

Terpenoids from Myrrh and Their Cytotoxic Activity against HeLa Cells

 and
and
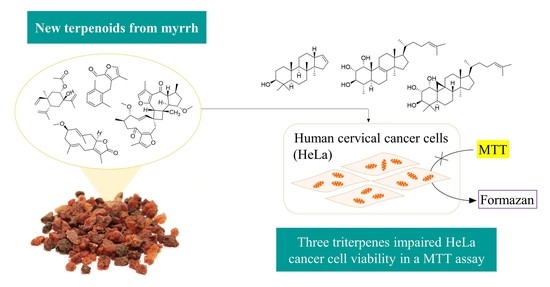
Abstract
:
1. Introduction
2. Results
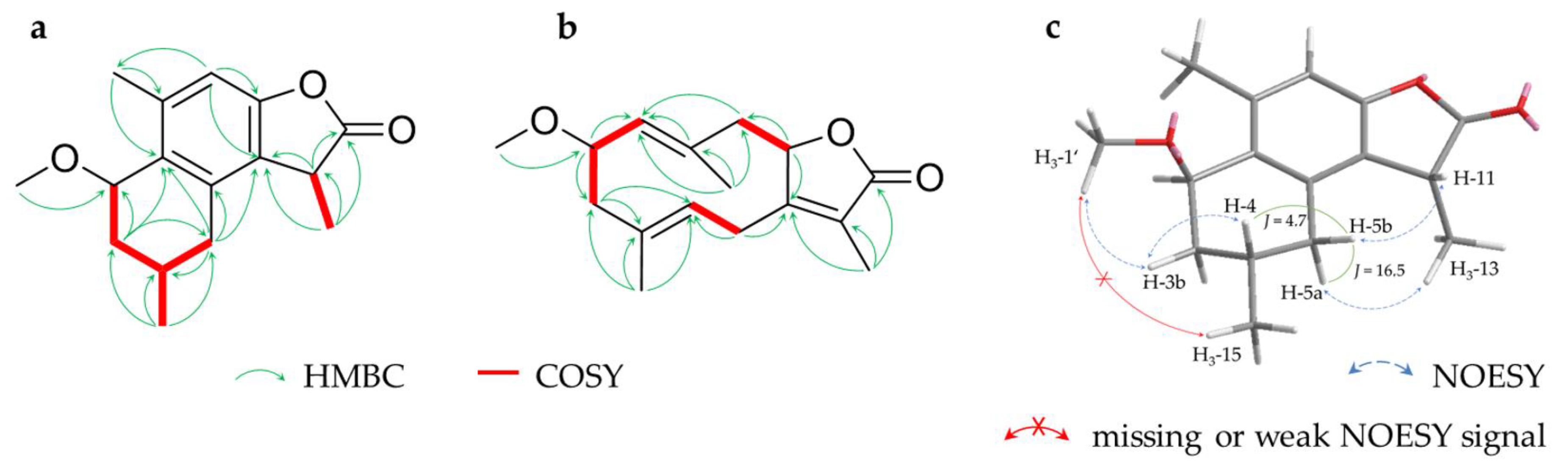
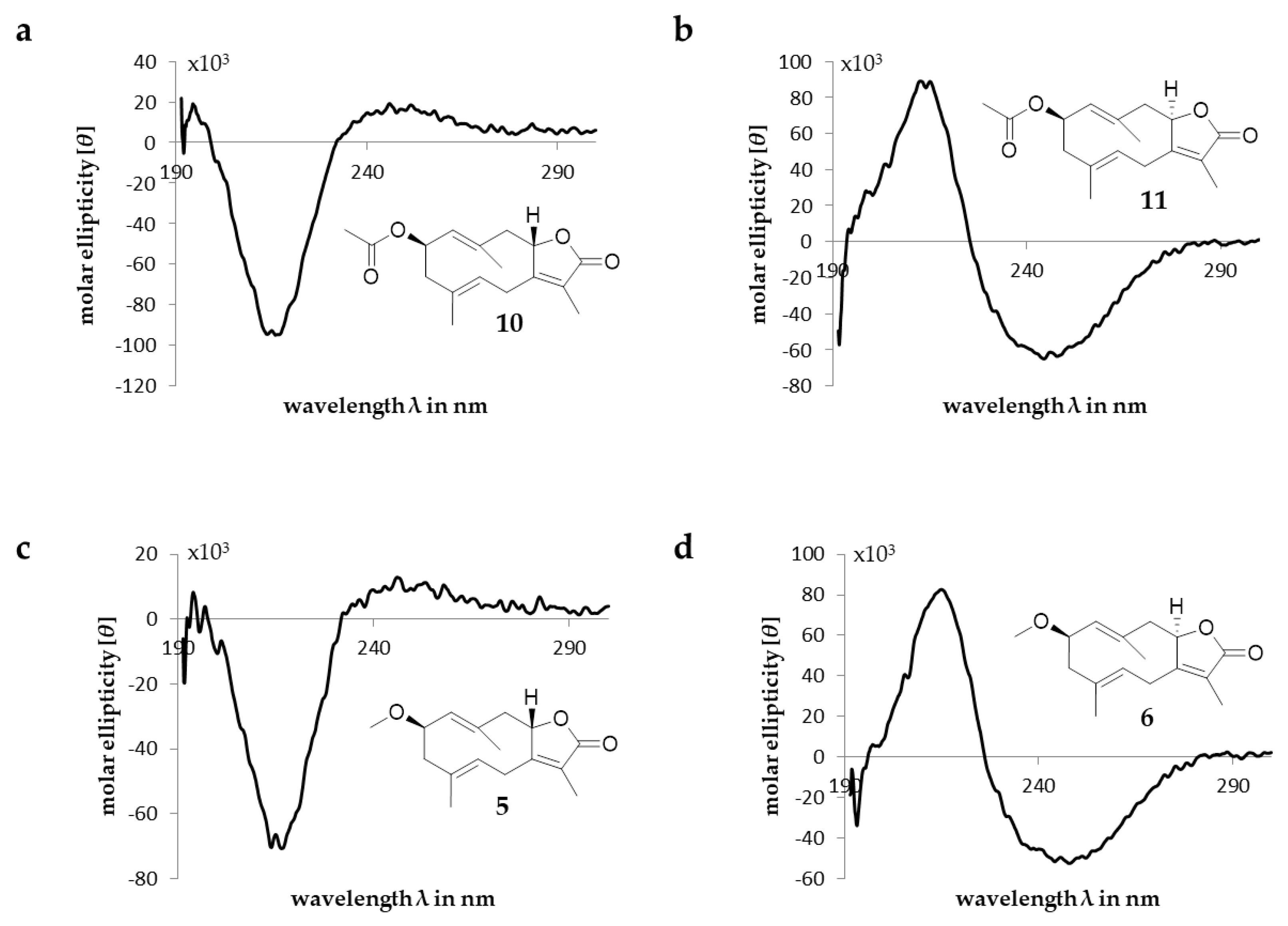
2.1. Sesquiterpenoids
2.2. Triterpenoids
2.3. The Cytotoxicity of Selected Compounds against HeLa Cells
3. Discussion and Conclusions
4. Materials and Methods
4.1. Chemicals
4.2. Plant Material and Extraction
4.3. Isolation
4.3.1. Liquid-Liquid Partition
4.3.2. Solid Phase Extraction: MeOH Fraction
4.3.3. Silica Flash Chromatography 1
HEP Fraction
MeOH Fraction M1
4.3.4. Centrifugal Partition Chromatography (CPC)
4.3.5. Silica Flash Chromatography 2
4.3.6. Thin-Layer Chromatography (TLC)
4.3.7. Preparative HPLC
4.4. NMR
4.5. UHPLC-MS
4.6. Optical Methods
4.7. Simulation of ECD Spectra
4.8. Purity
4.9. Isolated Compounds
4.9.1. 9,10-Seco-isolindestrenolide (1)
4.9.2. 9-Oxo-9,10-seco-isolindestrene (2)
4.9.3. rel-8S-Acetyloxy-7R-hydroxy-5R,10R-β-elemene (3)
4.9.4. Commiterpene E (4)
4.9.5. 2β-Methoxyglechomanolide (5)
4.9.6. 8-epi-2β-Methoxyglechomanolide (6)
4.9.7. Dehydroisolindestrenolide (7)
4.9.8. Commiphorine C (8)
4.9.9. Glechomanolide (9)
4.9.10. 2β-Acetyloxyglechomanolide (10)
4.9.11. 8-epi-2β-Acetyloxyglechomanolide (11)
4.9.12. 2α-Methoxy-6-oxogermacra-1(10),7(11)-dien-8,12-olide (12)
4.9.13. 1(10)Z,4E-Isofuranodienone (13)
4.9.14. 3S-Methoxy-4R-furano-1E,10(14)-germacradien-6-one (14)
4.9.15. 2-Methoxy-5-acetoxyfuranogermacr-1(10)-en-6-one (15)
4.9.16. rel-2R-Methoxy-5S-acetoxy-4R-furanogermacr-1(10)Z-en-6-one (16)
4.9.17. (1S,4S,5S,10S)-Germacron-1(10),4-diepoxide (17)
4.9.18. Dehydrolindestrenolide (18)
4.9.19. Tubipolide B (19)
4.9.20. Atractylenolide II (20)
4.9.21. 8-epi-Isogermafurenolide (21)
4.9.22. Myrrhterpenoid O (22)
4.9.23. 3β-Isovaleroyloxy-29-nor-lanost-8,24-diene-1α,2α-diol (23)
4.9.24. 29-Nor-1,2-cis-epoxylanost-8,24-diene-3β-triol (24)
4.9.25. rel-20S-Hydroxydammar-24-en-3,16-dione (25)
4.9.26. Mansumbinol (26)
4.9.27. 3,4-Seco-mansumbinoic acid (27)
4.9.28. Cycloartan-24-ene-1α,3β-diol (28)
4.9.29. Cycloartan-24-ene-1α,2α,3β-triol (29)
4.9.30. 1α-Acetoxy-9,19-cyclolanost-24-ene-3β-ol (30)
4.9.31. 1α-Acetoxycycloartan-24-ene-2α,3β-diol (31)
4.9.32. 3β-Isovaleroyloxycycloartan-24-ene-1α,2α-diol (32)
