
Electric Dipole Moments from Stark Effect in Supersonic Expansion: n-Propanol, n-Butanol, and n-Butyl Cyanide

Abstract
:1. Introduction
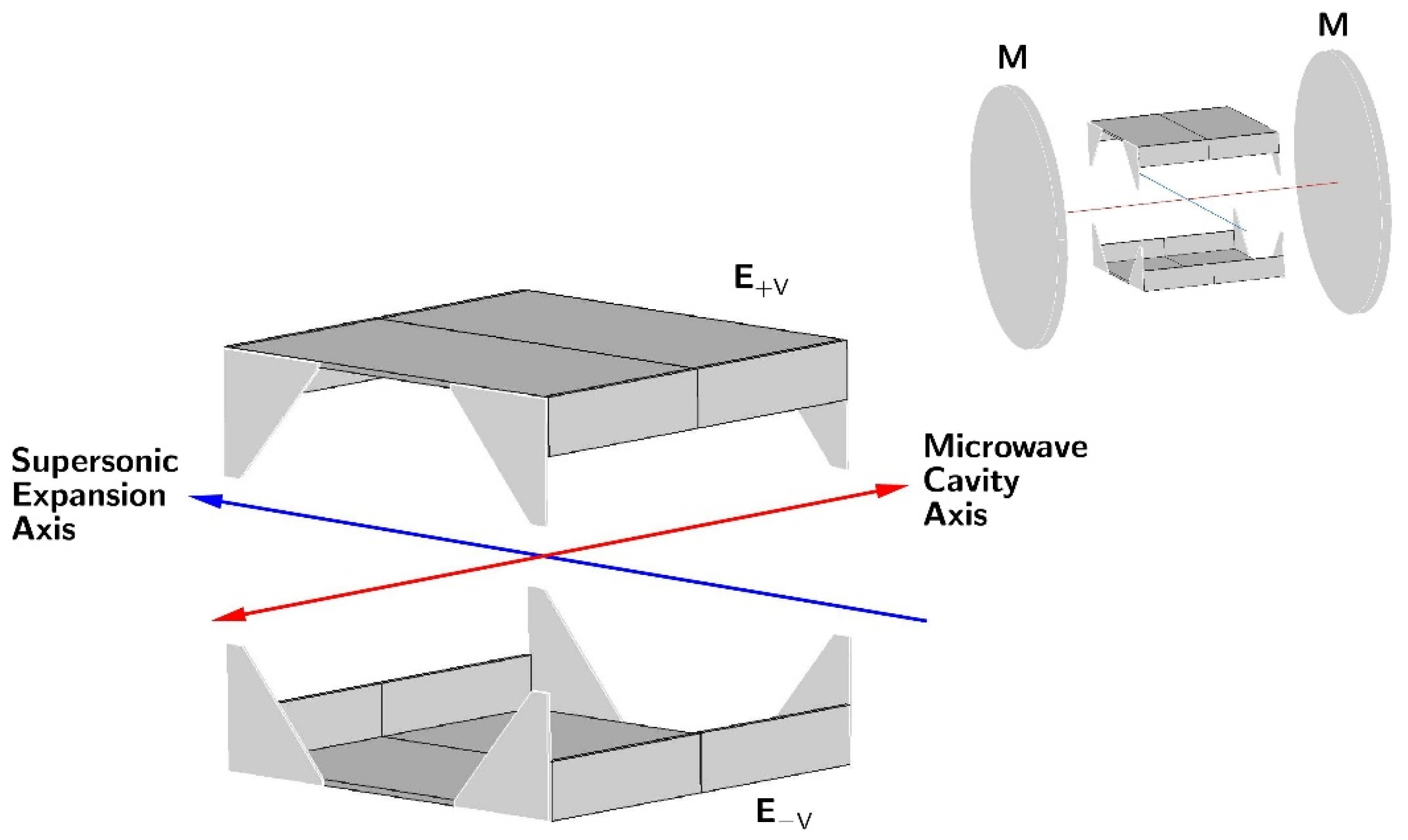
2. Dipole Moment Measurements in Supersonic Expansion
3. Results
3.1. Electric Dipole Moments of Ga and Aa Conformers of n-Propanol
3.2. Electric Dipole Moments of TGt and GTg’ Conformers of n-Butanol
3.3. Electric Dipole Moments of AA and GA Conformers of n-Butyl Cyanide
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| FTMW | Fourier Transform Microwave |
| CMW | Centimetre-wave |
| MMW | Millimetre-wave |
| SMM | Submillimetre-wave |
References
- McGuire, B.A. 2018 Census of Interstellar, Circumstellar, Extragalactic, Protoplanetary Disk, and Exoplanetary Molecules. Astrophys. J. Suppl. Ser. 2018, 239, 17. [Google Scholar] [CrossRef]
- Kisiel, Z. THz Molecular Spectroscopy. In Encyclopedia of Modern Optics Second Edition; Guenther, B.D., Steel, D.G., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; Volume 1, pp. 387–402. [Google Scholar] [CrossRef]
- Schlemmer, S. High-Resolution Laboratory Terahertz Spectroscopy and Applications to Astrophysics. In Frontiers and Advances in Molecular Spectroscopy; Laane, J., Ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2018; Chapter 15. [Google Scholar] [CrossRef]
- Müller, H.S.P.; Schlder, F.; Stutzki, J.; Winnewisser, G. The Cologne Database for Molecular Spectroscopy, CDMS: A useful tool for astronomers and spectroscopists. J. Mol. Struct. 2005, 742, 215–227. [Google Scholar] [CrossRef]
- Pickett, H.M.; Poynter, R.L.; Cohen, E.A.; Delitsky, M.L.; Pearson, J.C.; Müller, H.S.P. Submillimeter, millimeter, and microwave spectral line catalog. J. Quant. Spectrosc. Radiat. Transf. 1998, 60, 883–890. [Google Scholar] [CrossRef]
- Belloche, A.; Garrod, R.T.; Müller, H.S.P.; Menten, K.M.; Medvedev, I.; Thomas, J.; Kisiel, Z. Re-exploring Molecular Complexity with ALMA (ReMoCA): Interstellar detection of urea. Astron. Astrophys. 2019, 628, A10. [Google Scholar] [CrossRef]
