1H), 6.65 (s, 2H); 13C NMR (101 MHz, d6-DMSO): δ (ppm) 168.4, 144.5, 139.3, 135.0, 121.7, 121.5.
5-Amino-2-imidazolyl-pyridine-4-carboxamide (13b)
In a round-bottom 100 mL two-necked flask, equipped with a hydrogen-balloon, septum and stir bar, 2-imidazolyl-5-nitropyridine-4-carboxamide 12b (300 mg, 1.287 mmol, 1.00 eq.) and Pd/C (15 mg, 5 w%) were combined in THF (20 mL). The reaction mixture was stirred for 24 h at room temperature and filtered over Celite. The Celite was thoroughly washed with EtOAc. The solution was coated on Celite. Flash chromatography using 0–20% MeOH/EtOAc afforded the title compound (188 mg, 0.925 mmol, 72%) as an off-white solid. mp 248–251 °C.
1H NMR (400 MHz, d6-DMSO): δ (ppm) 8.28 (s, 1H), 8.14 (br. s, 1H), 8.05 (s, 1H), 7.79 (s, 1H), 7.75 (s, 1H), 7.67 (br. s, 1H), 7.08 (s, 1H), 6.67 (br. s, 2H); 13C NMR (101 MHz, d6-DMSO): δ (ppm) 168.9, 144.1, 138.2, 137.9, 134.3, 129.4, 120.8, 116.6, 111.1; HRMS (ESI-Q-TOF): m/z [M+H]+ calcd. for C9H9N5O1: 204.08798; found: 204.0871.
2-Thioxo-5-chloropyrido[3,4-d]pyrimidine-4-one (14a)
DBU (5.0 mL, 33.5 mmol, 2.3 eq.) was added dropwise to a suspension of 2-chloro-5-aminopyridine-4-carboxamide 13a (2.50 g, 14.6 mmol, 1.00 eq.) in dry DMF (25 mL) and CS2 (5.0 mL, 83 mmol, 5.7 eq.), under N2-atmosphere, while cooling in an ice bath. Following complete addition, the reaction mixture was heated to 60 °C for 3 h, and then cooled in an ice bath and poured over 100 mL ice. While stirring, the reaction mixture was acidified (pH = 2–3) with 1M HCl. The obtained precipitate was washed with H2O, MeOH, and EtOAc and dried under vacuum to afford the title compound (2.783 g, 13.0 mmol, 89%) as a green solid that was used without further purification.
6-Chloro-2-thioxopyrido[3,4-d]pyrimidine-4-one (14b)
A suspension of 5-amino-2-imidazolyl-pyridine-4-carboxamide 13b (150 mg, 0.738 mmol, 1.00 eq.) in dry DMF (1.2 mL), in a flame dried, N2-flushed reaction tube sealed with a septum screw cap, was cooled to 0 °C in an ice bath. Carbon disulfide (0.22 mL, 3.7 mmol, 5 eq.) was added in one portion and DBU (0.22 mL, 1.5 mmol, 1.5 eq.) was added dropwise. After heating to 60 °C for 3 h, the reaction mixture was cooled in an ice bath and poured onto 10 mL ice. The mixture was acidified with 1M HCl (pH 3–4) and filtered. The obtained solid was washed with water, methanol and EtOAc and dried under vacuum to afford the title compound as a yellow solid (142 mg, 0.579 mmol, 78%), which was used without further purification.
2-(2,3-Difluorobenzylmercapto)-6-chloropyrido[3,4-d]pyrimidine-4-one (15a)
2-Thioxo-6-chloropyrido[3,4-d]pyrimidine-4-one 14a (4.180 g, 19.57 mmol, 1.00 eq.) and dry DMF (40 mL) were added to a flame dried, N2-flushed 250 mL round-bottom flask under N2-atmosphere. The suspension was sonicated for 15 min, after which triethylamine (3.0 mL, 22 mmol, 1.1 eq.) was added, resulting in dissolution of the starting material. Next, a solution of 2,3-difluorobenzyl bromide (4.324 g, 20.88 mmol, 1.05 eq.) in dry DMF (20 mL) was added dropwise. The reaction mixture was stirred at room temperature for 2 h, concentrated in vacuo and 50 mL H2O was added. The formed precipitate was filtered, washed with water (2 × 10 mL), MeOH (10 mL) and Et2O (10 mL) and dried under vacuum to afford the title compound (4.996 g, 14.705 mmol, 74%) as a beige solid. mp 235–237 °C
1H NMR (400 MHz, d6-DMSO): δ (ppm) 13.11 (br. s, 1H), 8.82 (s, 1H), 7.88 (s, 1H), 7.51–7.43 (m, 1H), 7.41–7.29 (m, 1H), 7.21–7.12 (m, 1H), 4.56 (s, 2H); 19F NMR (377 MHz, d6-DMSO): δ (ppm) −139.17 (m, 1F), −142.14 (m, 1F) HRMS (ESI-Q-TOF): m/z [M+H]+ calcd. for C14H9Cl1F2N3O1S1: 340.0117; found: 340.0119.
2-(2,3-Difluorobenzylthio)-6-imidazolyl-pyrido[3,4-d]pyrimidine-4-one (15b)
To a flame dried, N2-flushed reaction tube equipped with a stirring bar and a septum screw cap, 6-chloro-2-thioxopyrido[3,4-d]pyrimidine-4-one 14b (123 mg, 0.50 mmol, 1.00 eq.), and dry DMF (2.0 mL) and Et3N (76 μL, 0.55 mmol, 1.1 eq.) were added. A solution of 2,3-difluorobenzyl bromide (109 mg, 0.525 mmol, 1.05 eq.) in dry DMF (1 mL) was added dropwise. After stirring at room temperature for 1 h, the reaction mixture was concentrated to dryness in vacuo, and coated on Celite. Flash chromatography using 0–10% MeOH/EtOAc afforded the title compound (95 mg, 0.26 mmol, 52%).
1H NMR (400 MHz, d6-DMSO): δ (ppm) 13.06 (br. s, 1H), 8.91 (s, 1H), 8.64 (s, 1H), 8.21 (s, 1H), 8.08 (s, 1H), 7.64–7.26 (m, 2H), 7.25–6.93 (m, 2H), 4.49 (s, 2H); 19F NMR (377 MHz, d6-DMSO): δ (ppm) −139.17 (m, 1F), −142.15 (m, 1F); HRMS (ESI-Q-TOF): m/z [M+H]+ calcd. for C17H11F2N5O1S1: 372.07250; found: 372.0727.
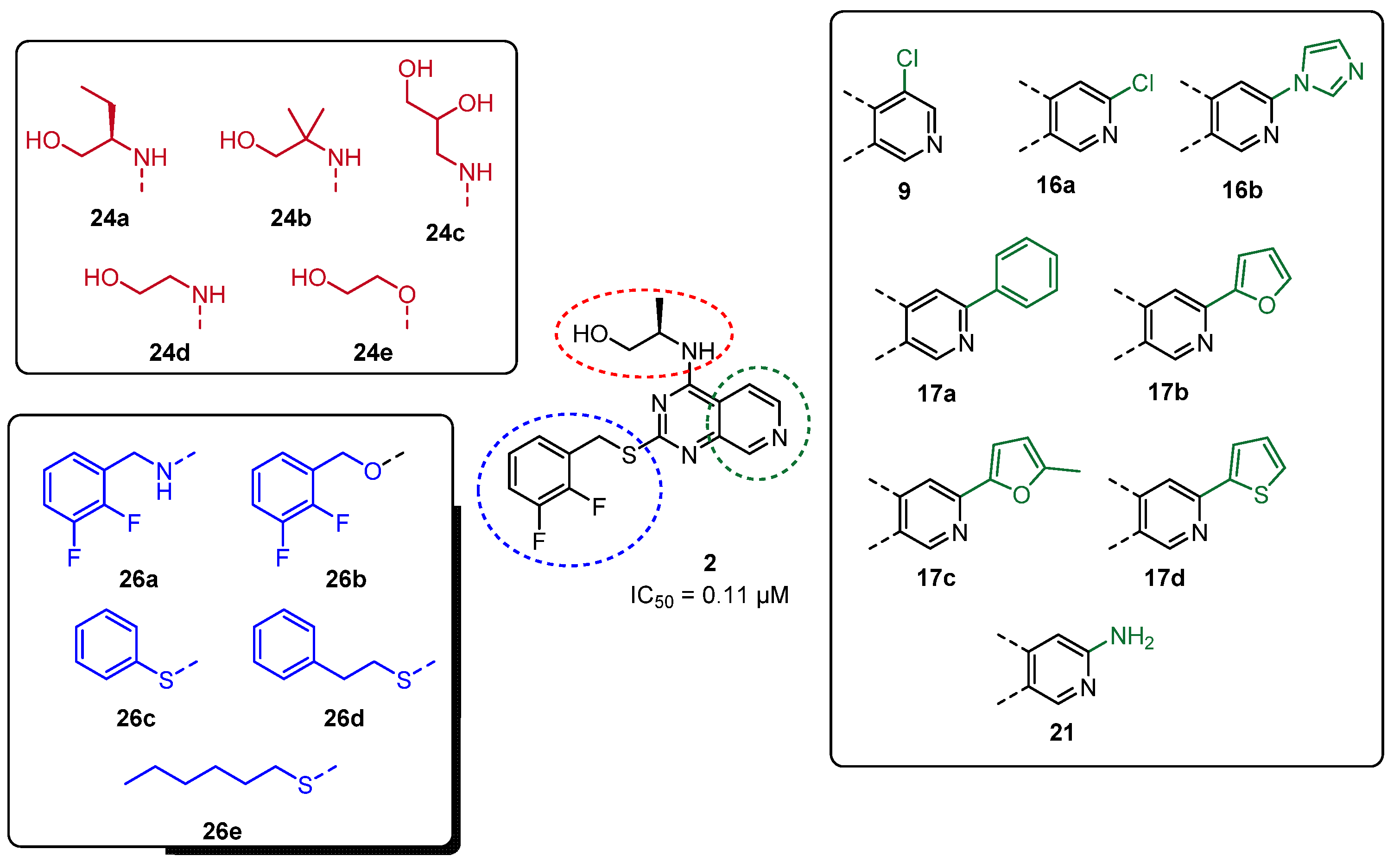
(2R)-2-[(6-Chloro-2-(2,3-difluorobenzylmercapto)-pyrido[3,4-d]pyrimidine-4-yl)amino]propanol (16a)
In a flame dried, N2-flushed screw capped reaction tube 2-(2,3-difluorobenzylmercapto)-6-chloropyrido[3,4-d]pyrimidine-4-one 15a (960 mg, 2.83 mmol, 1.00 eq.) and SOCl2 (10 mL) and 1 drop of DMF were added. After heating to 80 °C for 4 h, the mixture was cooled down to room temperature, and concentrated to dryness in vacuo to afford the crude intermediate as a yellow solid. The intermediate was then dissolved in dry DMF (10 mL), and Et3N (1.00 mL, 7.2 mmol, 2.5 eq.) and (R)-alaninol (0.50 mL, 6.4 mmol, 2.3 eq.) were added dropwise while cooling in an ice bath. After stirring for 1 h at room temperature, the reaction mixture was concentrated to dryness in vacuo, transferred to a separatory funnel along with water (10 mL) and EtOAc (20 mL). After extracting with EtOAc (3 × 20 mL), the combined organic phases were washed with water (3 × 20 mL) and brine (2 × 20 mL), dried over Na2SO4 and coated on Celite. Flash chromatography using 50% PE/Et2O–1% MeOH/Et2O, followed by trituration with 50% pentane/Et2O afforded the title compound (902 mg, 2.27 mmol, 80%) as a white solid. m.p. 211–213 °C.
