
Synthesis of 6-Alkynylated Purine-Containing DNA via On-Column Sonogashira Coupling and Investigation of Their Base-Pairing Properties

 ,
,
Abstract
:1. Introduction
2. Results
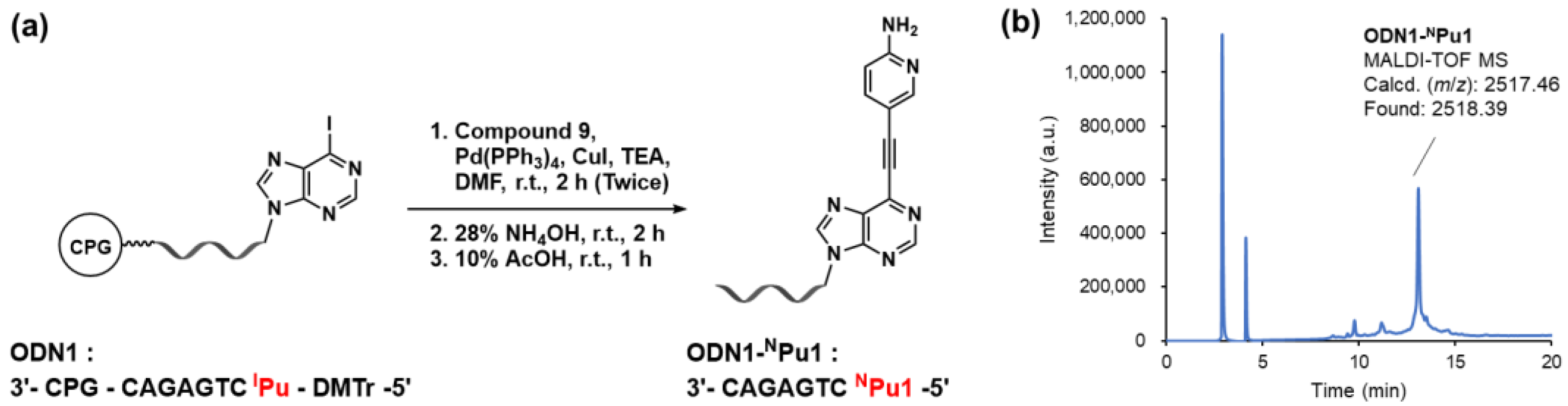
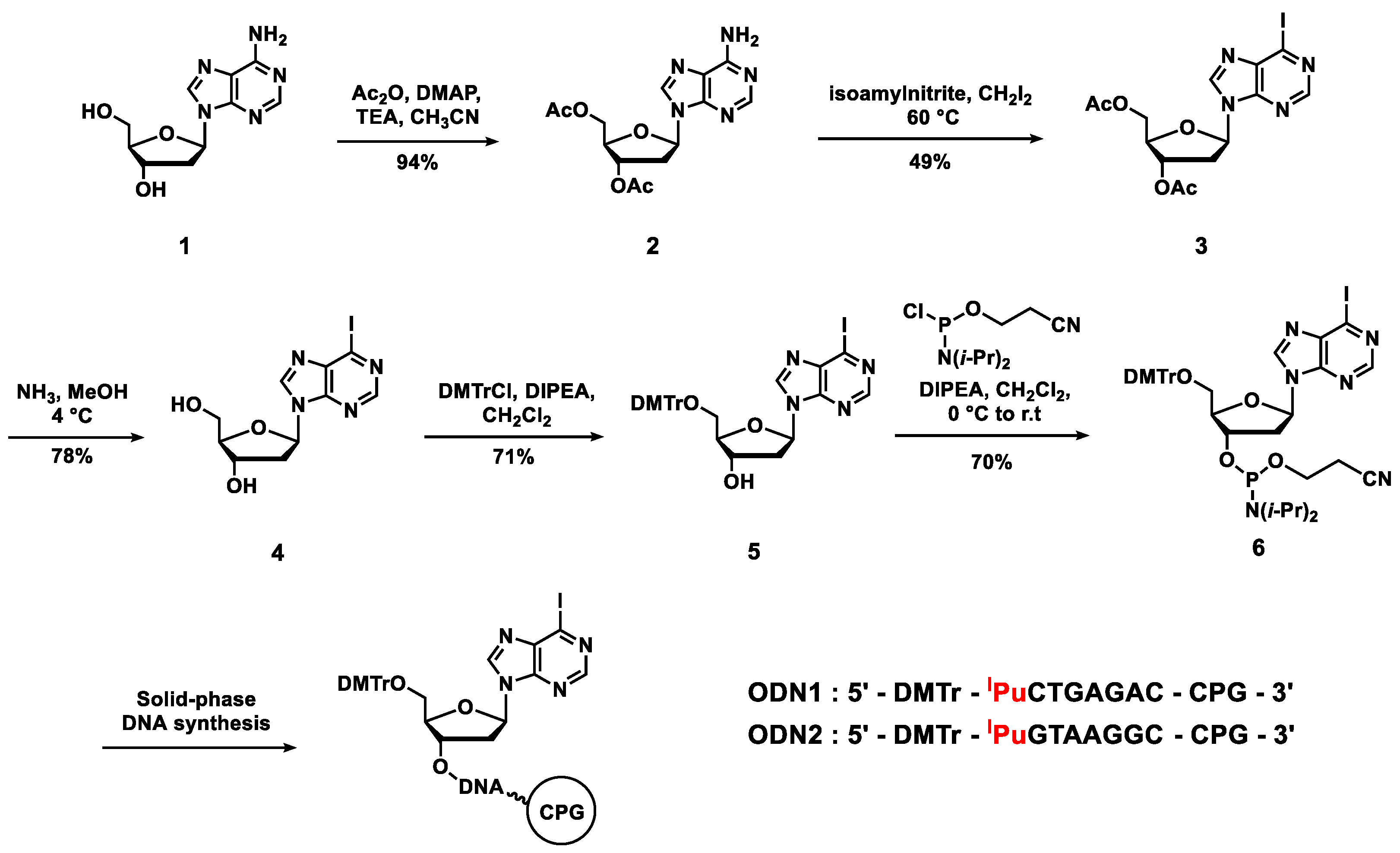
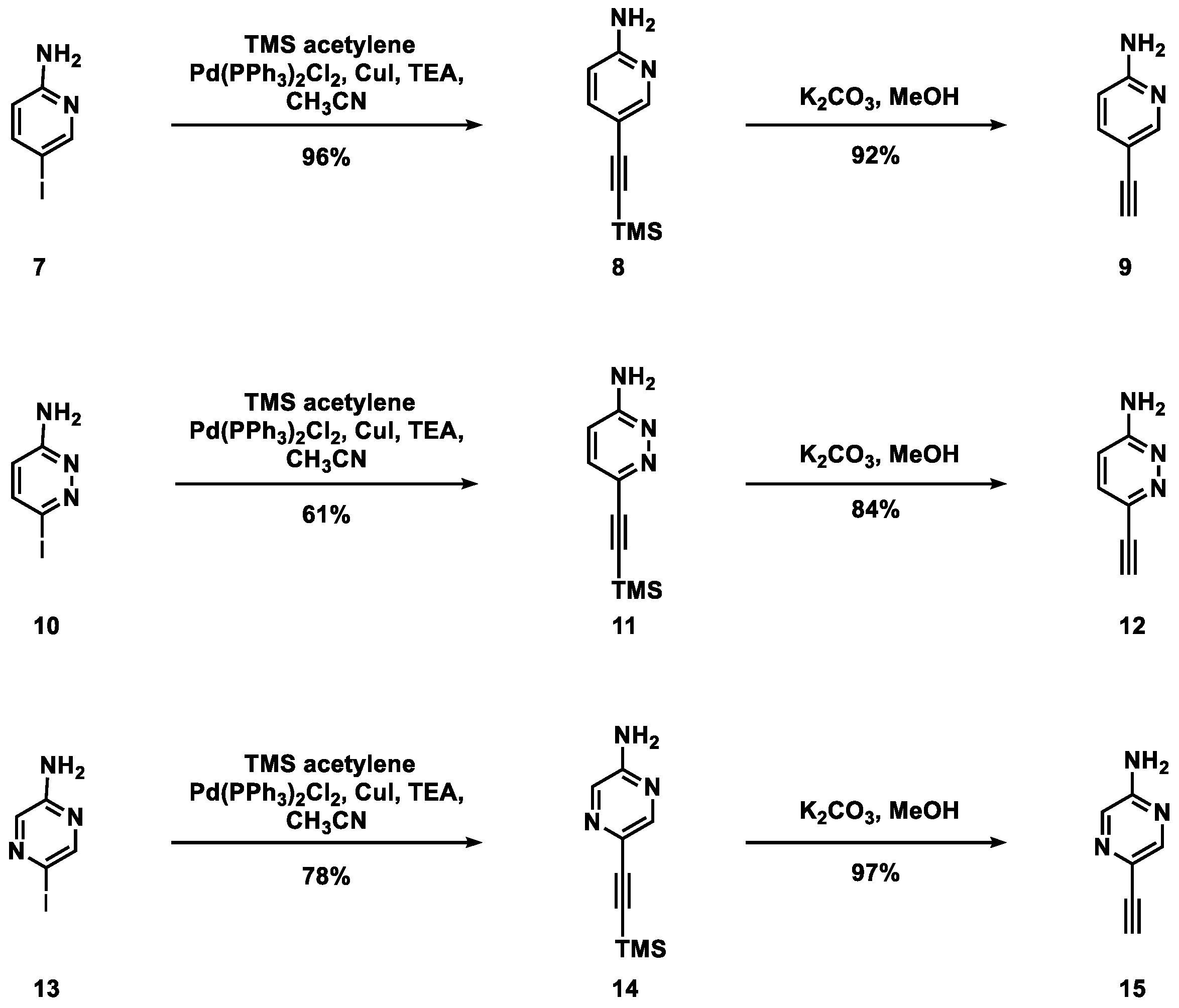
2.1. On-Column Synthesis of Oligodeoxynucleotides Containing the Alkynylated Purine Derivatives
2.2. Base-Pairing Properties of the Alkynylated Purine Derivatives
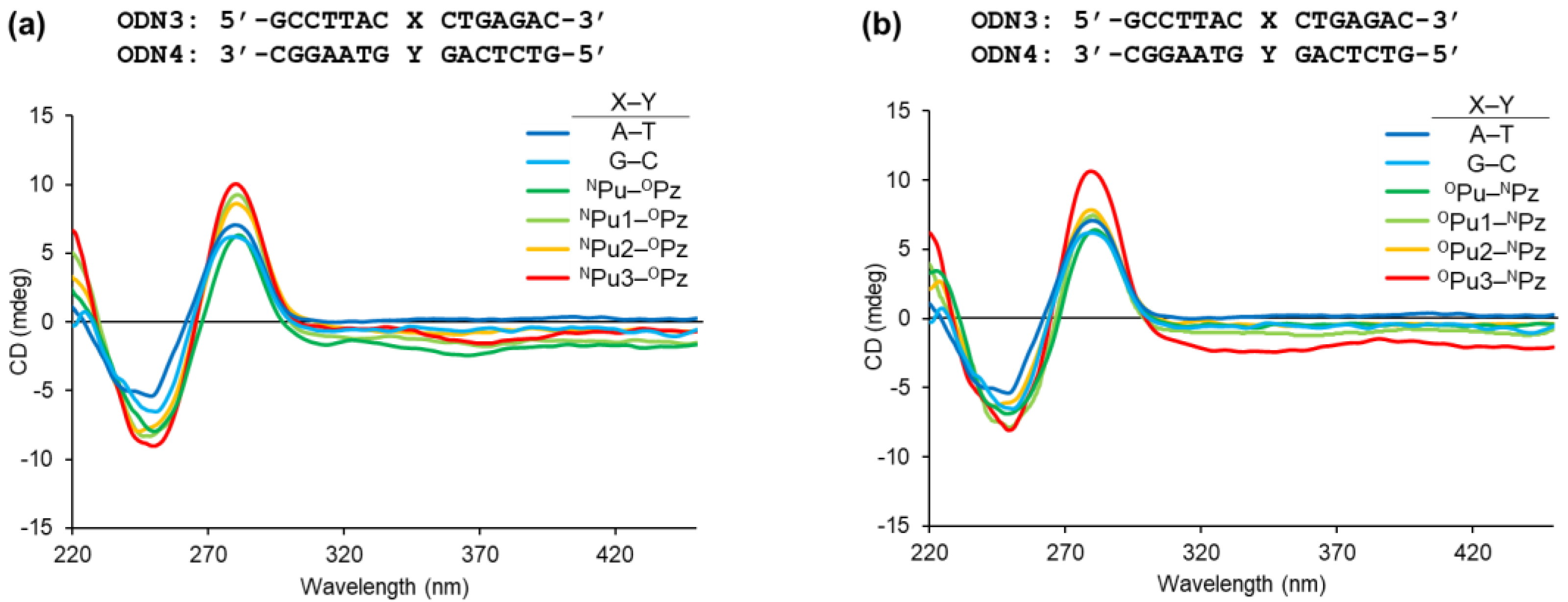
2.3. Structural Impact of the Alkynylated Purine Derivatives on DNA Duplex
3. Discussion
4. Materials and Methods
4.1. General Information
4.2. Synthesis of the Compounds Used in This Study
4.3. Solid-Phase DNA Synthesis
4.4. On-Column Synthesis of ODNs Incorporating the Alkynylated Purine Derivatives
4.5. Deprotection and Purification of the Synthesized ODNs
4.6. UV Melting Temperature Measurement of the Duplex DNAs
4.7. CD Spectroscopy Measurement of the Duplex DNAs
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Smanski, M.J.; Zhou, H.; Claesen, J.; Shen, B.; Fischbach, M.A.; Voigt, C.A. Synthetic biology to access and expand nature’s chemical diversity. Nat. Rev. Microbiol. 2016, 14, 135–149. [Google Scholar] [CrossRef] [PubMed]
- Chin, J.W. Expanding and reprogramming the genetic code. Nature 2017, 550, 53–60. [Google Scholar] [CrossRef]
- Chidchob, P.; Sleiman, H.F. Recent advances in DNA nanotechnology. Curr. Opin. Chem. Biol. 2018, 46, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Niemeyer, C.M. From DNA Nanotechnology to Material Systems Engineering. Adv. Mater. 2019, 31, e1806294. [Google Scholar] [CrossRef] [PubMed]
- Keller, A.; Linko, V. Challenges and Perspectives of DNA Nanostructures in Biomedicine. Angew. Chem. Int. Ed. 2020, 59, 15818–15833. [Google Scholar] [CrossRef]
- Li, X.; Liu, D.R. DNA-templated organic synthesis: Nature’s strategy for controlling chemical reactivity applied to synthetic molecules. Angew. Chem. Int. Ed. 2004, 43, 4848–4870. [Google Scholar] [CrossRef]