- Cernicharo, J.; Kisiel, Z.; Tercero, B.; Kolesnikova, L.; Medvedev, I.R.; Lopez, A.; Fortman, S.; Winnewisser, M.; deLucia, F.C.; Alonso, J.L.; et al. A rigorous detection of interstellar CH3NCO: An important missing species in astrochemical networks. Astron. Astrophys. 2016, 587, L4. [Google Scholar] [CrossRef]
- McGuire, B.A.; Burkhardt, A.M.; Kalenskii, S.; Shingledecker, C.N.; Remijan, A.J.; Herbst, E.; McCarthy, M.C. Detection of the aromatic molecule benzonitrile (c-C6H5CN) in the interstellar medium. Science 2018, 359, 202–205. [Google Scholar] [CrossRef]
- McGuire, B.A.; Loomis, R.A.; Burkhardt, A.M.; Lee, K.L.K.; Shingledecker, C.N.; Charnley, S.B.; Cooke, I.R.; Cordiner, M.A.; Herbst, E.; Kalenskii, S.; et al. Detection of two interstellar polycyclic aromatic hydrocarbons via spectral matched filtering. Science 2021, 371, 1265–1269. [Google Scholar] [CrossRef]
- Kisiel, Z.; Nixon, C.A.; Cordiner, M.A.; Thelen, A.E.; Charnley, S.B. Propionitrile in the two lowest excited vibrational states in the laboratory and on Titan. J. Mol. Spectrosc. 2020, 372, 111324. [Google Scholar] [CrossRef]
- Jimenez-Serra, I.; Rodriguez-Almeida, L.F.; Martin-Pintado, J.; Rivilla, V.M.; Melosso, M.; Zeng, S.S.; Colzi, L.; Kawashima, Y.; Hirota, E.; Puzzarini, C.; et al. Precursors of fatty alcohols in the ISM: Discovery of n-propanol. Astron. Astrophys. 2022, 663, A1181. [Google Scholar] [CrossRef]
- Hughes, R.H.; Wilson, E.B., Jr. A Microwave Spectrograph. Phys. Rev. 1947, 71, 562. [Google Scholar] [CrossRef]
- Kroto, H.W. Molecular Rotation Spectra; John Wiley & Sons, Ltd.: New York, NY, USA, 1975. [Google Scholar]
- Harrington, H.W.; Hearn, J.R.; Rauskolb, R.F. The Routine Rotational Microwave Spectrometer. Hewlett Packard J. 1971, 7, 1–12. Available online: http://hparchive.com/Journals/HPJ-1971-06.pdf (accessed on 1 January 2023).