1H NMR (400 MHz, d6-DMSO): δ (ppm) 8.79 (s, 1H), 8.39 (d, J = 7.9 Hz, 1H), 8.37 (s, 1H), 7.46–7.37 (app. t, J = 7.1 Hz, 1H), 7.34–7.23 (m, 1H), 7.17–7.07 (m, 1H), 4.81 (t, J = 5.8 Hz, 1H), 4.47 (s, 2H), 4.42–4.31 (m, 1H), 3.57–3.41 (m, 2H), 1.19 (d, J = 6.7 Hz, 3H); 13C NMR (101 MHz, d6-DMSO): δ (ppm) 168.0, 156.9, 150.2, 149.6 (dd, J = 12.5, 245.5 Hz), 148.1 (dd, J = 12.9, 246.8 Hz), 143.7, 143.0, 128.1 (d, J = 11.2 Hz), 126.3 (t, J = 2.7 Hz), 124.5 (dd, J = 4.7, 6.9 Hz), 119.9, 116.3, 116.2, 116.1, 63.5, 48.8, 27.3, 16.4; 19F NMR (377 MHz, d6-DMSO): δ (ppm) −139.30 (m, 1F), −142.77 (m, 1F); HRMS (ESI-Q-TOF): m/z [M+H]+ calcd. for C17H15Cl1F2N4O1S1: 397.06958; found: 397.0693.
(2R)-2-[(6-Imidazolyl-2-(2,3-difluorobenzylmercapto)-pyrido[3,4-d]pyrimidine-4-yl)amino]propanol (16b)
DBU (0.05 mL, 0.3 mmol, 1.3 eq.) was added dropwise to a solution of 6-chloro-2-(2,3-difluorobenzylthio)-pyrido[3,4-d]pyrimidine-4-one 15b (75 mg, 0.20 mol, 1.00 eq.) and BOP-reagent (116 mg, 0.26 mmol, 1.30 eq.) in dry DMF (1 mL) under N2-atmosphere. Following complete addition, the reaction mixture was stirred for 10 min, after which (R)-alaninol (0.05 mL, 0.6 mmol, 3 eq.) was added dropwise. The reaction mixture was stirred at room temperature overnight, and then heated to 60 °C for 2–3 h. Next, the reaction mixture was concentrated in vacuo and redissolved in 20 mL EtOAc and NH4Cl (10 mL) was added. The aqueous phase was extracted with EtOAc (2 × 20 mL). The combined organic phases were washed with saturated NH4Cl (3 × 10 mL), with water (2 × 10 mL) and brine (2 × 10 mL), and dried over Na2SO4. After coating on Celite, flash chromatography using 0–5% MeOH/DCM afforded the title compound (51 mg, 0.12 mmol, 60%) as a white-yellow solid. mp 245–247 °C.
1H NMR (400 MHz, d6-DMSO): δ (ppm) 8.89 (s, 1H), 8.53 (s, 1H), 8.48 (s, 1H), 8.32 (d, J = 7.2 Hz, 1H), 7.90 (s, 1H), 7.49–7.38 (m, 1H), 7.37–7.25 (m, 1H), 7.24–7.06 (m, 2H), 4.91 (s, 1H), 4.50 (s, 2H), 4.35–4.34 (m, 1H), 3.62–3.45 (m, 2H), 1.23 (d, J = 6.4 Hz, 3H); 13C NMR (101 MHz, d6-DMSO): δ (ppm) 167.1, 157.5, 149.6 (dd, J = 12.5, 245.3 Hz), 149.4, 148.1 (dd, J = 12.7, 246.7 Hz), 143.5, 143.4, 130.2, 128.2 (d, J = 11.1 Hz), 127.3, 126.4 (t, J = 2.7 Hz), 124.6 (dd, J = 4.5, 7.1 Hz), 119.6, 116.2 (d, J = 16.8 Hz), 109.6, 104.1, 63.5, 48.8, 27.3, 16.5; 19F NMR (377 MHz, d6-DMSO): δ (ppm) −139.33 (m, 1F), −142.79 (m, 1F); HRMS (ESI-Q-TOF): m/z [M+H]+ calcd. for C20H18F2N6O1S1: 429.13035; found: 429.1311.
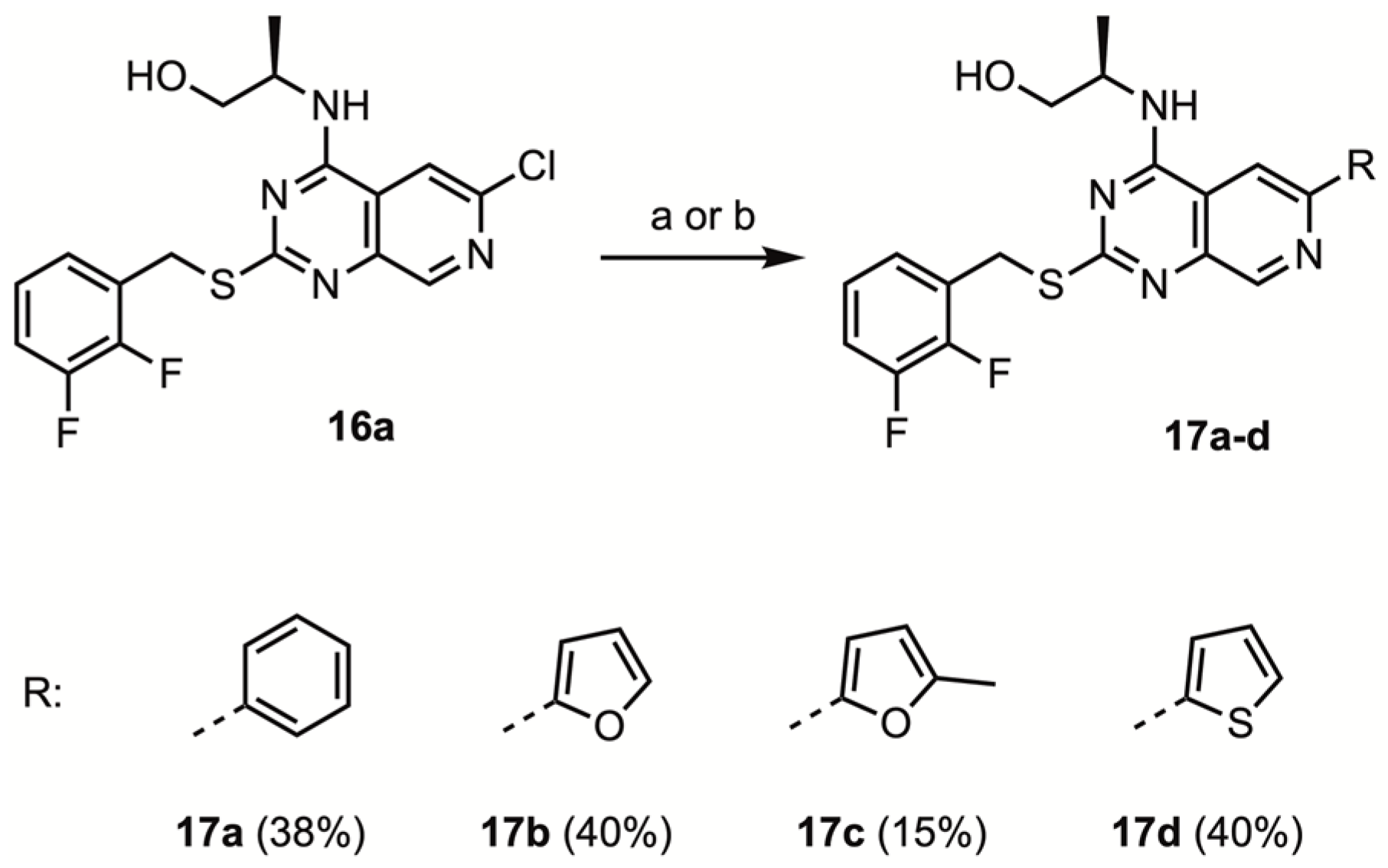
(2R)-2-[(2-(2,3-Difluorobenzylmercapto)-6-phenyl-pyrido[3,4-d]pyrimidine-4-yl)amino]propanol (17a)
To a flame dried, N2-flushed 8 mL reaction tube equipped with a stir bar and a septum screw cap, (2R)-2-[(6-chloro-2-(2,3-difluorobenzylmercapto)-pyrido[3,4-d]pyrimidine-4-yl)amino]propanol 16a (100 mg, 0.250 mmol, 1.00 eq.) K2CO3 (104 mg, 0.75 mmol, 3.00 eq.), 2-phenylboronic acid (56 mg, 0.50 mmol, 2.0 eq.) and Pd(PPh3)4 (30 mg, 0.025 mmol, 10 mol%) were added. Following three cycles of backfilling with nitrogen, 1,4-dioxane (3.0 mL) and water (1.0 mL) were added. After heating to 120 °C for 48 h, the reaction mixture was transferred to a separatory funnel and water (10 mL) was added. The aqueous phase was extracted with EtOAc (3 × 20 mL). The combined organic phases were washed with saturated aqueous NaHCO3, water and brine, dried over Na2SO4 and coated on Celite. Flash chromatography using 20–500% EtOAc/isohexane gave a mixture of starting material and title compound (70 mg, in a 1:9 ratio). This crude product was recrystallized by dissolving in EtOAc and layering with pentane. The obtained crystals were filtered and washed with 1:1 Et2O/pentane to afford the title compound (42 mg, 0.096 mmol, 38%) as an off-white/light-yellow solid. mp 202–204 °C.