- Kodadek, T.; Paciaroni, N.G.; Balzarini, M.; Dickson, P. Beyond protein binding: Recent advances in screening DNA-encoded libraries. Chem. Commun. 2019, 55, 13330–13341. [Google Scholar] [CrossRef]
- Malyshev, D.A.; Romesberg, F.E. The expanded genetic alphabet. Angew. Chem. Int. Ed. 2015, 54, 11930–11944. [Google Scholar] [CrossRef]
- Karalkar, N.B.; Benner, S.A. The challenge of synthetic biology. Synthetic Darwinism and the aperiodic crystal structure. Curr. Opin. Chem. Biol. 2018, 46, 188–195. [Google Scholar] [CrossRef]
- Kimoto, M.; Hirao, I. Genetic alphabet expansion technology by creating unnatural base pairs. Chem. Soc. Rev. 2020, 49, 7602–7626. [Google Scholar] [CrossRef]
- Handal-Marquez, P.; Anupama, A.; Pezo, V.; Marlière, P.; Herdewijn, P.; Pinheiro, V.B. Beneath the XNA world: Tools and targets to build novel biology. Curr. Opin. Syst. Biol. 2020, 24, 142–152. [Google Scholar] [CrossRef]
- Rich, A. Horizons in Biochemistry; Kasha, M., Pullman, B., Eds.; Academic Press: New York, NY, USA, 1962; pp. 103–126. [Google Scholar]
- Minakawa, N.; Kojima, N.; Hikishima, S.; Sasaki, T.; Kiyosue, A.; Atsumi, N.; Ueno, Y.; Matsuda, A. New base pairing motifs. The synthesis and thermal stability of oligodeoxynucleotides containing imidazopyridopyrimidine nucleosides with the ability to form four hydrogen bonds. J. Am. Chem. Soc. 2003, 125, 9970–9982. [Google Scholar] [CrossRef] [PubMed]
- Doi, Y.; Chiba, J.; Morikawa, T.; Inouye, M. Artificial DNA made exclusively of nonnatural C-nucleosides with four types of nonnatural bases. J. Am. Chem. Soc. 2008, 130, 8762–8768. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Chen, F.; Alvarado, J.B.; Benner, S.A. Amplification, mutation, and sequencing of a six-letter synthetic genetic system. J. Am. Chem. Soc. 2011, 133, 15105–15112. [Google Scholar] [CrossRef] [PubMed]
- Hoshika, S.; Leal, N.A.; Kim, M.J.; Kim, M.S.; Karalkar, N.B.; Kim, H.J.; Bates, A.M.; Watkins, N.E., Jr.; SantaLucia, H.A.; Meyer, A.J.; et al. Hachimoji DNA and RNA: A genetic system with eight building blocks. Science 2019, 363, 884–887. [Google Scholar] [CrossRef]
