
Evaluation of Rhenium and Technetium-99m Complexes Bearing Quinazoline Derivatives as Potential EGFR Agents
 ,
,  , , ,
, , ,
Abstract
:1. Introduction
2. Results and Discussion
2.1. Chemistry
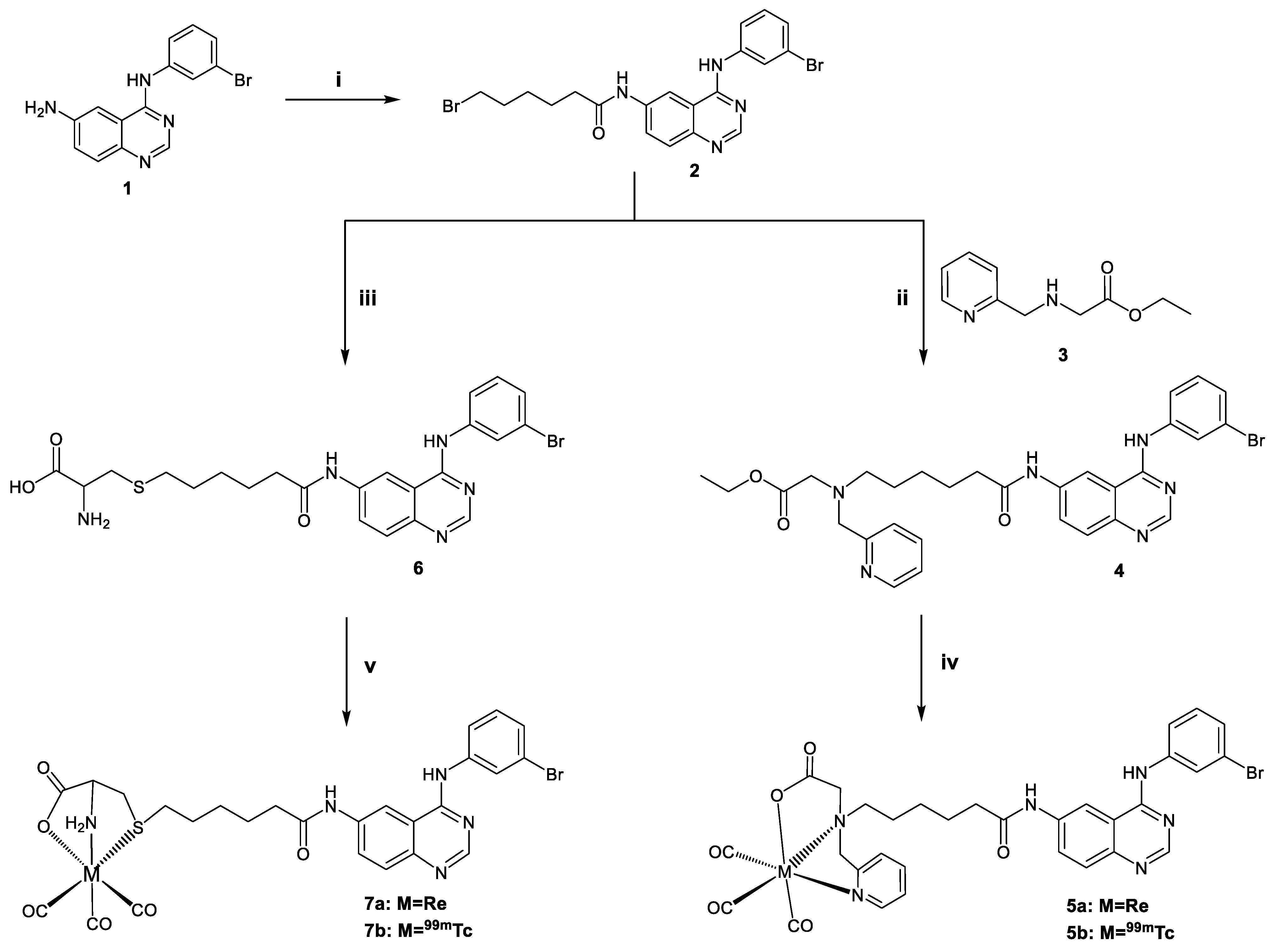
2.1.1. Synthesis of the Ligands
2.1.2. Synthesis of the Rhenium Complexes
2.2. Spectroscopy Studies
2.3. Radiochemistry
Preparation of 99mTc-Complexes
2.4. Biological Evaluation
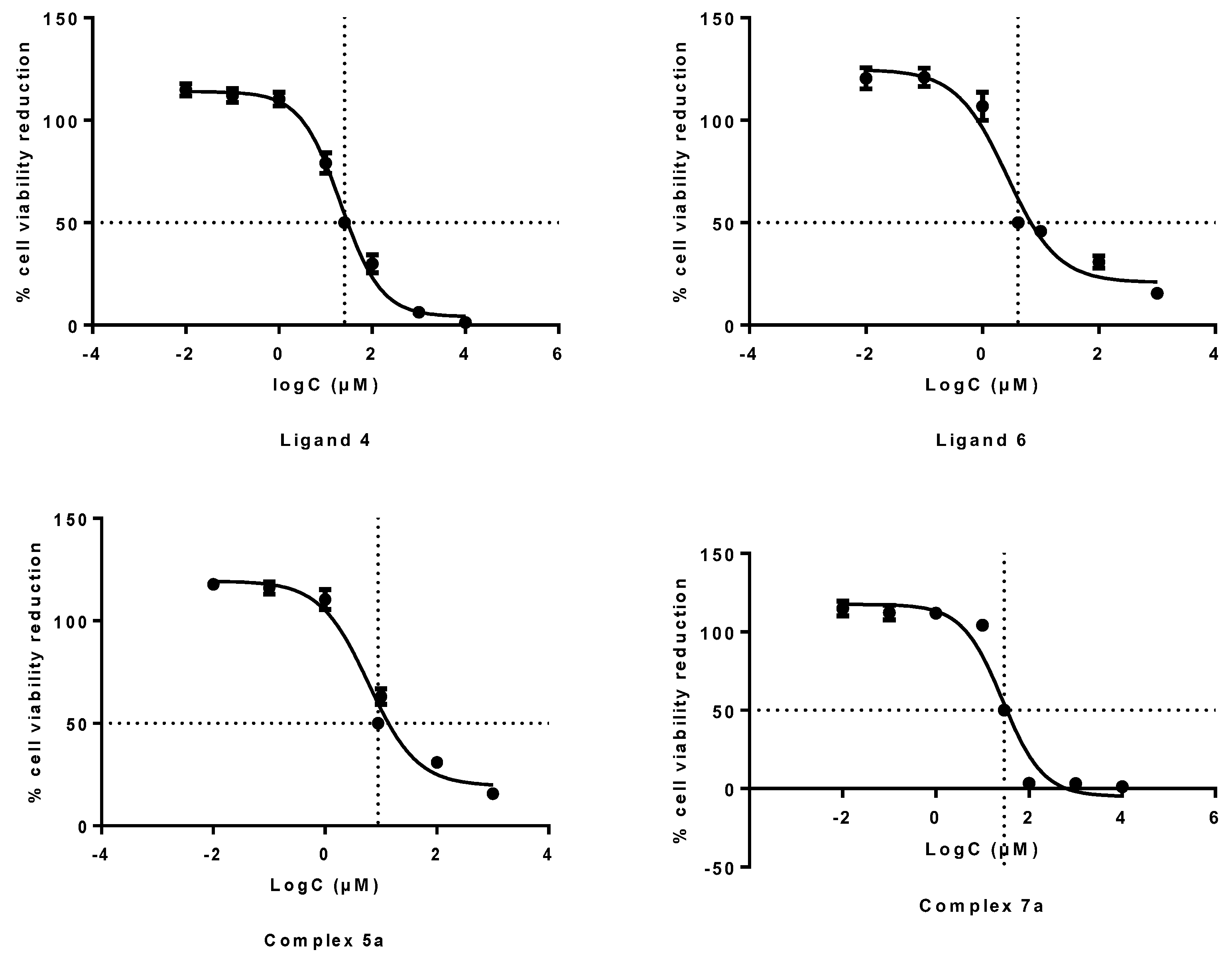
2.4.1. Inhibition of Cell Growth (MTT Assay)
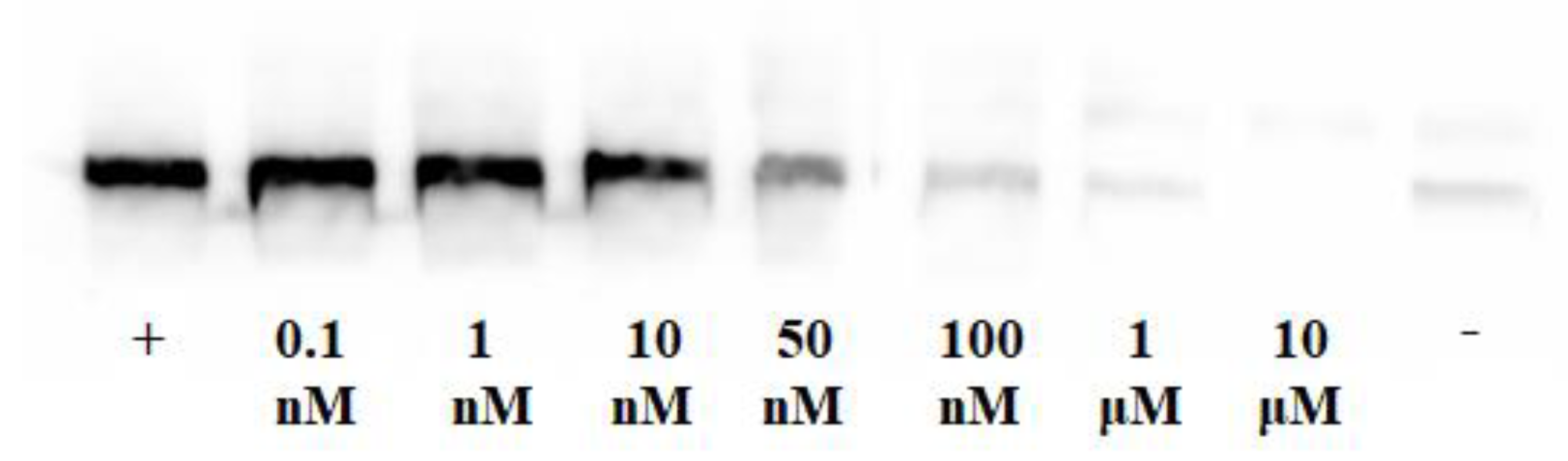
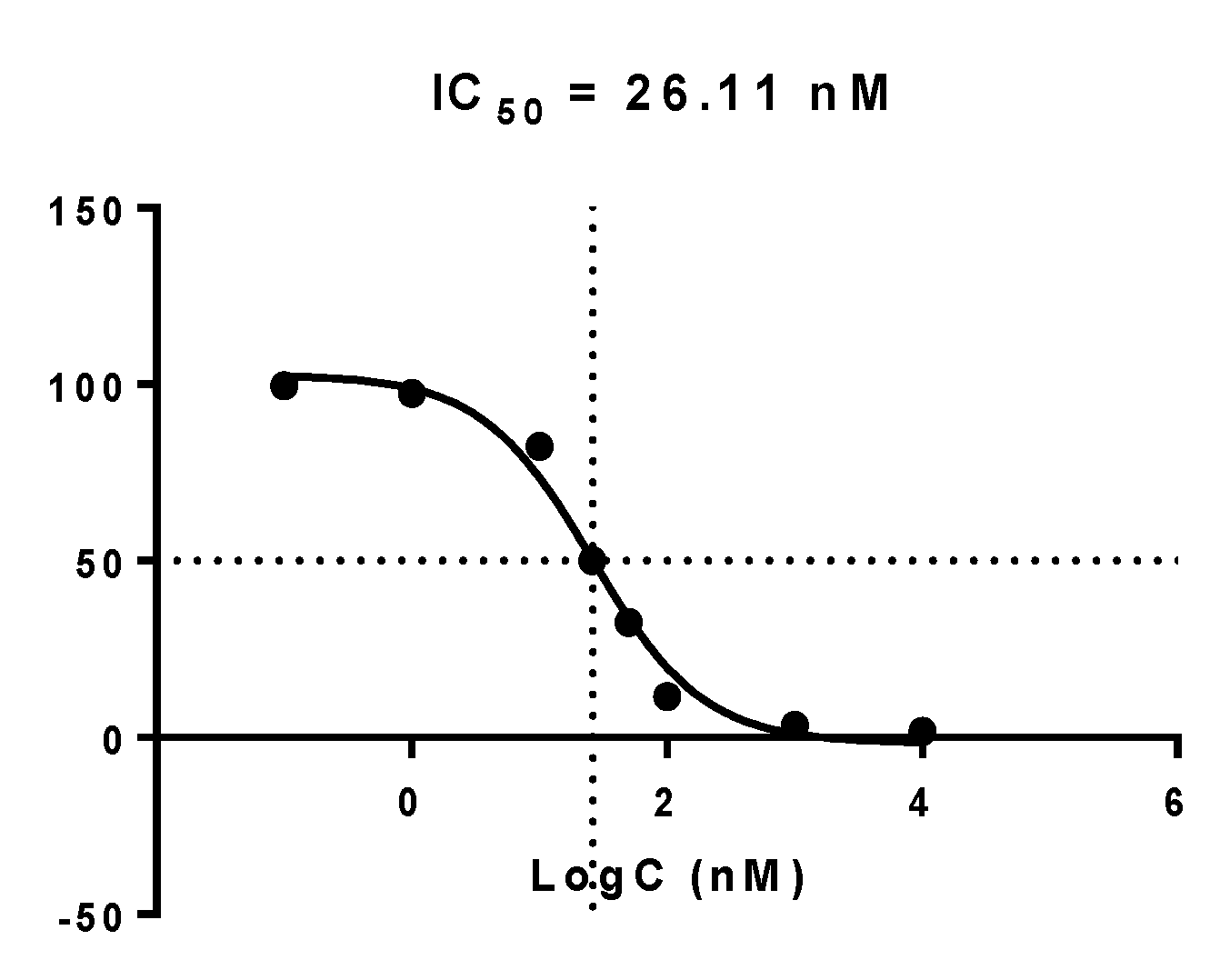
2.4.2. Inhibition of EGFR Phosphorylation
2.5. Biodistribution Studies
3. Materials and Methods
3.1. Synthesis
3.1.1. Synthesis of the Ligands
3.1.2. Synthesis of Rhenium Complexes
3.2. In Vitro Biological Studies
3.2.1. In Vitro Growth Inhibition Assay
3.2.2. Inhibition of Phosphorylation
3.3. Radiochemistry
3.3.1. Preparation of fac-[99mTc][Tc(OH2)3(CO)3]+ Precursor
3.3.2. Synthesis of 99mTc Complexes, 5b and 7b
3.3.3. Stability Studies of 99mTc Complexes, 5b and 7b
3.3.4. Lipophilicity
3.4. Biodistribution Studies in Mice
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Wieduwilt, M.J.; Moasser, M.M. The epidermal growth factor receptor family: Biology driving targeted therapeutics. Cell. Mol. Life Sci. 2008, 65, 1566–1584. [Google Scholar] [CrossRef]