1H NMR (400 MHz, d6-DMSO): δ (ppm) 9.02 (s, 1H), 8.75 (s, 1H), 8.47 (d, J = 7.8 Hz, 1H), 8.24–8.16 (m, 2H), 7.54 (t, J = 7.6 Hz, 2H), 7.49–7.41 (m, 2H), 7.38–7.27 (m, 1H), 7.20–7.10 (m, 1H), 4.87 (t, J = 5.7 Hz, 1H), 4.52 (s, 2H), 4.48–4.38 (m, 1H), 3.60–3.46 (m, 2H), 1.24 (d, J = 6.7 Hz, 3H); 13C NMR (101 MHz, d6-DMSO): δ (ppm) 167.2, 157.9, 149.9, 149.6 (dd, J = 12.3, 245.1 Hz), 148.2 (dd, J = 12.9, 246.6 Hz), 143.6, 138.2, 128.7, 128.3 (d, J = 11.0 Hz), 126.3, 124.6 (dd, J = 4.9, 6.7 Hz), 119.1, 116.1 (d, J = 16.8 Hz), 111.6, 63.6, 48.7, 27.2, 16.6;
19F NMR (377 MHz, d6-DMSO): δ (ppm) −139.35 (m, 1F), −142.82 (m, 1F); HRMS (ESI-Q-TOF): m/z [M+H]+ calcd. for C23H20F2N4O1S1: 439.13985; found: 439.1395.
(2R)-2-[(2-(2,3-Difluorobenzylmercapto)-6-(2-furanyl)-pyrido[3,4-d]pyrimidine-4-yl)amino]propanol (17b)
To a flame dried, N2-flushed 8 mL reaction tube equipped with a stir bar and a septum screw cap, (2R)-2-[(6-chloro-2-(2,3-difluorobenzylmercapto)-pyrido[3,4-d]pyrimidine-4-yl)amino]propanol 16a (100 mg, 0.250 mmol, 1.00 eq.) K2CO3 (104 mg, 0.75 mmol, 3.00 eq.), 2-furanyl boronic acid (56 mg, 0.50 mmol, 2.0 eq.) and Pd(PPh3)4 (30 mg, 0.025 mmol, 10 mol%) were added. Following three cycles of backfilling with nitrogen, 1,4-dioxane (3.0 mL) and water (1.0 mL) were added. After heating to 120 °C for 48 h, the reaction mixture was transferred to a separatory funnel, and water (10 mL) was added. The aqueous phase was extracted with EtOAc (3 × 20 mL). The combined organic phases were washed with saturated aqueous NaHCO3, water and brine, and dried over Na2SO4. After coating on Celite, flash chromatography using 70–100% Et2O/PE to 1–2 MeOH/Et2O afforded the title compound (43 mg, 0.10 mmol, 40%) as a light brown solid. mp 163–165 °C.
1H NMR (400 MHz, d6-DMSO): δ (ppm) 8.91 (s, 1H), 8.54 (d, J = 7.9 Hz, 1H), 8.50 (s, 1H), 7.86 (d, J = 1.0 Hz, 1H), 7.47–7.38 (m, 1H), 7.34–7. 23 (m, 1H), 7.17–7.09 (m, 1H), 7.06 (d, J = 3.2 Hz, 1H), 6.68 (d, J = 1.8 Hz, 1H), 6.68 (dd, J = 1.8, 3.3 Hz, 1H), 4.84 (t, J = 5.7 Hz, 1H), 4.49 (s, 2H), 4.45–4.36 (m, 1H), 3.63–3.53 (m, 1H), 3.53–3.44 (m, 1H), 1.21 (d, J = 6.7 Hz, 3H); 13C NMR (101 MHz, d6-DMSO): δ (ppm) 167.1, 157.7, 153.3, 150.2, 149.6 (dd, J = 12.5, 245.3 Hz), 148.2 (dd, J = 12.9, 246.8 Hz), 143.8, 143.5, 142.7, 128.3 (d, J = 11.4 Hz), 126.3 (t, J =2.7 Hz), 124.6 (dd, J = 4.7, 6.9 Hz), 117.9, 116.1 (d, J = 16.8 Hz), 112.5, 109.6, 108.4, 63.7, 48.7, 27.2, 16.5; 19F NMR (377 MHz, d6-DMSO): δ (ppm) −139.32 (m, 1F), −142.80 (m, 1F);
HRMS (ESI-Q-TOF): m/z [M+H]+ calcd. for C21H18F2N4O2S1: 429.1191; found: 429.1195.
(2R)-2-[(2-(2,3-Difluorobenzylmercapto)-6-(5-methylfuran-2-yl)-pyrido[3,4-d]pyrimidine-4-yl)amino]propanol (17c)
To a flame dried, N2-flushed 8 mL reaction tube equipped with a stir bar and a septum screw cap, (2R)-2-[(6-chloro-2-(2,3-difluorobenzylmercapto)-pyrido[3,4-d]pyrimidine-4-yl)amino]propanol 16a (100 mg, 0.250 mmol, 1.00 eq.) K2CO3 (104 mg, 0.75 mmol, 3.00 eq.), 2-furanyl boronic acid (56 mg, 0.50 mmol, 2.0 eq.) and Pd(PPh3)4 (30 mg, 0.025 mmol, 10 mol%) were added. Following three cycles of backfilling with nitrogen, 1,4-dioxane (3.0 mL) and water (1.0 mL) were added. After heating to 120 °C for 10 h, the reaction mixture was transferred to a separatory funnel, and water (10 mL) was added. The aqueous phase was extracted with EtOAc (3 × 20 mL). The combined organic phases were washed with saturated aqueous NaHCO3, water and brine, and dried over Na2SO4. After coating on Celite, flash chromatography using EtOAc/DCM afforded the title compound (17 mg, 0.038 mmol, 15%) as an off-white solid. mp 162–164 °C.
1H NMR (400 MHz, d6-DMSO): δ (ppm) 8.89 (s, 1H), 8.50 (d, J = 7.9 Hz, 1H), 8.37 (s, 1H), 7.48–7.38 (m, 1H), 7.36–7.25 (m, 1H), 7.19–7.09 (m, 1H), 6.96 (d, J = 3.1 Hz, 1H), 6.30 (dd, J = 0.8, 3.1 Hz, 1H), 4.83 (t, J = 5.7 Hz, 1H), 4.50 (s, 2H), 4.47–4.35 (m, 1H), 3.61–3.52 (m, 1H), 3.51–3.44 (m, 1H), 2.42 (s, 3H), 1.21 (d, J = 6.8 Hz, 3H); 13C NMR (101 MHz, d6-DMSO): δ (ppm) 166.7, 157.7, 153.0, 151.8, 150.2, 149.6 (dd, J = 12.5, 245.1 Hz), 148.2 (dd, J = 12.6, 246.8 Hz), 143.2, 142.9, 128.3 (d, J = 11.2 Hz), 126.3 (t, J = 2.8 Hz), 124.6 (dd, J = 4.5, 7.2 Hz), 118.0, 116.1 (d, J = 16.8 Hz), 109.5, 108.7 (d, J = 15.1 Hz), 63.6, 48.7, 27.2, 16.5, 13.7;
19F NMR (377 MHz, d6-DMSO): δ (ppm) −139.36 (m, 1F), −142.83 (m, 1F); HRMS (ESI-Q-TOF): m/z [M+H]+ calcd. for C22H20F2N4O2S1: 443.13476; found: 443.1345.
(2R)-2-[(2-(2,3-Difluorobenzylmercapto)-6-(2-thienyl)-pyrido[3,4-d]pyrimidine-4-yl)amino]propanol (17d)
To a flame dried, N2-flushed 8 mL reaction tube equipped with a stir bar and a screw cap septum, Pd2(dba)3 (6 mg, 7 μmol, 3 mol%), XPhos (24 mg, 0.05 mmol, 20 mol%) (2R)-2-[(6-chloro-2-(2,3-difluorobenzylmercapto)-pyrido[3,4-d]pyrimidine-4-yl)amino]propanol 16a (100 mg, 0.250 mmol, 1.00 eq.), 2-thienyl boronic acid (65 mg, 0.50 mmol, 2.0 eq.), and K3PO4 (107 mg, 0.50 mmol, 2.00 eq.) were added. Following three cycles of backfilling with nitrogen, degassed nBuOH (2.0 mL) was added. After heating to 120 °C for 20 h, the mixture was concentrated in vacuo, and transferred to a separatory funnel along with saturated NH4Cl (8 mL), H2O (2 mL) and EtOAc (20 mL). Following phase separation, the mixture was extracted with EtOAc (1 × 20 mL). The combined organic phases were washed with water (10 mL) and brine (10 mL), and dried over Na2SO4. After coating on Celite. Flash chromatography using 5–40% EtOAc/isohexane afforded the title compound (49 mg, 0.11 mmol, 44%) as an off-white solid. mp 176–178 °C.
1H NMR (400 MHz, d6-DMSO): δ (ppm) 8.89 (s, 1H), 8.65 (s, 1H), 8.35 (d, J = 7.8 Hz, 1H), 7.77 (dd, J = 0.7, 3.5 Hz, 1H), 7.63 (dd, J = 0.7, 5.0 Hz, 1H), 7.47–7.40 (m, 1H), 7.36–7.26 (m, 1H), 7.22 (d, J = 3.7, 5.0 Hz, 1H), 7.18–7.09 (m, 1H), 4.89 (t, J = 5.6 Hz, 1H), 4.50 (s, 2H), 4.46–4.35 (m, 1H), 3.62.–3.45 (m, 2H), 1.24 (d, J = 6.7 Hz, 3H); 13C NMR (101 MHz, d6-DMSO): δ (ppm) 166.9, 157.7, 149.8, 149.6 (dd, J = 12.6, 245.3 Hz), 148.2 (dd, J = 12.8, 246.6 Hz), 146.0, 144.3, 143.4, 128.4, 128.2 (d, J = 11.2 Hz), 127.9, 126.3 (t, J= 2.7 Hz), 124.6 (d, J = 4.6, 7.0 Hz), 124.4, 118.1, 116.2 (d, J = 16.8 Hz), 109.9, 63.6, 48.7, 27.3, 16.5; 19F NMR (377 MHz, d6-DMSO): δ (ppm) −139.32 (m, 1F), −142.80 (m, 1F); HRMS (ESI-Q-TOF): m/z [M+H]+ calcd. for C21H18F2N4O1S2: 445.09627; found: 445.0955.