- Morihiro, K.; Moriyama, Y.; Nemoto, Y.; Osumi, H.; Okamoto, A. Anti-syn Unnatural Base Pair Enables Alphabet-Expanded DNA Self-Assembly. J. Am. Chem. Soc. 2021, 143, 14207–14217. [Google Scholar] [CrossRef]
- Miyahara, R.; Taniguchi, Y. Selective Unnatural Base Pairing and Recognition of 2-Hydroxy-2’-deoxyadenosine in DNA Using Pseudo-dC Derivatives. J. Am. Chem. Soc. 2022, 144, 16150–16156. [Google Scholar] [CrossRef]
- Morales, J.C.; Kool, E.T. Minor Groove Interactions between Polymerase and DNA: More Essential to Replication than Watson-Crick Hydrogen Bonds? J. Am. Chem. Soc. 1999, 121, 2323–2324. [Google Scholar] [CrossRef]
- Malyshev, D.A.; Seo, Y.J.; Ordoukhanian, P.; Romesberg, F.E. PCR with an expanded genetic alphabet. J. Am. Chem. Soc. 2009, 131, 14620–14621. [Google Scholar] [CrossRef] [PubMed]
- Yamashige, R.; Kimoto, M.; Takezawa, Y.; Sato, A.; Mitsui, T.; Yokoyama, S.; Hirao, I. Highly specific unnatural base pair systems as a third base pair for PCR amplification. Nucleic Acids Res. 2012, 40, 2793–2806. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, K.; Clever, G.H.; Takezawa, Y.; Yamada, Y.; Kaul, C.; Shionoya, M.; Carell, T. Programmable self-assembly of metal ions inside artificial DNA duplexes. Nat. Nanotechnol. 2006, 1, 190–194. [Google Scholar] [CrossRef]
- Kaul, C.; Muller, M.; Wagner, M.; Schneider, S.; Carell, T. Reversible bond formation enables the replication and amplification of a crosslinking salen complex as an orthogonal base pair. Nat. Chem. 2011, 3, 794–800. [Google Scholar] [CrossRef] [PubMed]
- Takezawa, Y.; Hu, L.; Nakama, T.; Shionoya, M. Sharp Switching of DNAzyme Activity through the Formation of a Cu(II) -Mediated Carboxyimidazole Base Pair. Angew. Chem. Int. Ed. 2020, 59, 21488–21492. [Google Scholar] [CrossRef]