- Zingsheim, O.; Massen, J.; Müller, H.S.P.; Heyne, B.; Fatima, M.; Bonah, L.; Belloche, A.; Lewen, F.; Schlemmer, S. Rotational spectroscopy of n-propanol: Aa and Ag conformers. Astron. Astrophys. 2022, 662, A111. [Google Scholar] [CrossRef]
- Kawashima, Y.; Tanak, Y.; Uzuyama, T.; Hirota, E. Conformations and low-frequency intramolecular motions of 1-Butanol, 1-Butanethiol, Iso-butanol, and Iso-butanethiol Investigated by Fourier Transform Microwave Spectroscopy Combined with Quantum Chemical Calculations. J. Phys. Chem. A 2021, 125, 1166–1183. [Google Scholar] [CrossRef] [PubMed]
- Ordu, M.H.; Müller, H.S.P.; Walters, A.; Nunez, M.; Lewen, F.; Belloche, A.; Menten, K.M.; Schlemmer, S. The quest for complex molecules in space: Laboratory spectroscopy of n-butyl cyanide, n-C4H9CN, in the millimeter wave region and its astronomical search in Sagittarius B2(N). Astron. Astrophys. 2012, 541, A41. [Google Scholar] [CrossRef]
- Balle, T.J.; Flygare, W.H. Fabry-Perot cavity pulsed Fourier transform microwave spectrometer with a pulsed nozzle particle source. Rev. Sci. Instr. 1981, 52, 33–45. [Google Scholar] [CrossRef]
- Coudert, L.H.; Lovas, F.J.; Suenram, R.D.; Hougen, J.T. New measurements of microwave transitions in the water dimer. J. Chem. Phys. 1987, 87, 6290–6299. [Google Scholar] [CrossRef]
- Consalvo, D. Advances in Stark effect measurements in a molecular beam Fourier transform microwave spectrometer. Rev. Sci. Instrum. 1998, 69, 3136–3141. [Google Scholar] [CrossRef]
- Emilsson, T.; Gutowsky, H.S.; de Oliveira, G.; Dykstra, C.E. Rotational patches: Stark effect, dipole moment, and dynamics of water loosely bound to benzene. J. Chem. Phys. 2000, 112, 1287–1294. [Google Scholar] [CrossRef]
- Schnell, M.; Banser, D.; Grabow, J.-U. Coaxially aligned electrodes for Stark-effect applied in resonators using a supersonic jet Fourier transform microwave spectrometer. Rev. Sci. Instrum. 2004, 75, 2111–2115. [Google Scholar] [CrossRef]
- Kisiel, Z.; Kosarzewski, J.; Pietrewicz, B.A.; Pszczółkowski, L. Electric dipole moments of the cyclic trimers (H2O)2HCl and (H2O) 2HBr from Stark effects in their rotational spectra. Chem. Phys. Lett. 2000, 325, 523–530. [Google Scholar] [CrossRef]
- Kisiel, Z.; Białkowska-Jaworska, E.; Desyatnyk, O.; Pietrewicz, B.A.; Pszczółkowski, L. The gas-phase electric dipole moments of the symmetric top tertiary butyl moleculestBuX, X = F, Cl, Br, I, CN, and NC. J. Mol. Spectrosc. 2001, 208, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Ito, H.; Harada, K.; Tanaka, T. CO2 and CO laser microwave double resonance spectroscopy of OCS: Precise measurement of dipole moment and polarizability anisotropy. J. Chem. Phys. 1984, 80, 5893–5905. [Google Scholar] [CrossRef]
- Gadhi, J.; Wlodarczak, G.; Legrand, J.; Demaison, J. The dipole moments of methyl bromide and methyl iodide. Chem. Phys. Lett. 1989, 156, 401–404. [Google Scholar] [CrossRef]
- Gadhi, J.; Lahrouni, A.; Legrand, J.; Demaison, J. Moment dipolaire de CH3CN. J. Chim. Phys. 1995, 92, 1984–1992. [Google Scholar] [CrossRef]
- Dorosh, O.; Kisiel, Z. Electric Dipole Moments of Acetone and of Acetic Acid Measured in Supersonic Expansion. Acta. Phys. Pol. A 2007, 112, S95–S104. [Google Scholar] [CrossRef]
- Benz, H.P.; Bauder, A.; Gunthard, H.H. Exact Quadrupole Interaction Energies in Rotational Spectra. J. Mol. Spectrosc. 1966, 21, 156–164. [Google Scholar] [CrossRef]
- Kisiel, Z. Assignment and Analysis of Complex Rotational Spectra. In Spectroscopy from Space; Demaison, J., Sarka, K., Cohen, E.A., Eds.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2001; pp. 91–106. [Google Scholar] [CrossRef]
- PROSPE—Programs for ROtational SPEctroscopy. Available online: http://info.ifpan.edu.pl/~kisiel/prospe.htm/ (accessed on 6 February 2023).