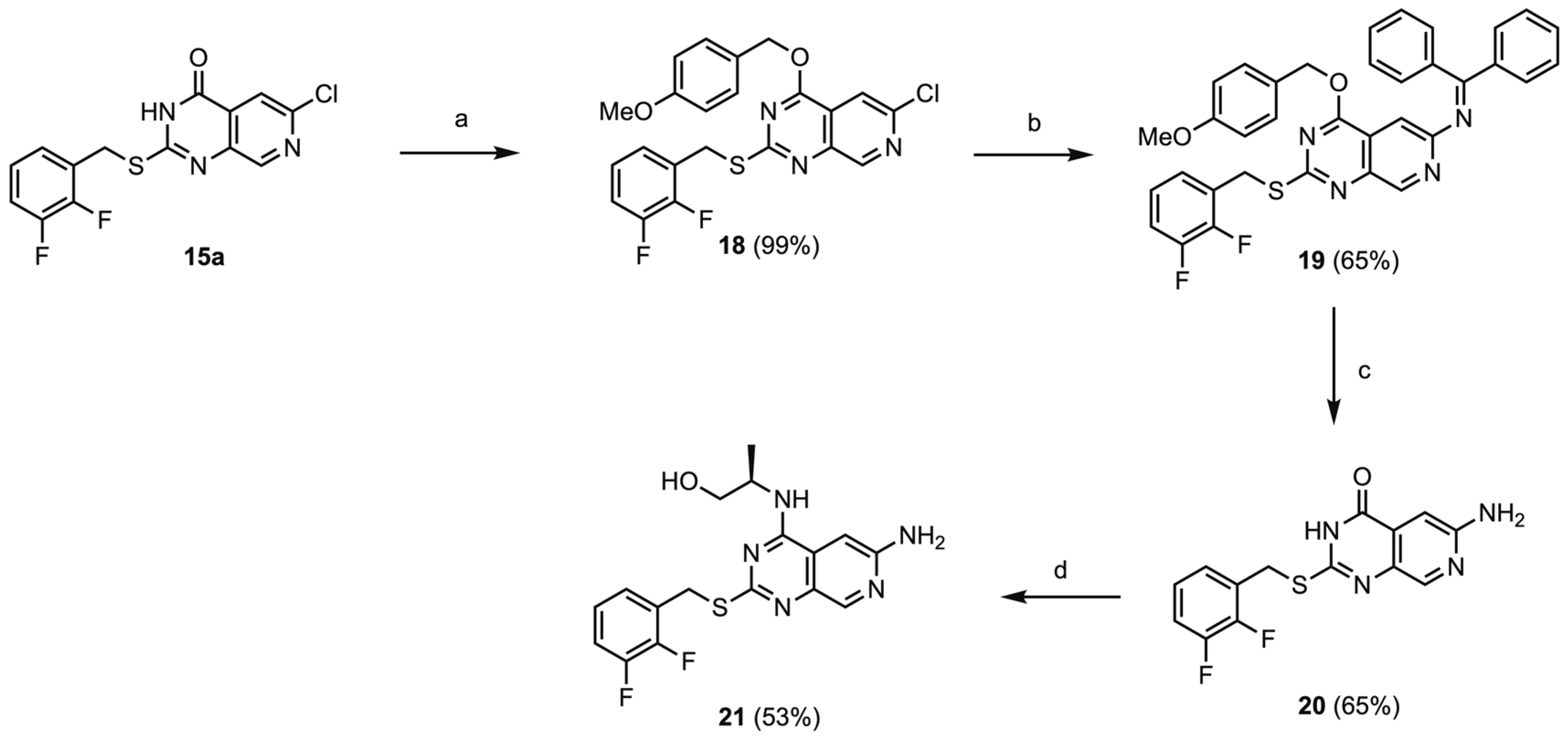
6-Chloro-2-(2,3-difluorobenzylmercapto)-4-(4-methoxybenzyl)-pyrido[3,4-d]pyrimidine (18)
To a flame dried N2-flushed reaction tube equipped with a screw-cap septum and a stirring bar, 2-(2,3-difluorobenzylmercapto)-6-chloropyrido[3,4-d]pyrimidin-4-one 15a (1.359 g, 4.00 mmol, 1.00 eq.), K2CO3 (0.829 g, 6.00 mmol, 1.5 eq.) and dry DMF (10 mL) were added, resulting in the formation of a clear orange solution. 4-Methoxybenzyl chloride (0.65 mL, 4.8 mmol, 1.2 eq.) was added dropwise while stirring at room temperature. The mixture was stirred at room temperature for 1 h. After a few minutes, a white precipitate started to form. After stirring at 60 °C for 22 h, the mixture was cooled to room temperature; 20 mL ice water was added. The resulting precipitate was filtered, and washed with water, methanol and diethyl ether. After drying in vacuo, the obtained solid was purified via flash chromatography using 40% EtOAc/PE to afford the title compound as a white solid (1.826, 3.970 mmol, 99%). mp 137–139 °C.
1H NMR (400 MHz, d6-DMSO): δ (ppm) 8.89 (s, 1H), 7.97 (s, 1H), 7.51–7.42 (app. t, J = 7.1 Hz, 1H), 7.40–7.29 (m, 1H), 7.21 (d, J = 8.6 Hz, 2H), 7.19–7.10 (m, 1H), 6.89 (d, J = 8.7 Hz, 2H), 5.21 (s, 2H), 4.61 (s, 2H), 3.71 (s, 3H); 13C NMR (101 MHz, d6-DMSO): δ (ppm) 159.2, 158.9, 158.7, 149.6, 145.2, 140.8, 128.5, 127.0, 126.7, 126.6, 126.2 (d, J = 9.9 Hz), 124.7, 119.0, 116.8 (d, J = 16.6 Hz), 113.9, 55.1, 48.9, 29.1; 19F NMR (377 MHz, d6-DMSO): δ (ppm) −139.15 (m, 1F), −141.77 (m, 1F); HRMS (ESI-Q-TOF): m/z [M+H]+ calcd. for C22H16Cl1F2N3O2S1: 460.0693; found: 460.0690.
2-(2,3-Difluorobenzylmercapto)-4-(4-methoxybenzyl)-(N-diphenylmethylene)-pyrido[3,4-d]pyrimidine-6-amine (19)
To a flame dried, N2-flushed 8 mL reaction tube equipped with a stir bar and a septum screw cap, Pd(OAc)2 (23 mg, 0.10 mmol, 10 mol%), (±)-BINAP (93 mg, mg, 0.15 mmol, 15 mol%), 6-chloro-2-(2,3-difluorobenzylmercapto)-4-(4-methoxybenzyl)-pyrido[3,4-d]pyrimidine 18 (460 mg, 1.00 mmol, 1.00 eq.) and Cs2CO3 (456 mg, 1.40 mmol, 1.40 eq.) were added. Following three cycles of backfilling with N2, dry 1,4-dioxane (6.0 mL) and benzophenone imine (370 mg, 2.04 mmol, 2.04 eq.) were added. The mixture was heated to 120 °C for 24 h, then cooled to room temperature, and transferred to a separatory funnel. Water (20 mL) was added, and the aqueous phase was extracted with EtOAc (3 × 20 mL). The combined organic phases were washed with water and brine, dried over Na2SO4 and coated on Celite. Flash chromatography using 0–40% EtOAc/isohexane afforded the title compound (392 mg, 0.648 mmol, 65%) as an orange solid. mp 188–191 °C.
1H NMR (400 MHz, d6-DMSO): δ (ppm) 8.73 (d, J = 0.8 Hz, 1H), 7.72 (d, J = 7.4 Hz, 2H), 7.63–7.55 (m, 1H), 7.55–7.47 (m, 2H), 7.46–7.40 (m, 1H), 7.39–7.27 (m, 4H), 7.25–7.09 (m, 6H), 6.90–6.83 (m, 2H), 5.14 (s, 2H), 4.56 (s, 2H), 3.71 (s, 3H); 1H NMR (400 MHz, CDCl3): δ (ppm) 8.80 (s, 1H), 7.85 (d, J = 7.2 Hz, 2H), 7.57–7.41 (m, 3H), 7.39–7.17 (m, 9H), 7.15–6.99 (m, 2H), 6.89–6.81 (m, 2H), 5.22 (s, 2H), 4.56 (s, 2H), 3.79 (s, 3H); 13C NMR (101 MHz, d6-DMSO): δ (ppm) 171.3, 160.9, 160.5, 159.4, 155.8, 150.6 (dd, J = 12.6, 248.6 Hz), 149.3 (dd, J = 13.0, 249.8 Hz), 148.6, 138.8, 138.2, 136.0, 132.5, 131.5, 130.1, 129.9, 129.6, 129.4, 129.1, 128.3, 128.2, 127.0, 126.15 (t, J = 3.2 Hz), 126.05 (d, J = 10.6 Hz), 124.08 (dd, J = 4.8, 6.7 Hz), 116.8 (d, J = 17.0 Hz), 114.0, 109.6, 55.3, 47.3, 29.7; 19F NMR (377 MHz, d6-DMSO): δ (ppm) −137.73 (m, 1F), −141.08 (m, 1F); HRMS (ESI-Q-TOF): m/z [M+H-Benzophenone]+ calcd. for C22H16Cl1F2N3O2S1: 441.1191; found: 441.1142.