- Matsunaga, K.I.; Kimoto, M.; Hirao, I. High-Affinity DNA Aptamer Generation Targeting von Willebrand Factor A1-Domain by Genetic Alphabet Expansion for Systematic Evolution of Ligands by Exponential Enrichment Using Two Types of Libraries Composed of Five Different Bases. J. Am. Chem. Soc. 2017, 139, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, S.; Yang, Z.; Hoshika, S.; Xie, S.; Li, J.; Chen, X.; Wan, S.; Li, L.; Benner, S.A.; et al. An Aptamer-Nanotrain Assembled from Six-Letter DNA Delivers Doxorubicin Selectively to Liver Cancer Cells. Angew. Chem. Int. Ed. 2020, 59, 663–668. [Google Scholar] [CrossRef] [PubMed]
- Bornewasser, L.; Domnick, C.; Kath-Schorr, S. Stronger together for in-cell translation: Natural and unnatural base modified mRNA. Chem. Sci. 2022, 13, 4753–4761. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.C.; Marshall, D.J.; Harms, G.; Miller, C.M.; Sherrill, C.B.; Beaty, E.L.; Lederer, S.A.; Roesch, E.B.; Madsen, G.; Hoffman, G.L.; et al. Multiplexed genetic analysis using an expanded genetic alphabet. Clin. Chem. 2004, 50, 2019–2027. [Google Scholar] [CrossRef] [PubMed]
- Hirao, I.; Ohtsuki, T.; Fujiwara, T.; Mitsui, T.; Yokogawa, T.; Okuni, T.; Nakayama, H.; Takio, K.; Yabuki, T.; Kigawa, T.; et al. An unnatural base pair for incorporating amino acid analogs into proteins. Nat. Biotechnol. 2002, 20, 177–182. [Google Scholar] [CrossRef]
- Fischer, E.C.; Hashimoto, K.; Zhang, Y.; Feldman, A.W.; Dien, V.T.; Karadeema, R.J.; Adhikary, R.; Ledbetter, M.P.; Krishnamurthy, R.; Romesberg, F.E. New codons for efficient production of unnatural proteins in a semisynthetic organism. Nat. Chem. Biol. 2020, 16, 570–576. [Google Scholar] [CrossRef]
- Okamura, H.; Trinh, G.H.; Dong, Z.; Masaki, Y.; Seio, K.; Nagatsugi, F. Selective and stable base pairing by alkynylated nucleosides featuring a spatially-separated recognition interface. Nucleic Acids Res. 2022, 50, 3042–3055. [Google Scholar] [CrossRef]
- Ferentz, A.E.; Keating, T.A.; Verdine, G.L. Synthesis and characterization of disulfide cross-linked oligonucleotides. J. Am. Chem. Soc. 2002, 115, 9006–9014. [Google Scholar] [CrossRef]
- MacMillan, A.M.; Verdine, G.L. Synthesis of functionally tethered oligodeoxynucleotides by the convertible nucleoside approach. J. Org. Chem. 2002, 55, 5931–5933. [Google Scholar] [CrossRef]