- Kuczkowski, R.L.; Suenram, R.D.; Lovas, F.J. Microwave Spectrum, Structure, and Dipole Moment of Sulfuric Acid. J. Amer. Chem. Soc. 1981, 103, 2561–2566. [Google Scholar] [CrossRef]
- Sego, G.; Schultz, J.; Leopold, K.R. Electric dipole moment of sulfuric acid from Fourier transform microwave spectroscopy. J. Mol. Spectrosc. 2008, 251, 4–8. [Google Scholar] [CrossRef]
- Kraśnicki, A.; Kisiel, Z. Electric dipole moments of acrylonitrile and of propionitrile measured in supersonic expansion. J. Mol. Spectrosc. 2011, 270, 83–87. [Google Scholar] [CrossRef]
- Kisiel, Z.; Pszczółkowski, L.; Drouin, B.J.; Brauer, C.S.; Yu, S.; Pearson, J.C. The rotational spectrum of acrylonitrile up to 1.67 THz. J. Mol. Spectrosc. 2009, 258, 26–34. [Google Scholar] [CrossRef]
- Watson, J.K.G. Aspects of quartic and sextic centrifugal effects on rotational energy levels. In Vibrational Spectra and Structure; During, J.R., Ed.; Elsevier: New York, NY, USA; Amsterdam, The Netherlands, 1977; Volume 6, pp. 1–89. [Google Scholar]
- Kisiel, Z.; Dorosh, O.; Maeda, A.; Medvedev, I.R.; De Lucia, F.; Herbst, E.; Drouin, B.J.; Pearson, J.C.; Shipman, S.T. Determination of precise relative energies of conformers of n-propanol by rotational spectroscopy. Phys. Chem. Chem. Phys. 2010, 12, 8329–8339. [Google Scholar] [CrossRef]
- Abdurakhmanov, A.A.; Ragimova, R.A.; Imanov, L.M. Microwave spectrum of normal propyl alcohol. Phys. Lett. A 1970, 32, 123–124. [Google Scholar] [CrossRef]
- Dreizler, H.; Scappini, F. Centrifugal Distortion and Internal Rotation Analysis in the Ground State of Trans N-Propanol. Z. Naturforsch. 1981, 36, 1187–1191. [Google Scholar] [CrossRef]
- Kisiel, Z.; Dorosh, O.; Maeda, A.; De Lucia, F.C.; Herbst, E. Assignment of GG, GG’, TT, and TG Conformers in the FASSST Rotational Spectrum of n-Propanol. Presentation RI14 at 61st OSU International Symposium on Molecular Spectroscopy, 19–23 June 2006. Available online: https://kb.osu.edu/bitstream/handle/1811/31042/nprop.ppt?sequence=20&isAllowed=y (accessed on 6 February 2023).