4-Amino-2-(2,3-difluorobenzylmercapto)-pyrido[3,4-d]pyrimidine-4-one (20)
In a screw-capped reaction tube equipped with a stir bar, 2-(2,3-difluorobenzylmercapto)-4-(4-methoxybenzyl)-(N-diphenylmethylene)-pyrido[3,4-d]pyrimidine-6-amine 19 (324 mg, 0.536 mmol) and TFA (6.0 mL) were combined and heated to 75 °C for 1 h. Next, the reaction mixture was concentrated in vacuo. Ethyl acetate and saturated aqueous NaHCO3 were added; the mixture was stirred and filtered. The obtained solid was washed with water and methanol, and dried under vacuum to afford impure 6-amino-2-(2,3-difluorobenzylmercapto)-pyrido[3,4-d]pyrimidine-4-one (112 mg, 0.350 mmol, 65%), which was used without further purification. mp > 300 °C.
(2R)-2-[(6-Amino-2-(2,3-difluorobenzylmercapto)-pyrido[3,4-d]pyrimidine-4-yl)amino]propanol (21)
The crude 6-amino-2-(2,3-difluorobenzylmercapto)-pyrido[3,4-d]pyrimidine-4-one 20 (100 mg, 0.312 mmol, 1.00 eq.), BOP (180 mg, 0.406 mmol, 1.30 eq.) and dry DMF (2.0 mL) were combined. DBU (0.07 mL, 0.5 mmol, 1.5 eq.) was added dropwise, and the reaction mixture was stirred for 10 min at room temperature. Next, (R)-alaninol (0.05 mL, 0.6 mmol, 2 eq.) was added dropwise. The reaction mixture was stirred for 23 h at room temperature and for 2 h at 60 °C. After concentrating in vacuo, EtOAc (40 mL) and saturated NH4Cl (10 mL) were added and the mixture was transferred to a separatory funnel. After phase separation, the aqueous phase was extracted with EtOAc (3 × 20 mL). The combined organic phases were washed with saturated NH4Cl (3 × 10 mL), water (2 × 10 mL) and brine (2 × 10 mL), dried over Na2SO4 and coated on Celite. Flash chromatography using 0–10% MeOH/DCM afforded the title compound (62 mg, 0.16 mmol, 53%) as an off-white solid. mp 174–176 °C.
1H NMR (400 MHz, d6-DMSO): δ (ppm) 8.47 (s, 1H), 7.84 (d, J = 7.9 Hz, 1H), 7.46–7.37 (m, 1H), 7.35–7.24 (m, 1H), 7.19–7.08 (m, 1H), 6.94 (s, 1H), 5.98 (s, 2H), 4.76 (t, J = 5.7 Hz, 1H), 4.42 (s, 2H), 4.37–4.27 (m, 1H), 3.56–3.47 (m, 1H), 3.46–3.37 (m, 1H), 1.16 (d, J = 6.7 Hz, 3H); 13C NMR (101 MHz, d6-DMSO): δ (ppm) 160.8, 156.9, 156.4, 149.6 (dd, J = 12.1, 245.7 Hz), 148.4, 148.1 (dd, J = 12.8, 246.5 Hz), 137.2, 128.7 (d, J = 11.1 Hz), 126.3 (t, J = 2.8 Hz), 124.5 (dd, J = 4.6, 6.9 Hz), 120.5, 116.0 (d, J = 16.9 Hz), 94.6, 63.7, 48.3, 27.0, 16.6; HRMS (ESI-Q-TOF): m/z [M+H]+ calcd. for C17H17F2N5O1S1: 378.11945; found: 378.1200.
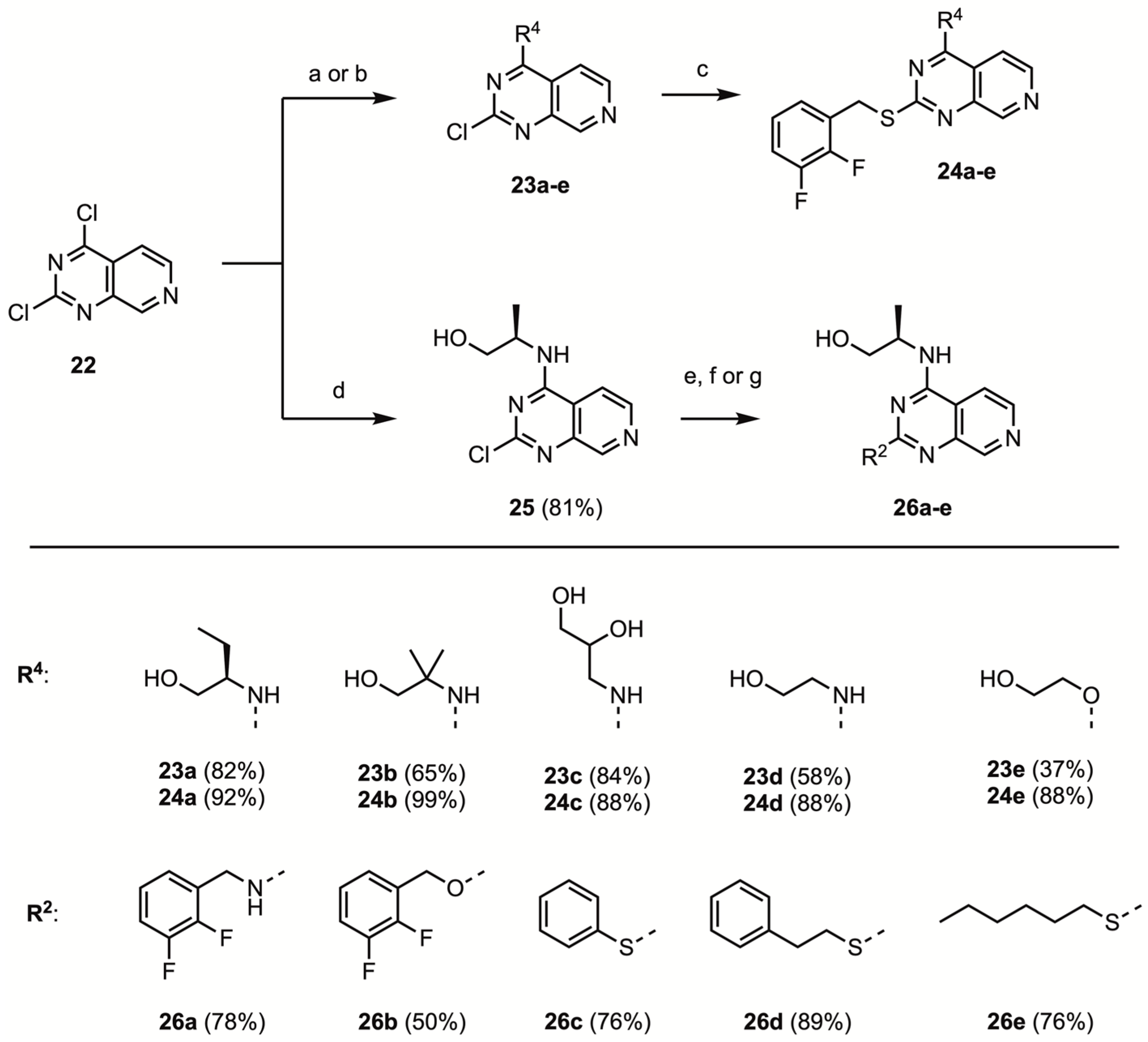
2,4-Dichloro-pyrido[3,4-d]pyrimidine (22)
This compound was prepared following a reported procedure [
54].
A mixture of 3-aminoisonicotinic acid (10.00 g, 72.4 mmol, 1.00 eq.) and urea (21.74 g, 181.0 mmol) in a screw-capped sealed 80 mL reaction tube was heated to 210 °C for 1 h. After cooling to room temperature, a solution of NaOH (14.2 g, 360 mmol) in 60 mL water was added and the mixture was heated to 100 °C for 1 h. Next, the reaction mixture was cooled down to room temperature, the solid was filtered and washed with water. The obtained crude product was suspended in AcOH (60 mL) and heated to 100 °C for 1 h. After cooling to room temperature, the reaction mixture was filtered and the obtained solid was washed with a large amount of water, and a small amount of methanol and diethyl ether. After drying in vacuo (40 °C) pyrido[3,4-d]pyrimidine-2,4-dione (9.948 g, 61.0 mmol, 84%) was obtained as a white solid.
Pyrido[3,4-d]pyrimidine-2,4-dione was first finely ground with a pestle and mortar. To a flame dried two-necked 500 mL round bottom flask, equipped with an Ar-balloon, septum and stir bar, pyrido[3,4-d]pyrimidine-2,4-dione (9.800 g, 60.07 mmol, 1.00 eq.), dry toluene (100 mL) and POCl3 (55 mL, 92 mmol, 10 eq.) were added. While cooling to 0 °C in an ice bath, DIPEA (21.5 mL, 15.9 mmol, 123.1 eq.) was added dropwise. After stirring at 0 °C for 30 min, the reaction mixture was stirred at room temperature for 24 h and concentrated in vacuo. To the obtained oil, diethyl ether and ice water were added. The mixture was neutralized with saturated aqueous NaHCO3 (pH = 7) while cooling in an ice bath. Due to extensive tar formation, phase separation of the aqueous and organic layers did not occur. This was solved by pouring the mixture into a large beaker (2L) and adding a lot of paper towel to adsorb the tar. This mixture was extracted 5 times with 300 mL diethyl ether, by manually stirring the contents of the beaker with a large spatula and decanting off the organic phase. The combined organic phases were washed with water (2 × 50 mL) and brine (2 × 50 mL), decolorized with activated charcoal and dried over Na2SO4. After concentrating to dryness in vacuo, while keeping the heating bath at room temperature, and drying in vacuo (30 °C), the title compound (6.544 g, 32.72 mmol, 55%) was obtained as a beige-brown solid which was used without further purification.
General procedure A: Nucleophilic aromatic substitution of 2,4-dichloro-pyrido[3,4-d]pyrimidine with amines
To a flame dried, N2-flushed 8 mL reaction tube equipped with a stirring bar 2,4-dichloro-pyrido[3,4-d]pyrimidine 22 (200 mg, 1.00 mmol, 1.00 eq.) and dry ACN (4.0 mL) were added. While cooling to 0 °C in an ice bath, Et3N (0.21 mL, 1.5 mmol, 1.5 eq.) and an amine nucleophile (1.5 mmol, 1.5 eq.) were added dropwise. After stirring for 10 min at 0 °C, the reaction mixture was stirred at room temperature for the specified time. During and after the addition of Et3N and nucleophile, generally, the formation of a yellow precipitate occurred. Next, the reaction mixture was transferred to a 100 mL round bottom flask with methanol; the entire sample was dissolved under gentle heating, and coated on Celite. Flash chromatography using 0–20% MeOH/DCM afforded the title compounds as white or beige solids.