4.9.33. 29-Nor-lanost-8,24-diene-lα,2α,3β-triol (33)
4.9.34. Hydroxydammarenone II (34)
4.10. Cell Culture
4.10.1. Cultivation
4.10.2. MTT Assay
4.10.3. ICAM-1 Assay
4.11. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Tucker, A.O. Frankincense and myrrh. Econ. Bot. 1986, 40, 425–433. [Google Scholar] [CrossRef]
- Shen, T.; Li, G.-H.; Wang, X.-N.; Lou, H.-X. The genus Commiphora: A review of its traditional uses, phytochemistry and pharmacology. J. Ethnopharmacol. 2012, 142, 319–330. [Google Scholar] [CrossRef]
- Abegaz, B.; Dagne, E.; Bates, C.; Waterman, P.G. Monoterpene-rich resins from two ethiopian species of Commiphora. Flavour Fragr. J. 1989, 4, 99–101. [Google Scholar] [CrossRef]
- Asres, K.; Tei, A.; Moges, G.; Sporer, F.; Wink, M. Terpenoid composition of the wound-induced bark exudate of Commiphora tenuis from Ethiopia. Planta Med. 1998, 64, 473–475. [Google Scholar] [CrossRef]
- Hanuš, L.O.; Řezanka, T.; Dembitsky, V.M.; Moussaieff, A. Myrrh-Commiphora chemistry. Biomed. Papers 2005, 149, 3–28. [Google Scholar] [CrossRef]
- Dekebo, A.; Dagne, E.; Sterner, O. Furanosesquiterpenes from Commiphora sphaerocarpa and related adulterants of true myrrh. Fitoterapia 2002, 73, 48–55. [Google Scholar] [CrossRef]
- Morteza-Semnani, K.; Saeedi, M. Constituents of the essential oil of Commiphora myrrha (Nees) Engl. var. molmol. J. Essent. Oil Res. 2003, 15, 50–51. [Google Scholar] [CrossRef]
- Madia, V.N.; de Angelis, M.; de Vita, D.; Messore, A.; de Leo, A.; Ialongo, D.; Tudino, V.; Saccoliti, F.; de Chiara, G.; Garzoli, S.; et al. Investigation of Commiphora myrrha (Nees) Engl. oil and its main components for antiviral activity. Pharmaceuticals 2021, 14, 243. [Google Scholar] [CrossRef]
- Marongiu, B.; Piras, A.; Porcedda, S.; Scorciapino, A. Chemical composition of the essential oil and supercritical CO2 extract of Commiphora myrrha (Nees) Engl. and of Acorus calamus L. J. Agric. Food Chem. 2005, 53, 7939–7943. [Google Scholar] [CrossRef]
- Nikolic, M.; Smiljkovic, M.; Markovic, T.; Ciric, A.; Glamoclija, J.; Markovic, D.; Sokovic, M. Sensitivity of clinical isolates of Candida to essential oils from Burseraceae family. EXCLI J. 2016, 15, 280–289. [Google Scholar] [CrossRef]