- Ito, Y.; Matsuo, M.; Yamamoto, K.; Yamashita, W.; Osawa, T.; Hari, Y. Post-Synthetic modification of oligonucleotides containing 5-trifluoromethylpyrimidine bases. Tetrahedron 2018, 74, 6854–6860. [Google Scholar] [CrossRef]
- Ito, Y.; Hayashi, H.; Fuchi, Y.; Hari, Y. Post-Synthetic modification of oligonucleotides containing 5-mono- and 5-di-fluoromethyluridines. Tetrahedron 2021, 77, 131769. [Google Scholar] [CrossRef]
- Ravi Kumara, G.S.; Pandith, A.; Seo, Y.J. Direct and selective metal-free N6-arylation of adenosine residues for simple fluorescence labeling of DNA and RNA. Chem. Commun. 2021, 57, 5450–5453. [Google Scholar] [CrossRef]
- Wang, J.; Shang, J.; Xiang, Y.; Tong, A. General Method for Post-Synthetic Modification of Oligonucleotides Based on Oxidative Amination of 4-Thio-2’-deoxyuridine. Bioconjug. Chem. 2021, 32, 721–728. [Google Scholar] [CrossRef]
- Xie, Y.; Fang, Z.; Yang, W.; He, Z.; Chen, K.; Heng, P.; Wang, B.; Zhou, X. 6-Iodopurine as a Versatile Building Block for RNA Purine Architecture Modifications. Bioconjugate Chem. 2022, 33, 353–362. [Google Scholar] [CrossRef]
- Gierlich, J.; Burley, G.A.; Gramlich, P.M.; Hammond, D.M.; Carell, T. Click chemistry as a reliable method for the high-density postsynthetic functionalization of alkyne-modified DNA. Org. Lett. 2006, 8, 3639–3642. [Google Scholar] [CrossRef]
- Tolle, F.; Brandle, G.M.; Matzner, D.; Mayer, G. A Versatile Approach Towards Nucleobase-Modified Aptamers. Angew. Chem. Int. Ed. 2015, 54, 10971–10974. [Google Scholar] [CrossRef]
- Kahl, J.D.; Greenberg, M.M. Introducing Structural Diversity in Oligonucleotides via Photolabile, Convertible C5-Substituted Nucleotides. J. Am. Chem. Soc. 1999, 121, 597–604. [Google Scholar] [CrossRef]
- Raiber, E.A.; Beraldi, D.; Ficz, G.; Burgess, H.E.; Branco, M.R.; Murat, P.; Oxley, D.; Booth, M.J.; Reik, W.; Balasubramanian, S. Genome-wide distribution of 5-formylcytosine in embryonic stem cells is associated with transcription and depends on thymine DNA glycosylase. Genome Biol. 2012, 13, R69. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.I.; Grinstaff, M.W. Palladium(0)-Catalyzed Modification of Oligonucleotides during Automated Solid-Phase Synthesis. J. Am. Chem. Soc. 1999, 121, 4704–4705. [Google Scholar] [CrossRef]