- Goubet, M.; Motiyenko, R.A.; Real, F.; Margules, L.; Huet, T.T.; Asselin, P.; Soulard, P.; Kraśnicki, A.; Kisiel, Z.; Alekseev, E.A. Influence of the geometry of a hydrogen bond on conformational stability: A theoretical and experimental study of ethyl carbamate. Phys. Chem. Chem. Phys. 2009, 11, 1719–1728. [Google Scholar] [CrossRef] [PubMed]
- Bohn, R.K.; Pardus, J.L.; August, J.; Brupbacher, T.; Jager, W. Microwave studies of the three conformers of butyl cyanide. J. Mol. Struct. 1997, 413-414, 293–300. [Google Scholar] [CrossRef]
- Kisiel, Z.; Kosarzewski, J.; Pszczółkowski, L. Nuclear Quadrupole Coupling Tensor of CH2Cl2: Comparison of Quadrupolar and Structural Angles in Methylene Halides. Acta. Phys. Pol. A 1997, 92, 507–516. [Google Scholar] [CrossRef]
- Kraśnicki, A.; Pszczółkowski, L.; Kisiel, Z. Analysis of the rotational spectrum of pyruvonitrile up to 324 GHz. J. Mol. Spectrosc. 2010, 260, 57–65. [Google Scholar] [CrossRef]
- Krüger, M.; Dreizler, H. A Microwave Fourier Transform Spectrometer with a Single Microwave Source. Z. Nat. 1990, 45, 724–726. [Google Scholar] [CrossRef]
- Krüger, M.; Harder, H.; Gerke, C.; Dreizler, H. An Automatic Scan Waveguide Microwave Fourier Transform Spectrometer. Z. Naturforsch. 1993, 48, 737–738. [Google Scholar] [CrossRef]
- Kisiel, Z. Further rotational spectroscopy of phenol: Sextic centrifugal distortion and vibrational satellites. J. Mol. Spectrosc. 2022, 386, 111630. [Google Scholar] [CrossRef]
- Medvedev, I.; Winnewisser, M.; De Lucia, F.C.; Herbst, E.; Białkowska-Jaworska, E.; Pszczółkowski, L.; Kisiel, Z. The millimeter- and submillimeter-wave spectrum of the trans–gauche conformer of diethyl ether. J. Mol. Spectrosc. 2004, 228, 314–328. [Google Scholar] [CrossRef]
- Kisiel, Z.; Kraśnicki, A. The millimetre-wave rotational spectrum of phenylacetylene. J. Mol. Spectrosc. 2010, 262, 82–88. [Google Scholar] [CrossRef]
- Kisiel, Z.; Białkowska-Jaworska, E. Sextic centrifugal distortion in fluorobenzene and phenylacetylene from cm-wave rotational spectroscopy. J. Mol. Spectrosc. 2019, 359, 16–21. [Google Scholar] [CrossRef]
- Pickett, H. The Fitting and Prediction of Vibration-Rotation Spectra with Spin Interactions. J. Mol. Spectrosc. 1991, 148, 371–377. [Google Scholar] [CrossRef]
- JPL Molecular Spectroscopy. Available online: https://spec.jpl.nasa.gov (accessed on 6 February 2023).
- Frisch, M.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Petersson, G.; Nakatsuji, H.; et al. Gaussian 16; Revision, A.03; Gaussian, Inc.: Wallingford, CT, UK, 2016; Available online: https://gaussian.com/g09citation (accessed on 6 February 2023).
- CFOUR, a Quantum Chemical Program Package written by J.F. Stanton, J. Gauss, M.E. Harding, P.G. Szalay with contributions from A.A. Auer, R.J. Bartlett, U.Benedikt, C. Berger, D.E. Bernholdt, Y.J. Bomble, O. Christiansen, M. Heckert, O.Heun, C. Huber, T.-C. Jagau, D. Jonsson, J. Jusélius, K. Klein, W.J. Lauderdale, D.A.Matthews, T. Metzroth, D.P. O’Neill, D.R. Price, E. Prochnow, K. Ruud, F.Schiffmann, S. Stopkowicz, J. Vázquez, F. Wang, J.D. Watts and the integral packages MOLECULE (J. Almlöf and P.R. Taylor), PROPS (P.R. Taylor), ABACUS (T. Helgaker, H.J. Aa. Jensen, P. Jørgensen, and J. Olsen), and ECP routines by A.V. Mitin and C. van Wüllen. Available online: http://www.cfour.de (accessed on 6 February 2023).