- An, Z.; Aksoy, O.; Zheng, T.; Fan, Q.-W.; Weiss, W.A. Epidermal growth factor receptor and EGFRvIII in glioblastoma: Signaling pathways and targeted therapies. Oncogene 2018, 37, 1561–1575. [Google Scholar] [CrossRef]
- Sebastian, S.; Settleman, J.; Reshkin, S.J.; Azzariti, A.; Bellizzi, A.; Paradiso, A. The complexity of targeting EGFR signalling in cancer: From expression to turnover. Biochim. Biophys. Acta BBA Rev. Cancer 2006, 1766, 120–139. [Google Scholar] [CrossRef]
- Han, W.; Du, Y. Recent Development of the Second and Third Generation Irreversible Epidermal Growth Factor Receptor Inhibitors. Chem. Biodivers. 2017, 14, 1600372. [Google Scholar] [CrossRef]
- Seshacharyulu, P.; Ponnusamy, M.P.; Haridas, D.; Jain, M.; Ganti, A.K.; Batra, S.K. Targeting the EGFR signaling pathway in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 15–31. [Google Scholar] [CrossRef]
- Flynn, J.F.; Wong, C.; Wu, J.M. Anti-EGFR Therapy: Mechanism and Advances in Clinical Efficacy in Breast Cancer. J. Oncol. 2009, 2009, 526963. [Google Scholar] [CrossRef] [PubMed]
- Hicks, J.W.; VanBrocklin, H.F.; Wilson, A.A.; Houle, S.; Vasdev, N. Radiolabeled Small Molecule Protein Kinase Inhibitors for Imaging with PET or SPECT. Molecules 2010, 15, 8260–8278. [Google Scholar] [CrossRef]
- Johnston, J.B.; Navaratnam, S.; Pitz, M.W.; Maniate, J.M.; Wiechec, E.; Baust, H.; Gingerich, J.; Skliris, G.P.; Murphy, L.C.; Los, M. Targeting the EGFR pathway for cancer therapy. Curr. Med. Chem. 2006, 13, 3483–3492. [Google Scholar] [CrossRef]