General procedure B: Nucleophilic aromatic substitution of 2,4-dichloro-pyrido[3,4-d]pyrimidine with alcohols
NaH (60% dispersion) was added to a solution of alcohol nucleophile (1.1 eq.) in dry acetonitrile (2.0 mL), in a flame dried N2-flushed 8 mL screw capped reaction tube, and the mixture was sonicated for 30 min. In a separate flame dried, N2-flushed 8 mL reaction tube equipped with a stir bar, 2,4-dichloro-pyrido[3,4-d]pyrimidine 22 (200 mg, 1.00 mmol, 1.00 eq.) and dry ACN (3.0 mL) were added. While cooling to 0 °C in an ice bath, the solution of alcohol sodium salt was added dropwise to the 2,4-dichloro-pyrido[3,4-d]pyrimidine solution. After stirring for 10 min at 0 °C, the reaction mixture was stirred at room temperature for the specified time. Next, the reaction mixture was transferred to a 100 mL round bottom flask with methanol; the entire sample was dissolved under gentle heating, and coated on Celite. Flash chromatography using 0–100% EtOAc/DCM, followed by trituration with pentane and drying under vacuum (40 °C), afforded the title compounds as white or beige solids.
General procedure C: Nucleophilic aromatic substitution of 2-chloropyrido[3,4-d]pyrimidines with thiols
To a flame dried, N2-flused 8 mL reaction tube equipped with a stir bar, the appropriate 2-chloropyrido[3,4-d]pyrimidine (0.25 mmol, 1.00 eq.), thiol nucleophile (0.50 mmol, 2.00 eq.) and dry 1,4-dioxane (1 mL) were combined. NaH 60% dispersion in mineral oil (20 mg, 0.50 mmol, 0.50 eq.) was added in one portion. After stirring at room temperature overnight, methanol (1.0 mL) was added and the mixture was heated to 60 °C for 30 min. After cooling to room temperature, the reaction mixture was transferred to a 100 mL round bottom flask with methanol; the entire sample was dissolved under gentle heating, and coated on Celite. Flash chromatography using 0–20 MeOH/DCM, followed by trituration with pentane and drying under vacuum (40 °C), afforded the title compounds as white solids.
The following compounds were made according to these procedures:
(2R)-2-[(6-Chloro-2-pyrido[3,4-d]pyrimidine-4-yl)amino]butanol (23a)
This compound was prepared following general procedure A. (R)-2-Aminobutan-1-ol (0.14 mL, 1.5 mmol, 1.5 eq.) was reacted for 3h at room temperature. Flash chromatography was performed using 0–20% MeOH/DCM to afford the title compound (206 mg, 0.815 mmol, 82%) as an off-white solid. mp> 300 °C.
1H NMR (400 MHz, d6-DMSO): δ (ppm) 8.98 (s, 1H), 8.72–8.55 (m, 2H), 8.29 (d, J = 5.4 Hz, 1H), 4.82 (t, J = 5.2 Hz, 1H), 4.35–4.19 (m, 1H), 3.65–3.50 (m, 2H), 1.83–1.68 (m, 1H), 1.67–1.51 (m, 1H), 0.90 (t, J = 7.3 Hz, 3H); 13C NMR (400 MHz, d6-DMSO): δ (ppm) 160.8, 158.3, 150.5, 145.1, 144.2, 118.0, 115.9, 62.1, 54.9, 23.2, 10.6; HRMS (ESI-Q-TOF): m/z [M+H]+ calcd. for C11H13Cl1N4O1: 253.0851; found: 253.0848.
2-[(2-Chloro-pyrido[3,4-d]pyrimidine-4-yl)amino]-2-methyl-propan-1-ol (23b)
This compound was prepared following general procedure A. 2-Amino-2-methylpropan-1-ol (0.14 mL, 1.5 mmol, 1.5 eq.) was reacted for 4h at room temperature. Flash chromatography was performed using 0–20% MeOH/DCM, followed by a second column using 0–10 MeOH/Et2O to afford the title compound (165 mg, 0.653 mmol, 65%) as an off-white solid. mp > 300 °C.
1H NMR (400 MHz, d6-DMSO): δ (ppm) 8.96 (s, 1H), 8.59 (d, J = 5.6 Hz, 1H), 8.41 (d, J = 5.6 Hz, 1H), 5.16 (br. s, 1H), 3.70 (s, 2H), 1.44 (s, 6H); 13C NMR (101 MHz, d6-DMSO): δ (ppm) 160.0, 157.2, 150.5, 144.9, 144.1, 118.5, 116.3, 65.8, 57.5, 23.1; HRMS (ESI-Q-TOF): m/z [M+H]+ calcd. for C11H13Cl1N4O1: 253.08505; found: 253.0854.
3-[(2-Chloro-pyrido[3,4-d]pyrimidine-4-yl)amino]propan-1,2-diol (23c)
This compound was prepared following general procedure A. 3-Aminopropan-1,2-diol (0.12 mL, 1.5 mmol, 1.5 eq.) was reacted for 1h30 at room temperature. Flash chromatography was performed using 0–20% MeOH/DCM to afford the title compound (214 mg, 0.840 mmol, 84%) as a beige solid. mp > 300 °C.
1H NMR (400 MHz, d6-DMSO): δ (ppm) 9.06 (t, J = 5.0 Hz, 1H), 8.98 (s, 1H), 8.61 (d, J = 5.5 Hz, 1H), 8.19 (d, J = 5.5 Hz, 1H), 4.96 (d, J = 4.9 Hz, 1H), 4.68 (t, J = 5.5 Hz, 1H), 3.90–3.75 (m, 1H), 3.65 (dt, J = 4.9, 13.2 Hz, 1H), 3.51–3.38 (m, 3H); 13C NMR (101 MHz, d6-DMSO): δ (ppm) 160.7, 158.2, 150.5, 144.9, 144.3, 118.1, 115.8, 69.1, 64.0, 44.8; HRMS (ESI-Q-TOF): m/z [M+H]+ calcd. for C10H11Cl1N4O2: 255.06432; found: 255.0643.
2-[(2-Chloro-pyrido[3,4-d]pyrimidine-4-yl)amino]ethanol (23d)
This compound was prepared following general procedure A. Aminoethanol (0.1 mL, 1.7 mmol, 1.7 eq.) was reacted for 2h at room temperature. Flash chromatography was performed using 0–20% MeOH/DCM followed by a second manual column using 0–10% MeOH/Et2O to afford the title compound (131 mg, 0.583 mmol, 58%) as an off-white solid. mp > 300 °C.
1H NMR (400 MHz, d6-DMSO): δ(ppm) 9.14 (s, 1H), 8.99 (d, J = 0.5 Hz, 1H), 8.63 (d, J = 5.6 Hz, 1H), 8.17 (dd, J = 0.7, 5.6 Hz, 1H), 4.87 (t, J = 5.4 Hz, 1H), 3.69–3.55 (m, 2H); 13C NMR (400 MHz, d6-DMSO): δ(ppm) 160.6, 158.2, 150.5, 144.9, 118.1, 115.7, 58.5, 43.8; HRMS (ESI-Q-TOF): m/z [M+H]+ calcd. for C9H9Cl1N4O1: 225.05376; found: 225.0539.
3-[(2-(2,3-Difluorobenzylmercapto)-pyrido[3,4-d]pyrimidine-4-yl)oxy]ethanol (23e)
This compound was prepared following general procedure B. 3-[(2-Chloro-pyrido[3,4-d]pyrimidine-4-yl)amino]ethanol (56 mg, 0.25 mmol, 1.00 eq.) and 2,3-difluorobenzylthiol (80 mg mL, 0.50 mmol, 2.00 eq.) were used and reacted at 60 °C for 30 min. Flash chromatography was performed using 0–10% MeOH/DCM to afford the title compound (77 mg, 0.22 mmol, 88%) as a beige-brown solid. mp 181–183 °C.
1H NMR (400 MHz, DMSO): δ(ppm) 8.94 (s, 1H), 8.76 (t, J = 4.9 Hz, 1H), 8.50 (d, J = 5.5 Hz, 1H), 8.07 (d, J = 5.2 Hz, 1H), 7.47–7.38 (m, 1H), 7.35–7.23 (m, 1H), 7.18–7.08 (m, 1H), 4.83 (t, J = 5.3 Hz, 1H), 4.49 (s, 2H), 3.66–3.52 (m, 2H); 13C NMR (101 MHz, DMSO): δ(ppm) 163.4, 158.2, 150.1, 149.6 (dd, J = 12.6, 245.3 Hz), 148.2 (dd, J = 12.8, 246.8 Hz), 144.3, 143.0, 128.3 (d, J = 11.4 Hz), 126.3 (t, J = 11.4 Hz), 126.3 (t, J = 2.8 Hz), 124.6 (dd, J = 4.7, 6.9 Hz), 117.3, 116.3 (d, J = 16.9 Hz), 115.6, 58.7, 43.5, 27.2; 19F NMR (377 MHz, DMSO): δ(ppm) −139.35 (m, 1F), −148.50 (m, 1F) HRMS (ESI-Q-TOF): m/z [M+H]+ calcd. for C16H14F2N4O1S1: 349.09290; found: 349.0922.
(2R)-2-[(2-(2,3-Difluorobenzylmercapto)-pyrido[3,4-d]pyrimidine-4-yl)amino]butanol (24a)
This compound was prepared following general procedure C. (2R)-2-[(6-Chloro-2-pyrido[3,4-d]pyrimidine-4-yl)amino]butanol (63 mg, 0.25 mmol, 1.00 eq.) and 2,3-difluorobenzylthiol (80 mg mL, 0.50 mmol, 2.00 eq.) were used and reacted at 60 °C for 30 min. Flash chromatography was performed using 0–10% MeOH/DCM to afford the title compound (87 mg, 0.23 mmol, 92%) as a white solid. mp 174–176 °C.