- Hanuš, L.O.; Rosenthal, D.; Řezanka, T.; Dembitsky, V.M.; Moussaief, A. Fast and easy GC/MS identification of myrrh resins. Pharm. Chem. J. 2008, 42, 719–720. [Google Scholar] [CrossRef]
- Su, S.L.; Duan, J.A.; Tang, Y.P.; Zhang, X.; Yu, L.; Jiang, F.R.; Zhou, W.; Luo, D.; Ding, A.W. Isolation and biological activities of neomyrrhaol and other terpenes from the resin of Commiphora myrrha. Planta Med. 2009, 75, 351–355. [Google Scholar] [CrossRef]
- Xu, J.; Guo, Y.; Zhao, P.; Xie, C.; Jin, D.; Hou, W.; Zhang, T. Neuroprotective cadinane sesquiterpenes from the resinous exudates of Commiphora myrrha. Fitoterapia 2011, 82, 1198–1201. [Google Scholar] [CrossRef]
- Xu, J.; Guo, Y.; Li, Y.; Zhao, P.; Liu, C.; Ma, Y.; Gao, J.; Hou, W.; Zhang, T. Sesquiterpenoids from the resinous exudates of Commiphora myrrha and their neuroprotective effects. Planta Med. 2011, 77, 2023–2028. [Google Scholar] [CrossRef]
- Greve, H.L.; Kaiser, M.; Schmidt, T.J. Investigation of antiplasmodial effects of terpenoid compounds isolated from myrrh. Planta Med. 2020, 86, 643–654. [Google Scholar] [CrossRef]
- Shen, T.; Wan, W.-Z.; Wang, X.-N.; Yuan, H.-Q.; Ji, M.; Lou, H.-X. A triterpenoid and sesquiterpenoids from the resinous exudates of Commiphora myrrha. Helv. Chim. Acta 2009, 92, 645–652. [Google Scholar] [CrossRef]
- Ayyad, S.-E.N.; Hoye, T.R.; Alarif, W.M.; Al Ahmadi, S.M.; Basaif, S.A.; Ghandourah, M.A.; Badria, F.A. Differential cytotoxic activity of the petroleum ether extract and its furanosesquiterpenoid constituents from Commiphora molmol resin. Z. Naturforsch. 2015, 70, 87–92. [Google Scholar] [CrossRef]
- Kuck, K.; Jürgenliemk, G.; Lipowicz, B.; Heilmann, J. Sesquiterpenes from myrrh and their ICAM-1 inhibitory activity in vitro. Molecules 2020, 26, 42. [Google Scholar] [CrossRef]
- Ahmed, F.; Ali, M.; Singh, O. New compounds from Commiphora myrrha (Nees) Engl. Pharmazie 2006, 61, 728–731. [Google Scholar] [CrossRef]
- Wang, C.-C.; Liang, N.-Y.; Xia, H.; Wang, R.-Y.; Zhang, Y.-F.; Huo, H.-X.; Zhao, Y.-F.; Song, Y.-L.; Zheng, J.; Tu, P.-F. Cytotoxic sesquiterpenoid dimers from the resin of Commiphora myrrha Engl. Phytochemistry 2022, 204, 113443. [Google Scholar] [CrossRef]