- Jain, A.; Kameswaran, M.; Pandey, U.; Prabhash, K.; Sarma, H.D.; Dash, A. 68 Ga labeled Erlotinib: A novel PET probe for imaging EGFR over-expressing tumors. Bioorg. Med. Chem. Lett. 2017, 27, 4552–4557. [Google Scholar] [CrossRef]
- Fernandes, C.; Santos, I.C.; Santos, I.; Pietzsch, H.-J.; Kunstler, J.-U.; Kraus, W.; Rey, A.; Margaritis, N.; Bourkoula, A.; Chiotellis, A.; et al. Rhenium and technetium complexes bearing quinazoline derivatives: Progress towards a 99mTc biomarker for EGFR-TK imaging. Dalton Trans. 2008, 24, 3215–3225. [Google Scholar] [CrossRef] [PubMed]
- Bourkoula, A.; Paravatou-Petsotas, M.; Papadopoulos, A.; Santos, I.; Pietzsch, H.-J.; Livaniou, E.; Pelecanou, M.; Papadopoulos, M.; Pirmettis, I. Synthesis and characterization of rhenium and technetium-99m tricarbonyl complexes bearing the 4-[3-bromophenyl]quinazoline moiety as a biomarker for EGFR-TK imaging. Eur. J. Med. Chem. 2009, 44, 4021–4027. [Google Scholar] [CrossRef]
- Si, Z.; Hu, P.; Zhou, J.; Lin, Q.; Xiu, Y.; Cheng, D. Evaluation of EGFR-TK Expression with a 99mTc-Labeled Complex Bearing Quinazoline Pharmacophore. Cancer Biother. Radiopharm. 2019, 34, 551–558. [Google Scholar] [CrossRef]
- Kiritsis, C.; Shegani, A.; Makrypidi, K.; Roupa, I.; Lazopoulos, A.; Panagiotopoulou, A.; Triantopoulou, S.; Paravatou-Petsotas, M.; Pietzsch, H.-J.; Pelecanou, M.; et al. Synthesis and preclinical evaluation of rhenium and technetium-99m “4 + 1” mixed-ligand complexes bearing quinazoline derivatives as potential EGFR imaging agents. Bioorg. Med. Chem. 2022, 73, 117012. [Google Scholar] [CrossRef]