- Minakawa, N.; Ono, Y.; Matsuda, A. A versatile modification of on-column oligodeoxynucleotides using a copper-catalyzed oxidative acetylenic coupling reaction. J. Am. Chem. Soc. 2003, 125, 11545–11552. [Google Scholar] [CrossRef]
- Rist, M.; Amann, N.; Wagenknecht, H.-A. Preparation of 1-Ethynylpyrene-Modified DNA via Sonogashira-Type Solid-Phase Couplings and Characterization of the Fluorescence Properties for Electron-Transfer Studies. Eur. J. Org. Chem. 2003, 2003, 2498–2504. [Google Scholar] [CrossRef]
- Schiemann, O.; Piton, N.; Mu, Y.; Stock, G.; Engels, J.W.; Prisner, T.F. A PELDOR-based nanometer distance ruler for oligonucleotides. J. Am. Chem. Soc. 2004, 126, 5722–5729. [Google Scholar] [CrossRef]
- Grunewald, C.; Kwon, T.; Piton, N.; Forster, U.; Wachtveitl, J.; Engels, J.W. RNA as scaffold for pyrene excited complexes. Bioorg. Med. Chem. 2008, 16, 19–26. [Google Scholar] [CrossRef]
- Omumi, A.; Beach, D.G.; Baker, M.; Gabryelski, W.; Manderville, R.A. Postsynthetic guanine arylation of DNA by Suzuki-Miyaura cross-coupling. J. Am. Chem. Soc. 2011, 133, 42–50. [Google Scholar] [CrossRef]
- Wicke, L.; Engels, J.W. Postsynthetic on column RNA labeling via Stille coupling. Bioconjug. Chem. 2012, 23, 627–642. [Google Scholar] [CrossRef]
- Lercher, L.; McGouran, J.F.; Kessler, B.M.; Schofield, C.J.; Davis, B.G. DNA modification under mild conditions by Suzuki-Miyaura cross-coupling for the generation of functional probes. Angew. Chem. Int. Ed. 2013, 52, 10553–10558. [Google Scholar] [CrossRef]
- Krause, A.; Hertl, A.; Muttach, F.; Jaschke, A. Phosphine-free Stille-Migita chemistry for the mild and orthogonal modification of DNA and RNA. Chem. Eur. J. 2014, 20, 16613–16619. [Google Scholar] [CrossRef]
- Ejlersen, M.; Lou, C.; Sanghvi, Y.S.; Tor, Y.; Wengel, J. Modification of oligodeoxynucleotides by on-column Suzuki cross-coupling reactions. Chem. Commun. 2018, 54, 8003–8006. [Google Scholar] [CrossRef] [PubMed]