- Dong, L.; Qin, D.-P.; Di, Q.-Q.; Liu, Y.; Chen, W.-L.; Wang, S.-M.; Cheng, Y.-X. Commiphorines A and B, unprecedented sesquiterpenoid dimers from Resina Commiphora with striking activities on anti-inflammation and lipogenesis inhibition. Org. Chem. Front. 2019, 6, 3825–3833. [Google Scholar] [CrossRef]
- Liu, J.-W.; Liu, Y.; Yan, Y.-M.; Yang, J.; Lu, X.-F.; Cheng, Y.-X. Commiphoratones A and B, two sesquiterpene dimers from Resina Commiphora. Org. Lett. 2018, 20, 2220–2223. [Google Scholar] [CrossRef]
- Hu, B.-Y.; Qin, D.-P.; Wang, S.-X.; Qi, J.-J.; Cheng, Y.-X. Novel terpenoids with potent cytotoxic activities from Resina Commiphora. Molecules 2018, 23, 3239. [Google Scholar] [CrossRef]
- Zhu, C.-Z.; Hu, B.-Y.; Liu, J.-W.; Cai, Y.; Chen, X.-C.; Qin, D.-P.; Cheng, Y.-X.; Zhang, Z.-D. Anti-Mycobacterium tuberculosis terpenoids from Resina Commiphora. Molecules 2019, 24, 1475. [Google Scholar] [CrossRef]
- Liu, J.-W.; Zhang, M.-Y.; Yan, Y.-M.; Wei, X.-Y.; Dong, L.; Zhu, Y.-X.; Cheng, Y.-X. Characterization of sesquiterpene dimers from Resina Commiphora that promote adipose-derived stem cell proliferation and differentiation. J. Org. Chem. 2018, 83, 2725–2733. [Google Scholar] [CrossRef]
- Dong, L.; Jiang, J.-B.; Yan, Y.-M.; Wang, S.-M.; Cheng, Y.-X. Commiphoroids G1—G3, H and I, Five Terpenoid Dimers as Extracellular Matrix Inhibitors from Resina Commiphora. Chin. J. Chem. 2021, 39, 2172–2180. [Google Scholar] [CrossRef]
- Ma, Y.-H.; Dou, X.-X.; Tian, X.-H. Natural disesquiterpenoids: An overview of their chemical structures, pharmacological activities, and biosynthetic pathways. Phytochem. Rev. 2020, 19, 983–1043. [Google Scholar] [CrossRef]
- Zhan, Z.-J.; Ying, Y.-M.; Ma, L.-F.; Shan, W.-G. Natural disesquiterpenoids. Nat. Prod. Rep. 2011, 28, 594–629. [Google Scholar] [CrossRef]
- Cao, B.; Wei, X.-C.; Xu, X.-R.; Zhang, H.-Z.; Luo, C.-H.; Feng, B.; Xu, R.-C.; Zhao, S.-Y.; Du, X.-J.; Han, L.; et al. Seeing the unseen of the combination of two natural resins, frankincense and myrrh: Changes in chemical constituents and pharmacological activities. Molecules 2019, 24, 3076. [Google Scholar] [CrossRef]
- Langhorst, J.; Varnhagen, I.; Schneider, S.B.; Albrecht, U.; Rueffer, A.; Stange, R.; Michalsen, A.; Dobos, G.J. Randomised clinical trial: A herbal preparation of myrrh, chamomile and coffee charcoal compared with mesalazine in maintaining remission in ulcerative colitis—A double-blind, double-dummy study. Aliment. Pharmacol. Ther. 2013, 38, 490–500. [Google Scholar] [CrossRef]