- He, H.; Lipowska, M.; Xu, X.; Taylor, A.T.; Carlone, M.; Marzilli, L.G. Re(CO)(3) complexes synthesized via an improved preparation of aqueous fac-[Re(CO)(3)(H(2)O)(3)](+) as an aid in assessing (99m)Tc imaging agents. Structural characterization and solution behavior of complexes with thioether-bearing amino acids as tridentate ligands. Inorg. Chem. 2005, 44, 5437–5446. [Google Scholar] [CrossRef] [PubMed]
- Müller, C.; Dumas, C.; Hoffmann, U.; Schubiger, P.A.; Schibli, R. Organometallic 99mTc-technetium(I)- and Re-rhenium(I)-folate derivatives for potential use in nuclear medicine. J. Organomet. Chem. 2004, 689, 4712–4721. [Google Scholar] [CrossRef]
- Pirmettis, I.; Arano, Y.; Tsotakos, T.; Okada, K.; Yamaguchi, A.; Uehara, T.; Morais, M.; Correia, J.D.G.; Santos, I.; Martins, M.; et al. New 99m Tc(CO)3 Mannosylated Dextran Bearing S-Derivatized Cysteine Chelator for Sentinel Lymph Node Detection. Mol. Pharm. 2012, 9, 1681–1692. [Google Scholar] [CrossRef]
- Spingler, B.; Mundwiler, S.; Ruiz-Sánchez, P.; van Staveren, D.R.; Alberto, R. Structures of the b- and d-Acid Derivatives of Vitamin B12 and Their Complexes with [M(CO)3]+ (M = 99mTc, Re). Eur. J. Inorg. Chem. 2007, 2007, 2641–2647. [Google Scholar] [CrossRef]