- Probst, N.; Lartia, R.; Thery, O.; Alami, M.; Defrancq, E.; Messaoudi, S. Efficient Buchwald-Hartwig-Migita Cross-Coupling for DNA Thioglycoconjugation. Chem. Eur. J. 2018, 24, 1795–1800. [Google Scholar] [CrossRef] [PubMed]
- Walunj, M.B.; Srivatsan, S.G. Nucleic Acid Conformation Influences Postsynthetic Suzuki−Miyaura Labeling of Oligonucleotides. Bioconjug. Chem. 2020, 31, 2513–2521. [Google Scholar] [CrossRef] [PubMed]
- Sýkorová, V.; Tichý, M.; Hocek, M. Polymerase Synthesis of DNA Containing Iodinated Pyrimidine or 7-Deazapurine Nucleobases and Their Post-synthetic Modifications through the Suzuki-Miyaura Cross-Coupling Reactions. ChemBioChem 2021, 23, e202100608. [Google Scholar] [CrossRef]
- Filichev, V.V.; Pedersen, E.B. Stable and selective formation of hoogsteen-type triplexes and duplexes using twisted intercalating nucleic acids (TINA) prepared via postsynthetic Sonogashira solid-phase coupling reactions. J. Am. Chem. Soc. 2005, 127, 14849–14858. [Google Scholar] [CrossRef]
- Hari, Y.; Akabane, M.; Hatanaka, Y.; Nakahara, M.; Obika, S. A 4-[(3R,4R)-dihydroxypyrrolidino]pyrimidin-2-one nucleobase for a CG base pair in triplex DNA. Chem. Commun. 2011, 47, 4424–4426. [Google Scholar] [CrossRef]
- Hari, Y.; Nakahara, M.; Pang, J.; Akabane, M.; Kuboyama, T.; Obika, S. Synthesis and triplex-forming ability of oligonucleotides bearing 1-substituted 1H-1,2,3-triazole nucleobases. Bioorg. Med. Chem. 2011, 19, 1162–1166. [Google Scholar] [CrossRef]
- Okamura, H.; Taniguchi, Y.; Sasaki, S. N-(guanidinoethyl)-2′-deoxy-5-methylisocytidine exhibits selective recognition of a CG interrupting site for the formation of anti-parallel triplexes. Org. Biomol. Chem. 2013, 11, 3918–3924. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sequence Name | Sequence | Calcd. for [M − H]− | Found (m/z) |
|---|---|---|---|
| ODN3−NPu1 | 5′-GCCTTACNPu1 CTGAGAC | 4634.77 | 4634.59 |
| ODN3−NPu2 | 5′-GCCTTACNPu2 CTGAGAC | 4635.76 | 4636.06 |