- Boffa, L.; Binello, A.; Boscaro, V.; Gallicchio, M.; Amisano, G.; Fornasero, S.; Cravotto, G. Commiphora myrrha (Nees) Engl. extracts: Evaluation of antioxidant and antiproliferative activity and their ability to reduce microbial growth on fresh-cut salad. Int. J. Food Sci. Technol. 2016, 51, 625–632. [Google Scholar] [CrossRef]
- Shoemaker, M.; Hamilton, B.; Dairkee, S.H.; Cohen, I.; Campbell, M.J. In vitro anticancer activity of twelve chinese medicinal herbs. Phytother. Res. 2005, 19, 649–651. [Google Scholar] [CrossRef]
- Tipton, D.A.; Lyle, B.; Babich, H.; Dabbous, M.K. In vitro cytotoxic and anti-inflammatory effects of myrrh oil on human gingival fibroblasts and epithelial cells. Toxicol. In Vitro 2003, 17, 301–310. [Google Scholar] [CrossRef]
- Al-Harbi, M.M.; Qureshi, S.; Raza, M.; Ahmed, M.M.; Giangreco, A.B.; Shah, A.H. Anticarcinogenic effect of Commiphora molmol on solid tumors induced by Ehrlich carcinoma cells in mice. Chemotherapy 1994, 40, 337–347. [Google Scholar] [CrossRef]
- Liaw, C.-C.; Chen, Y.-C.; Huang, G.-J.; Tsai, Y.-C.; Chien, S.-C.; Wu, J.-H.; Wang, S.-Y.; Chao, L.K.; Sung, P.-J.; Huang, H.-C.; et al. Anti-inflammatory lanostanoids and lactone derivatives from Antrodia camphorata. J. Nat. Prod. 2013, 76, 489–494. [Google Scholar] [CrossRef]
- Silva, M.d.L.e.; David, J.P.; Silva, L.C.R.C.; Santos, R.A.F.; David, J.M.; Lima, L.S.; Reis, P.S.; Fontana, R. Bioactive oleanane, lupane and ursane triterpene acid derivatives. Molecules 2012, 17, 12197–12205. [Google Scholar] [CrossRef]
- Baltina, L.A.; Flekhter, O.B.; Nigmatullina, L.R.; Boreko, E.I.; Pavlova, N.I.; Nikolaeva, S.N.; Savinova, O.V.; Tolstikov, G.A. Lupane triterpenes and derivatives with antiviral activity. Bioorg. Med. Chem. Lett. 2003, 13, 3549–3552. [Google Scholar] [CrossRef]
- Ahmed, Y.; Sohrab, M.H.; Al-Reza, S.M.; Tareq, F.S.; Hasan, C.M.; Sattar, M.A. Antimicrobial and cytotoxic constituents from leaves of Sapium baccatum. Food Chem. Toxicol. 2010, 48, 549–552. [Google Scholar] [CrossRef]
- Mokoka, T.A.; McGaw, L.J.; Mdee, L.K.; Bagla, V.P.; Iwalewa, E.O.; Eloff, J.N. Antimicrobial activity and cytotoxicity of triterpenes isolated from leaves of Maytenus undata (Celastraceae). BMC Complement. Altern. Med. 2013, 13, 111. [Google Scholar] [CrossRef]