- Chiotellis, A.; Tsoukalas, C.; Pelecanou, M.; Pirmettis, I.; Papadopoulos, M. New 99mTc(CO)3(NNO) complexes in the development of 5HT1A receptor imaging agents. Radiochim. Acta 2011, 99, 307–315. [Google Scholar] [CrossRef]
- Chiotellis, A.; Tsoukalas, C.; Pelecanou, M.; Raptopoulou, C.; Terzis, A.; Papadopoulos, M.; Papadopoulou-Daifoti, Z.; Pirmettis, I. Convenient Route Leading to Neutral fac-M(CO)3(NNO) Complexes (M = Re, 99mTc) Coupled to Amine Pharmacophores. Inorg. Chem. 2008, 47, 2601–2607. [Google Scholar] [CrossRef] [PubMed]
- Van Staveren, D.R.; Benny, P.D.; Waibel, R.; Kurz, P.; Pak, J.-K.; Alberto, R. S-Functionalized Cysteine: Powerful Ligands for the Labelling of Bioactive Molecules with Triaquatricarbonyltechnetium-99m(1+) ([99mTc(OH2)3(CO)3]+). Helv. Chim. Acta 2005, 88, 447–460. [Google Scholar] [CrossRef]
- Morley, J.S.; Simpson, J.C.E. 88. The chemistry of simple heterocyclic systems. Part I. Reactions of 6- and 7-nitro-4-hydroxyquinazoline and their derivatives. J. Chem. Soc. 1948, 360–366. [Google Scholar] [CrossRef]
- Rewcastle, G.W.; Denny, W.A.; Bridges, A.J.; Zhou, H.; Cody, D.R.; McMichael, A.; Fry, D.W. Tyrosine kinase inhibitors. 5. Synthesis and structure-activity relationships for 4-[(phenylmethyl)amino]- and 4-(phenylamino)quinazolines as potent adenosine 5′-triphosphate binding site inhibitors of the tyrosine kinase domain of the epidermal growth factor receptor. J. Med. Chem. 1995, 38, 3482–3487. [Google Scholar] [CrossRef]