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| n-Propanol | n-Butanol | n-Butyl Cyanide | |||
|---|---|---|---|---|---|
| Parameter a | Aa | TGt | GTg’ | AA | GA |
| A/MHz | 26,401.4765(25) | 12,467.74156(60) | 12,305.00007(57) | 15,028.68713(43) | 7635.62481(21) |
| B/MHz | 3802.15318(36) | 2371.51771(10) | 2330.57664(34) | 1334.107310(22) | 1788.636927(33) |
| C/MHz | 3549.45833(49) | 2189.47940(11) | 2146.23454(32) | 1263.856699(21) | 1554.218399(37) |
| ∆J/kHz | 0.9788(27) | 0.75626(66) | 0.7131(16) | 0.1458342(27) | 0.878123(12) |
| ∆JK/kHz | −0.658(58) | −6.180(10) | −2.543(16) | −7.54678(13) | −10.00176(14) |
| ∆K/kHz | 151.28(54) | 60.25(11) | 73.01(23) | 214.3805(81) | 52.4857(12) |
| δJ/kHz | 0.12547(94) | 0.12484(13) | 0.13705(59) | 0.0220207(19) | 0.2333427(65) |
| δK/kHz | −6.32(16) | 2.063(35) | 2.45(15) | 0.48160(39) | 1.72956(61) |
| Nlines | 103 | 72 | 53 | 1481 | 1464 |
| σrms | 1.060 b | 0.944 | 0.697 | 0.939 b | 0.937 b |
| n-Propanol Ga | n-Propanol Aa | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Parameter a | Ref. [38] b | Ref. [37] c | Calc. d | Calc. e | Calc. f | Ref. [38] b | This work c | Calc. d | Calc. e | Calc. f |
| |µa|/D | 0.32(6) | 0.4914(4) | 0.529 | 0.580 | 0.570 | 0.21(7) | 0.3589(7) | 0.319 | 0.321 | 0.282 |
| |µb|/D | 1.23(2) | 0.9705(13) | 1.020 | 1.079 | 1.071 | 1.54(2) | 1.2820(13) | 1.422 | 1.500 | 1.549 |
| |µc|/D | 0.94(2) | 0.9042(12) | 0.894 | 0.929 | 1.009 | 0 | 0 | 0 | 0 | 0 |
| µtot/D | 1.58(3) | 1.4145(17) | 1.456 | 1.538 | 1.578 | 1.55(3) | 1.3312(13) | 1.458 | 1.534 | 1.575 |
| Nlines | 43 | 37 | ||||||||
| σfit/kHz | 2.08 | 2.29 | ||||||||
| n-Butanol TGt | n-Butanol GTg’ | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Parameter a | Ref. [16] b | This Work | Calc. c | Calc. d | Calc. e | Ref. [16] b | This Work | Calc. c | Calc. d | Calc. e |
| |µa|/D | 0.89 | 0.7137(10) | 0.752 | 0.813 | 0.805 | 1.26 | 1.0881(11) | 1.175 | 1.215 | 1.168 |
| |µb|/D | 1.05 | 0.8989(8) | 0.934 | 0.988 | 0.990 | 1.35 | 1.0304(12) | 1.164 | 1.203 | 1.315 |
| |µc|/D | 0.94 | 0.8071(8) | 0.801 | 0.829 | 0.910 | 0.80 | 0.6980(16) | 0.719 | 0.764 | 0.730 |
| µtot/D | 1.67 | 1.4032(11) | 1.442 | 1.525 | 1.567 | 2.01 | 1.6532(16) | 1.804 | 1.873 | 1.904 |
| Nlines | 68 + 6 | 31 + 5 | ||||||||
| σfit/kHz | 3.15 | 3.12 | ||||||||
| n-Butyl Cyanide AA | n-Butyl Cyanide GA | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Parameter a | Ref. [42] b | This Work | Calc. c | Calc. d | Calc. e | Ref. [42] | This Work | Calc. c | Calc. d | Calc. e |
| |µa|/D | 3.40 | 3.9047(32) | 3.885 | 3.914 | 4.076 | 2.3 | 2.6856(15) | 2.656 | 2.669 | 2.894 |
| |µb|/D | 1.90 | 1.6662(16) | 1.665 | 1.688 | 1.714 | 3.1 | 2.8521(36) | 2.841 | 2.882 | 2.919 |
| |µc|/D | 0 | 0 | 0 | 0 | 0 | 0 | 0.390(17) | 0.417 | 0.419 | 0.442 |
| µtot/D | 3.89 | 4.2454(36) | 4.226 | 4.262 | 4.421 | 3.86 | 3.9368(36) | 3.912 | 3.950 | 4.134 |
| Nlines | 70 + 7 | 85 + 14 | ||||||||
| σfit/kHz | 3.02 | 3.47 | ||||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kisiel, Z.; Habdas, K. Electric Dipole Moments from Stark Effect in Supersonic Expansion: n-Propanol, n-Butanol, and n-Butyl Cyanide. Molecules 2023, 28, 1692. https://doi.org/10.3390/molecules28041692
Kisiel Z, Habdas K. Electric Dipole Moments from Stark Effect in Supersonic Expansion: n-Propanol, n-Butanol, and n-Butyl Cyanide. Molecules. 2023; 28(4):1692. https://doi.org/10.3390/molecules28041692
Chicago/Turabian StyleKisiel, Zbigniew, and Krzysztof Habdas. 2023. "Electric Dipole Moments from Stark Effect in Supersonic Expansion: n-Propanol, n-Butanol, and n-Butyl Cyanide" Molecules 28, no. 4: 1692. https://doi.org/10.3390/molecules28041692