- Chudzik, M.; Korzonek-Szlacheta, I.; Król, W. Triterpenes as potentially cytotoxic compounds. Molecules 2015, 20, 1610–1625. [Google Scholar] [CrossRef] [Green Version]
- Weber, L.; Kuck, K.; Jürgenliemk, G.; Heilmann, J.; Lipowicz, B.; Vissiennon, C. Anti-inflammatory and barrier-stabilising effects of myrrh, coffee charcoal and chamomile flower extract in a co-culture cell model of the intestinal mucosa. Biomolecules 2020, 10, 1033. [Google Scholar] [CrossRef]
- Provan, G.J.; Gray, A.I.; Waterman, P.G. Sesquiterpenes from the myrrh-type resins of some kenyan Commiphora species. Flavour Fragr. J. 1987, 2, 109–113. [Google Scholar] [CrossRef]
- Brieskorn, C.H.; Noble, P. Drei neue Furanogermacrene aus Myrrhe. Tetrahedron Lett. 1980, 21, 1511–1514. [Google Scholar] [CrossRef]
- Santoro, E.; Messina, F.; Marcotullio, M.C.; Superchi, S. Absolute configuration of bioactive furanogermacrenones from Commiphora erythraea (Ehrenb) Engl. by computational analysis of their chiroptical properties. Tetrahedron 2014, 70, 8033–8039. [Google Scholar] [CrossRef]
- Brieskorn, C.H.; Noble, P. Furanosesquiterpenes from the essential oil of myrrh. Phytochemistry 1983, 22, 1207–1211. [Google Scholar] [CrossRef]
- Lange, G.L.; Lee, M. 13C NMR determination of the configuration of methyl-substituted double bonds in medium- and large-ring terpenoids. Magn. Reson. Chem. 1986, 24, 656–658. [Google Scholar] [CrossRef]
- Hikino, H.; Konno, C.; Agatsuma, K.; Takemoto, T.; Horibe, I.; Tori, K.; Ueyama, M.; Takeda, K. Sesquiterpenoids. Part XLVII. Structure, configuration, conformation, and thermal rearrangement of furanodienone, isofuranodienone, curzerenone, epicurzerenone, and pyrocurzerenone, sesquiterpenoids of Curcuma zedoaria. J. Chem. Soc. Perkin Trans. 1975, 1, 478–484. [Google Scholar] [CrossRef]
- Stahl, E.; Datta, S.N. Neue sesquiterpenoide Inhaltsstoffe der Gundelrebe (Glechoma hederacea L.). Justus Liebigs Ann. Chem. 1972, 757, 23–32. [Google Scholar] [CrossRef]
- Shen, T.; Wan, W.-Z.; Wang, X.-N.; Sun, L.-M.; Yuan, H.-Q.; Wang, X.-L.; Ji, M.; Lou, H.-X. Sesquiterpenoids from the resinous exudates of Commiphora opobalsamum (Burseraceae). Helv. Chim. Acta 2008, 91, 881–887. [Google Scholar] [CrossRef]