| ODN3−NPu3 | 5′-GCCTTACNPu3 CTGAGAC | 4635.76 | 4635.93 |
| ODN3−OPu1 | 5′-GCCTTACOPu1 CTGAGAC | 4636.75 | 4636.97 |
| ODN3−OPu2 | 5′-GCCTTAC OPu2 CTGAGAC | 4636.75 | 4636.35 |
| ODN3−OPu3 | 5′-GCCTTAC OPu3 CTGAGAC | 4636.75 | 4636.22 |
| ODN4−NPu1 | 5′-GTCTCAG NPu1 GTAAGGC | 4714.79 | 4714.96 |
| ODN4−NPu2 | 5′-GTCTCAGNPu2 GTAAGGC | 4715.78 | 4715.39 |
| ODN4−NPu3 | 5′-GTCTCAG NPu3 GTAAGGC | 4715.78 | 4716.29 |
| ODN4−OPu1 | 5′-GTCTCAG OPu1 GTAAGGC | 4718.97 | 4719.41 |
| ODN4−OPu2 | 5′-GTCTCAG OPu2 GTAAGGC | 4716.77 | 4716.46 |
| ODN4−OPu3 | 5′-GTCTCAG OPu3 GTAAGGC | 4716.77 | 4716.07 |
| ODN3: 5’-GCCTTAC X CTGAGAC-3’ ODN4: 3’-CGGAATG Y GACTCTG-5’ | Base Pair (X–Y) | Tm (°C) |
|---|---|---|
 | A–T | 51.0 ± 0.1 |
| G–C | 54.3 ± 0.1 | |
| NPu–OPz | 52.1 ± 0.1 | |
| NPu1–OPz | 52.2 ± 0.2 | |
| NPu2–OPz | 49.3 ± 0.1 | |
| NPu3–OPz | 52.7 ± 0.2 | |
| NPu–NPz | 48.4 ± 0.1 | |
| NPu1–NPz | 50.2 ± 0.2 | |
| NPu2–NPz | 49.0 ± 0.1 | |
| NPu3–NPz | 49.6 ± 0.1 | |
| OPu–NPz | 51.3 ± 0.1 | |
| OPu1–NPz | 53.2 ± 0.1 | |
| OPu2–NPz | 49.2 ± 0.1 | |
| OPu3–NPz | 51.9 ± 0.2 | |
| OPu–OPz | 48.8 ± 0.1 | |
| OPu1–OPz | 49.5 ± 0.7 | |
| OPu2–OPz | 48.3 ± 0.1 | |
| OPu3–OPz | 48.8 ± 0.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Okamura, H.; Trinh, G.H.; Dong, Z.; Fan, W.; Nagatsugi, F. Synthesis of 6-Alkynylated Purine-Containing DNA via On-Column Sonogashira Coupling and Investigation of Their Base-Pairing Properties. Molecules 2023, 28, 1766. https://doi.org/10.3390/molecules28041766
Okamura H, Trinh GH, Dong Z, Fan W, Nagatsugi F. Synthesis of 6-Alkynylated Purine-Containing DNA via On-Column Sonogashira Coupling and Investigation of Their Base-Pairing Properties. Molecules. 2023; 28(4):1766. https://doi.org/10.3390/molecules28041766
Chicago/Turabian StyleOkamura, Hidenori, Giang Hoang Trinh, Zhuoxin Dong, Wenjue Fan, and Fumi Nagatsugi. 2023. "Synthesis of 6-Alkynylated Purine-Containing DNA via On-Column Sonogashira Coupling and Investigation of Their Base-Pairing Properties" Molecules 28, no. 4: 1766. https://doi.org/10.3390/molecules28041766