- Policar, C.; Lambert, F.; Cesario, M.; Morgenstern-Badarau, I. An Inorganic Helix [Mn(IPG)(MeOH)]n[PF6]n[‡]: Structural and Magnetic Properties of a syn-anti Carboxylate-Bridged Manganese(II) Chain Involving a Tetradentate Ligand. Eur. J. Inorg. Chem. 1999, 1999, 2201–2207. [Google Scholar] [CrossRef]
- Alberto, R.; Egli, A.; Abram, U.; Hegetschweiler, K.; Gramlich, V.; Schubiger, P.A. Synthesis and reactivity of [NEt4]2[ReBr3(CO)3]. Formation and structural characterization of the clusters [NEt4][Re3(µ3-OH)(µ-OH)3(CO)9] and [NEt4][Re2(µ-OH)3(CO)6] by alkaline titration. J. Form. Struct. Charact. Clust. 1994, 3, 2815–2820. [Google Scholar] [CrossRef]
- Alberto, R.; Schibli, R.; Egli, A.; Schubiger, A.P.; Abram, U.; Kaden, T.A. A Novel Organometallic Aqua Complex of Technetium for the Labeling of Biomolecules: Synthesis of [99mTc(OH2)3(CO)3]+ from [99mTcO4]- in Aqueous Solution and Its Reaction with a Bifunctional Ligand. J. Am. Chem. Soc. 1998, 120, 7987–7988. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | tR (min) | %RCP in PBS a for 24 h | %RCP in Cysteine a for 24 h | %RCP in Histidine a for 24 h |
|---|---|---|---|---|
| 4 | 13.1 | - | - | - |
| 5a | 14.2 | - | - | - |
| 5b | 14.6 | 99 ± 2 | 99 ± 1 | 99 ± 1 |
| 6 | 12.8 | - | - | - |
| 7a | 13.9 | - | - | - |
| 7b | 14.2 | 99 ± 1 | 95 ± 1 | 95 ± 2 |
| Compound | IC50 Values (μM) ± SD a |
|---|---|