1H NMR (400 MHz, d6-DMSO): δ (ppm) 8.93 (s, 1H), 8.50 (d, J = 5.5 Hz, 1H), 8.25 (d, J = 8.1 Hz, 1H), 8.19 (d, J = 5.5 Hz, 1H), 7.51–7.37 (m, 1H), 7.36–7.23 (m, 1H), 7.20–7.07 (m, 1H), 7.77 (t, J = 5.4 Hz, 1H), 4.49 (d, J = 1.9 Hz, 2H), 4.37–4.17 (m, 1H), 3.59–3.43 (m, 2H), 1.79–1.65 (m, 1H), 1.64–1.49 (m, 1H), 0.86 (t, J = 7.4 Hz, 3H); 13C NMR (101 MHz, d6-DMSO): δ (ppm) 167.4, 158.3, 150.1, 149.6 (dd, J = 12.5, 245.4 Hz), 148.1 (dd, J = 12.9, 246.6 Hz), 144.4, 142.8, 128.3 (d, J = 11.1 Hz), 126.3 (t, J = 2.7 Hz), 124.6 (dd, J = 4.7, 6.9 Hz), 117.3, 116.1 (d, J = 16.9 Hz), 115.8, 62.2, 54.4, 27.2, 23.3, 10.6; 19F NMR (377 MHz, d6-DMSO): δ (ppm) −139.39 (m, 1F), −142.85 (m, 1F); HRMS (ESI-Q-TOF): m/z [M+H]+ calcd. for C18H18F2N4O1S1: 377.12420; found: 377.1239.
2-[(2-(2,3-Difluorobenzylmercapto)-pyrido[3,4-d]pyrimidine-4-yl)amino]-2-methyl-propan-1-ol (24b)
This compound was prepared following general procedure C. 2-[(2-Chloro-pyrido[3,4-d]pyrimidine-4-yl)amino]-2-methyl-propan-1-ol (63 mg, 0.25 mmol, 1.00 eq.) and 2,3-difluorobenzylthiol (80 mg mL, 0.50 mmol, 2.00 eq.) were used and reacted at 60 °C for 30 min. Flash chromatography was performed using 0–10% MeOH/DCM to afford the title compound (93 mg, 0.247 mmol, 99%) as a white solid. mp 166–168 °C.
1H NMR (400 MHz, d6-DMSO): δ (ppm) 8.94 (s, 1H), 8.49 (d, J = 5.5 Hz, 1H), 8.23 (d, J = 5.5 Hz, 1H), 7.49 (s, 1H), 7.47–7.40 (m, 1H), 7.35- 7.25 (m, 1H), 7.20–7.10 (m, 1H), 4.88 (t, J = 5.9 Hz, 1H), 4.49 (s, 2H), 3.66 (d, J = 5.8 Hz, 2H), 1.40 (s, 6H); 13C NMR (101 MHz, d6-DMSO): δ (ppm) 166.7, 157.7, 150.1, 149.6 (dd, J = 12.6, 245.4 Hz), 148.2 (dd, J = 12.8, 246.7 Hz), 144.2, 142.9, 127.9 (d, J = 11.2 Hz), 126.3 (t, J = 2.9 Hz), 124.6 (dd, J = 4.7, 6.9 Hz), 117.7, 116.2 (d, J = 16.9 Hz), 116.0, 65.9, 56.9, 27.2, 23.3; 19F NMR (377 MHz, d6-DMSO): δ (ppm) −139.33 (m, 1F), −142.78 (m, 1F); HRMS (ESI-Q-TOF): m/z [M+H]+ calcd. for C18H18F2N4O1S1: 377.12420; found: 377.1238.
3-[(2-(2,3-Difluorobenzylmercapto)-pyrido[3,4-d]pyrimidine-4-yl)amino]propan-1,2-diol (24c)
This compound was prepared following general procedure C. 3-[(2-Chloro-pyrido[3,4-d]pyrimidine-4-yl)amino]propan-1,2-diol (64 mg, 0.25 mmol, 1.00 eq.) and 2,3-difluorobenzylthiol (80 mg mL, 0.50 mmol, 2.00 eq.) were used and reacted at 60 °C for 30 min. Flash chromatography was performed using 0–10% MeOH/DCM to afford the title compound (84 mg, 0.22 mmol, 88%) as an off-white solid. mp = 156–158 °C.
1H NMR (400 MHz, d6-DMSO): δ (ppm) 8.94 (s, 1H), 8.72 (t, J = 5.5 Hz, 1H), 8.50 (d, J = 5.4 Hz, 1H), 8.11 (d, J = 5.4 Hz, 1H), 7.50–7.37 (app. t, J = 6.7 Hz, 1H), 7.36–7.23 (m, 1H), 7.19–7.07 (m, 1H), 4.92 (d, J = 4.7 Hz, 1H), 4.65 (t, J = 5.4 Hz, 1H), 4.50 (s, 2H), 3.88–3.75 (m, 1H), 3.74–3.61 (m, 2H), 3.45–3.37 (m, 2H); 13C NMR (101 MHz, d6-DMSO): δ (ppm) 167.3, 158.2, 150.1, 149.6 (dd, J = 12.5, 245.2 Hz), 148.2 (dd, J = 12.8, 246.8 Hz), 144.3, 143.0, 128.3 (d, J = 11.2 Hz), 126.3 (t, J = 2.6 Hz), 124.6 (dd, J = 4.7, 7.0 Hz), 117.3, 116.2 (d, J = 16.9 Hz), 115.7, 69.3, 64.0, 44.5, 27.3; 19F NMR (377 MHz, d6-DMSO): δ (ppm) −139.31 (m, 1F), −142.70 (m, 1F); HRMS (ESI-Q-TOF): m/z [M+H]+ calcd. for C17H16F2N4O2S1: 379.10347; found: 379.1031.
3-[(2-(2,3-Difluorobenzylmercapto)-pyrido[3,4-d]pyrimidine-4-yl)amino]ethanol (24d)
This compound was prepared following general procedure C. 3-[(2-Chloro-pyrido[3,4-d]pyrimidine-4-yl)amino]ethanol (56 mg, 0.25 mmol, 1.00 eq.) and 2,3-difluorobenzylthiol (80 mg mL, 0.50 mmol, 2.00 eq.) were used and reacted at 60 °C for 30 min. Flash chromatography was performed using 0–10% MeOH/DCM to afford the title compound (77 mg, 0.22 mmol, 88%) as a white solid. mp 181–183 °C.
1H NMR (400 MHz, d6-DMSO): δ (ppm) 8.94 (s, 1H), 8.76 (t, J = 4.9 Hz, 1H), 8.50 (d, J = 5.5 Hz, 1H), 8.07 (d, J = 5.2 Hz, 1H), 7.47–7.38 (m, 1H), 7.35–7.23 (m, 1H), 7.18–7.08 (m, 1H), 4.83 (t, J = 5.3 Hz, 1H), 4.49 (s, 2H), 3.66–3.52 (m, 2H); 13C NMR (101 MHz, d6-DMSO): δ (ppm) 163.4, 158.2, 150.1, 149.6 (dd, J = 12.6, 245.3 Hz), 148.2 (dd, J = 12.8, 246.8 Hz), 144.3, 143.0, 128.3 (d, J = 11.4 Hz), 126.3 (t, J = 11.4 Hz), 126.3 (t, J = 2.8 Hz), 124.6 (dd, J = 4.7, 6.9 Hz), 117.3, 116.3 (d, J = 16.9 Hz), 115.6, 58.7, 43.5, 27.2; 19F NMR (377 MHz, d6-DMSO): δ (ppm) −139.35 (m, 1F), −148.50 (m, 1F); HRMS (ESI-Q-TOF): m/z [M+H]+ calcd. for C16H14F2N4O1S1: 349.09290; found: 349.0922.
3-[(2-(2,3-Difluorobenzylmercapto)-pyrido[3,4-d]pyrimidine-4-yl)oxy]ethanol (24e)
This compound was prepared following general procedure B. 3-[(2-Chloro-pyrido[3,4-d]pyrimidine-4-yl)amino]ethanol (56 mg, 0.25 mmol, 1.00 eq.) and 2,3-difluorobenzylthiol (80 mg mL, 0.50 mmol, 2.00 eq.) were used and reacted at 60 °C for 30 min. Flash chromatography was performed using 0–10% MeOH/DCM to afford the title compound (77 mg, 0.22 mmol, 88%) as a beige-brown solid. mp 181–183 °C.
1H NMR (400 MHz, d6-DMSO): δ (ppm) 8.94 (s, 1H), 8.76 (t, J = 4.9 Hz, 1H), 8.50 (d, J = 5.5 Hz, 1H), 8.07 (d, J = 5.2 Hz, 1H), 7.47–7.38 (m, 1H), 7.35–7.23 (m, 1H), 7.18–7.08 (m, 1H), 4.83 (t, J = 5.3 Hz, 1H), 4.49 (s, 2H), 3.66–3.52 (m, 2H); 13C NMR (101 MHz, d6-DMSO): δ (ppm) 163.4, 158.2, 150.1, 149.6 (dd, J = 12.6, 245.3 Hz), 148.2 (dd, J = 12.8, 246.8 Hz), 144.3, 143.0, 128.3 (d, J = 11.4 Hz), 126.3 (t, J = 11.4 Hz), 126.3 (t, J = 2.8 Hz), 124.6 (dd, J = 4.7, 6.9 Hz), 117.3, 116.3 (d, J = 16.9 Hz), 115.6, 58.7, 43.5, 27.2; 19F NMR (377 MHz, d6-DMSO): δ (ppm) −139.35 (m, 1F), −148.50 (m, 1F); HRMS (ESI-Q-TOF): m/z [M+H]+ calcd. for C16H14F2N4O1S1: 349.09290; found: 349.0922.
(2R)-2-[(6-Chloro-2-pyrido[3,4-d]pyrimidine-4-yl)amino]propanol (25)
This compound was prepared following general procedure A. 2,4-Dichloropyrido[3,4-d]pyrimidine (1 g, 5.00 mmol, 1.00 eq.) and (R)-alaninol (0.59 mL, 7.6 mmol, 1.5 eq.) was reacted for 3h at room temperature. Flash chromatography was performed using 0–20% MeOH/DCM, followed by trituration with pentane afforded the title compound (970 mg, 4.06 mmol, 81%) as an off-white solid. mp > 300 °C.