| 1 | 2.3 ± 0.3 |
| 4 | 25.64 ± 3.87 |
| 5a | 8.85 ± 2.62 |
| 6 | 4.12 ± 0.92 |
| 7a | 29.56 ± 3.63 |
| Compound | Inhibition of Cell Growth IC50 | Inhibition of Phosphorylation IC50 |
|---|---|---|
| IC50 Values (μM) ± SD | IC50 Values (nM) ± SD | |
8 | 23.8 ± 5.1 | - |
9 | 41.2 ± 13.6 | 756 ± 137 |
10 | 2.0 ± 0.98 | 114 ± 23 |
11 | 2.9 ± 1.6 | 108 ± 13 |
12 | 8.24 ± 1.86 | 70 |
13 | 42.51 ± 5.04 | - |
14 | 33.29 ± 7.03 | - |
| Organ | %Injected Dose/g | |||||
|---|---|---|---|---|---|---|
| Complex 5b | Complex 7b | |||||
| 5 min | 60 min | 240 min | 5 min | 60 min | 240 min | |
| Blood | 0.85 ± 0.23 | 0.14 ± 0.03 | 0.08 ± 0.01 | 1.06 ± 0.29 | 0.30 ± 0.09 | 0.18 ± 0.04 |
| Liver | 27.68 ± 2.32 | 1.24 ± 0.24 | 0.39 ± 0.08 | 23.33 ± 3.63 | 4.88 ± 0.47 | 4.46 ± 0.16 |
| Heart | 0.80 ± 0.12 | 0.13 ± 0.03 | 0.05 ± 0.01 | 0.97 ± 0.29 | 0.18 ± 0.03 | 0.12 ± 0.00 |
| Kidneys | 8.49 ± 1.33 | 1.28 ± 0.17 | 0.06 ± 0.01 | 13.04 ± 2.34 | 1.94 ± 0.28 | 1.12 ± 0.03 |
| Stomach | 0.72 ± 0.25 | 0.64 ± 0.27 | 0.60 ± 0.18 | 1.04 ± 0.10 | 0.97 ± 0.22 | 0.96 ± 0.06 |
| Intestines | 9.22 ± 2.19 | 34.05 ± 2.05 | 37.28 ± 3.50 | 10.62 ± 1.17 | 26.81 ± 1.61 | 29.89 ± 2.41 |
| Spleen | 0.63 ± 0.09 | 0.11 ± 0.01 | 0.07 ± 0.02 | 0.92 ± 0.32 | 0.11 ± 0.02 | 0.09 ± 0.02 |
| Muscle | 0.19 ± 0.05 | 0.08 ± 0.01 | 0.07 ± 0.07 | 0.29 ± 0.11 | 0.13 ± 0.12 | 0.03 ± 0.00 |
| Lungs | 0.97 ± 0.20 | 0.13 ± 0.03 | 0.08 ± 0.01 | 1.19 ± 0.22 | 0.26 ± 0.04 | 0.25 ± 0.08 |
| Pancreas | 0.63 ± 0.09 | 0.10 ± 0.03 | 0.09 ± 0.00 | 1.10 ± 0.11 | 0.24 ± 0.05 | 0.12 ± 0.02 |
| Brain | 0.05 ± 0.02 | 0.03 ± 0.00 | 0.02 ± 0.00 | 0.06 ± 0.02 | 0.02 ± 0.01 | 0.07 ± 0.06 |
| Urine * | 0.09 ± 0.06 | 1.02 ± 0.17 | 1.33 ± 0.14 | 0.08 ± 0.06 | 5.83 ± 0.02 | 7.88 ± 0.71 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Makrypidi, K.; Kiritsis, C.; Roupa, I.; Triantopoulou, S.; Shegani, A.; Paravatou-Petsotas, M.; Chiotellis, A.; Pelecanou, M.; Papadopoulos, M.; Pirmettis, I. Evaluation of Rhenium and Technetium-99m Complexes Bearing Quinazoline Derivatives as Potential EGFR Agents. Molecules 2023, 28, 1786. https://doi.org/10.3390/molecules28041786
Makrypidi K, Kiritsis C, Roupa I, Triantopoulou S, Shegani A, Paravatou-Petsotas M, Chiotellis A, Pelecanou M, Papadopoulos M, Pirmettis I. Evaluation of Rhenium and Technetium-99m Complexes Bearing Quinazoline Derivatives as Potential EGFR Agents. Molecules. 2023; 28(4):1786. https://doi.org/10.3390/molecules28041786
Chicago/Turabian StyleMakrypidi, Konstantina, Christos Kiritsis, Ioanna Roupa, Sotiria Triantopoulou, Antonio Shegani, Maria Paravatou-Petsotas, Aristeidis Chiotellis, Maria Pelecanou, Minas Papadopoulos, and Ioannis Pirmettis. 2023. "Evaluation of Rhenium and Technetium-99m Complexes Bearing Quinazoline Derivatives as Potential EGFR Agents" Molecules 28, no. 4: 1786. https://doi.org/10.3390/molecules28041786