1H NMR (400 MHz, d6-DMSO): δ (ppm) 8.97 (s, 1H), 8.81 (d, J = 7.8 Hz, 1H), 8.61 (d, J = 5.6 Hz, 1H), 8.32 (d, J = 5.6 Hz, 1H), 4.91 (br. s, 1H), 4.47–4.30 (m, 1H), 3.61–3.46 (m, 2H), 1.23 (d, J = 6.7 Hz, 3H); 13C NMR (101 MHz, d6-DMSO): δ (ppm) 160.2 158.3, 150.5, 145.1, 144.2, 118.1, 116.0, 63.5, 49.1, 16.3; HRMS (ESI-Q-TOF): m/z [M+Na]+ calcd. for C10H11Cl1N4O1: 261.0514 found: 261.0512.
(2R)-2-[(2-(2,3-Difluorobenzylamino)-pyrido[3,4-d]pyrimidine-4-yl)amino]propanol (26a)
In a flame dried, N2-flushed 8 mL screw-capped reaction tube equipped with a stirring bar (2R)-2-[(6-chloro-2-pyrido[3,4-d]pyrimidine-4-yl)amino]propanol 25 (119 mg, 0.500 mmol, 1.00 eq.), 2,3-difluorobenzylamine (161 mg, 1.13 mmol, 2.25 eq.) and dry 1,4-dioxane (5 mL) were added. The mixture was stirred at 100 °C for 22 h. After cooling to room temperature, the reaction mixture was dissolved in methanol and coated on Celite. Flash chromatography using 0–20% MeOH/DCM, followed by trituration with pentane, afforded the title compound (135 mg, 0.391 mmol, 78%) as a white-yellow solid. mp 136–140 °C.
1H NMR (400 MHz, d6-DMSO): δ (ppm) 8.59 (s, 1H), 8.15 (d, J = 4.9 Hz, 1H), 7.93 (d, J = 5.4 Hz, 1H), 7.75 (br. s, 1H), 7.39 (br. s, 1H),7.31–7.17 (m, 2H), 7.16–7.08 (m, 2H), 4.74 (br. s, 1H), 4.62 (d, J = 6.0 Hz, 2H), 4.35 (br. s, 1H), 3.60–3.39 (m, 2H), 1.27–1.02 (m, 3H); 13C NMR (101 MHz, d6-DMSO): δ (ppm) 160.1, 15.9, 149.5 (dd, J = 12.7, 245.0 Hz), 148.9, 147.7 (dd, J = 12.6, 245.3 Hz), 146.8, 138.9, 130.3, 124.4 (dd, J = 4.5, 7.0 Hz), 115.6, 115.4 (d, J = 17.0 Hz), 63.8, 48.1, 37.6, 16.8; 19F NMR (377 MHz, d6-DMSO): δ (ppm) −140.15 (m, 1F), −145.26 (m, 1F); HRMS (ESI-Q-TOF): m/z [M+H]+ calcd. for C17H17F2N5O1: 346.1474; found: 346.145.
(2R)-2-[(2-(2,3-Difluorobenzyloxy)-pyrido[3,4-d]pyrimidine-4-yl)amino]propanol (26b)
In a flame dried, N2-flushed 8 mL screw-capped reaction tube equipped with a stir bar (2R)-2-[(6-chloro-2-pyrido[3,4-d]pyrimidine-4-yl)amino]propanol 25 (119 mg, 0.500 mmol, 1.00 eq.), 2,3-difluorobenzylalcohol (0.56 mL, 5.0 mmol, 10 eq.), dry 1,4-dioxane (4 mL) and K2CO3 (138 mg, 1.00 mmol, 2.00 eq.) were added. The mixture was stirred at 120 °C for 48 h. After cooling to room temperature, the reaction mixture was dissolved in methanol and coated on Celite. Flash chromatography using 0–10% MeOH/DCM, followed by trituration with pentane, afforded the title compound (87 mg, 0.25 mmol, 50%) as a light-brown solid. mp 167–169 °C.
1H NMR (400 MHz, d6-DMSO): δ (ppm) 8.88 (s, 1H), 8.43 (d, J = 5.2 Hz, 1H), 8.38 (d, J = 7.6 Hz, 1H), 8.15 (d, J = 5.3 Hz, 1H), 7.70–7.33 (m, 2H), 7.32–7.12 (m, 1H), 5.48 (s, 2H), 4.82 (t, J = 5.5 Hz, 1H), 4.47–4.26 (m, 2H), 3.62–3.50 (m, 1H), 3.50–3.40 (m, 1H), 1.21 (d, J = 6.6 Hz, 3H); 13C NMR (101 MHz, d6-DMSO): δ (ppm) 162.7, 160.9, 149.9, 149.6 (dd, J = 12.3, 245.4 Hz), 148.2 (dd, J = 12.9, 247.7 Hz), 145.6, 141.7, 126.7 (d, J = 11.4 Hz), 125.9 (t, J = 2.6 Hz), 124.9 (dd, J = 4.7, 6.7 Hz), 117.3 (d, J = 16.8 Hz), 116.8, 115.8, 63.7, 61.4, 48.7, 16.5; 19F NMR (377 MHz, d6-DMSO): δ (ppm) −139.34 (m, 1F), −143.8 (m, 1F); HRMS (ESI-Q-TOF): m/z [M+H]+ calcd. for C17H16F2N4O2: 347.1314; found: 347.1319.
(2R)-2-[(2-(Phenylmercapto)-pyrido[3,4-d]pyrimidine-4-yl)amino]propanol (26c)
This compound was prepared following general procedure C. (2R)-2-[(6-Chloro-2-pyrido[3,4-d]pyrimidine-4-yl)amino]propanol (60 mg, 0.25 mmol, 1.00 eq.) and thiophenol (55 mg, 0.50 mmol, 2.00 eq.) were used and reacted at 60 °C for 1 h 30 min. Flash chromatography was performed using 0–20% MeOH/DCM to afford the title compound (62 mg, 0.19 mmol, 76%) as an off-white solid. mp 193–196 °C.
1H NMR (400 MHz, d6-DMSO): δ (ppm) 8.79 (s, 1H), 8.47 (d, J = 5.5 Hz, 1H), 8.28 (d, J = 7.4 Hz, 1H), 8.13 (d, J = 5.5 Hz, 1H), 7.70–7.60 (m, 2H), 7.51–7.40 (m, 3H), 4.73 (t, J = 5.6 Hz, 1H), 4.12–3.96 (m, 1H), 3.45–3.37 (m, 2H), 1.08 (d, J = 6.7 Hz, 3H); 13C NMR (101 MHz, d6-DMSO): δ (ppm) 168.4, 157.8, 150.1, 144.7, 142.7, 135.0, 129.4, 128.94, 128.85, 117.3, 115.7, 63.3, 48.8, 16.3; HRMS (ESI-Q-TOF): m/z [M+H]+ calcd. for C16H16N4O1S1: 347.1314; found: 347.1319.
(2R)-2-[(2-(Phenylethylmercapto)-pyrido[3,4-d]pyrimidine-4-yl)amino]propanol (26d)
This compound was prepared following general procedure C. (2R)-2-[(6-Chloro-2-pyrido[3,4-d]pyrimidine-4-yl)amino]propanol (60 mg, 0.25 mmol, 1.00 eq.) and 2-phenylethanethiol (69 mg, 0.50 mmol, 2.00 eq.) were used and reacted at 60 °C for 1 h 30 min. Flash chromatography was performed using 0–20% MeOH/DCM, followed by trituration with pentane, to afford the title compound (76 mg, 0.223 mmol, 89%) as a white solid. mp 182–184 °C.
1H NMR (400 MHz, d6-DMSO): δ (ppm) 8.91 (s, 1H), 8.48 (d, J = 5.6 Hz, 1H), 8.26 (d, J = 7.8 Hz, 8.15 (d, J = 5.5 Hz, 1H), 7.44–7.26 (m, 4H), 7.26–7.17 (m, 1H), 4.83 (br. s, 1H), 4.50–4.35 (m, 1H), 3.62–3.44 (m, 2H), 3.42–3.35 (m, 2H), 3.01 (t, J = 7.7 Hz, 2H), 0.122 (d, J = 6.7 Hz, 3H); 13C NMR (101 MHz, d6-DMSO): δ (ppm) 168.5, 157.6, 150.1, 144.6, 142.6, 140.5, 128.6, 128.4, 126.2, 117.1, 115.7, 63.7, 48.6, 35.6, 31.5, 16.6; HRMS (ESI-Q-TOF): m/z [M+H]+ calcd. for C18H20N4O1S1: 341.1431; found: 341.1427.
(2R)-2-[(2-(Hexylmercapto)-pyrido[3,4-d]pyrimidine-4-yl)amino]propanol (26e)
This compound was prepared following general procedure C. (2R)-2-[(6-Chloro-2-pyrido[3,4-d]pyrimidine-4-yl)amino]propanol (60 mg, 0.25 mmol, 1.00 eq.) and hexane thiol (59 mg, 0.50 mmol, 2.00 eq.) were used and reacted at 60 °C for 30 min. Flash chromatography was performed using 0–20% MeOH/DCM to afford the title compound (62 mg, 0.19 mmol, 76%) as an off-white solid. mp 147–149 °C.
1H NMR (400 MHz, d6-DMSO): δ (ppm) 8.85 (s, 1H), 8.46 (d, J = 5.5 Hz, 1H), 8.23 (d, J = 7.8 Hz, 1H), 8.13 (d, J = 5.4 Hz, 1H), 4.81 (t, J = 5.7 Hz, 1H), 4.39 (septet, J = 6.6 Hz, 1H), 6.62–3.42 (m, 2H), 3.20–3.00 (m, 2H), 1.67 (quintet, J = 7.4 Hz, 2H), 1.48–1.35 (m, 2H), 1.34–1.24 (m, 4H), 1.23–1.17 (m, 3H), 0.85 (t, J = 7.0 Hz, 3H); 13C NMR (101 MHz, d6-DMSO): δ (ppm) 168.7, 157.5, 150.0, 144.6, 142.4, 117.0, 115.7, 63.6, 48.6, 30.8, 30.0, 29.4, 28.0, 22.0, 16.5, 13.8; HRMS (ESI-Q-TOF): m/z [M+H]+ calcd. for C16H24N4O1S1: 321.1744; found: 321.1747.

 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
