- Zhao, N.; Yang, G.-C.; Li, D.-H.; Li, X.-Y.; Li, Z.-L.; Bai, J.; Liu, X.-Q.; Hua, H.-M. Two new sesquiterpenes from myrrh. Helv. Chim. Acta 2015, 98, 1332–1336. [Google Scholar] [CrossRef]
- Zhu, N.; Kikuzaki, H.; Sheng, S.; Sang, S.; Rafi, M.M.; Wang, M.; Nakatani, N.; DiPaola, R.S.; Rosen, R.T.; Ho, C.-T. Furanosesquiterpenoids of Commiphora myrrha. J. Nat. Prod. 2001, 64, 1460–1462. [Google Scholar] [CrossRef]
- Ulubelen, A.; Gören, N.; Jakupovic, J. Germacrane derivatives from the fruits of Smyrnium creticum. Phytochemistry 1986, 26, 312–313. [Google Scholar] [CrossRef]
- Gao, J.-F.; Xie, J.-H.; Iitaka, Y.; Inayama, S. The stereostructure of wenjine and related (1S,10S),(4S,5S)-germacrone-1(10),4-diepoxide isolated from Curcuma wenyujin. Chem. Pharm. Bull. 1989, 37, 233–236. [Google Scholar] [CrossRef]
- Tada, H.; Minato, H.; Takeda, K. Components of the root of Lindera strychnifolia Vill. Part XVIII. Neosericenyl acetate and dehydrolindestrenolide. J. Chem. Soc. C 1971, 1070–1073. [Google Scholar] [CrossRef]
- Duh, C.-Y.; Chen, K.-J.; El-Gamal, A.A.H.; Dai, C.-F. Sesquiterpenes from the formosan stolonifer Tubipora musica. J. Nat. Prod. 2001, 64, 1430–1433. [Google Scholar] [CrossRef]
- Endo, K.; Taguchi, T.; Taguchi, F.; Hikino, H.; Yamahara, J.; Fujimura, H. Antiinflammatory principles of Atractylodes rhizomes. Chem. Pharm. Bull. 1979, 27, 2954–2958. [Google Scholar] [CrossRef]
- Friedrich, D.; Bohlmann, F. Total synthesis of various elemanolides. Tetrahedron 1988, 44, 1369–1392. [Google Scholar] [CrossRef]
- Provan, G.J.; Waterman, P.G. Major triterpenes from the resins of Commiphora incisa and C. kua and their potential chemotaxonomic significance. Phytochemistry 1988, 27, 3841–3843. [Google Scholar] [CrossRef]
- Provan, G.J.; Waterman, P.G. The mansumbinanes: Octanordammaranes from the resin of Commiphora incisa. Phytochemistry 1986, 25, 917–922. [Google Scholar] [CrossRef]
- Rahman, M.M.; Garvey, M.; Piddock, L.J.V.; Gibbons, S. Antibacterial terpenes from the oleo-resin of Commiphora molmol (Engl.). Phytother. Res. 2008, 22, 1356–1360. [Google Scholar] [CrossRef]
- Shen, T.; Yuan, H.-Q.; Wan, W.-Z.; Wang, X.-L.; Wang, X.-N.; Ji, M.; Lou, H.-X. Cycloartane-type triterpenoids from the resinous exudates of Commiphora opobalsamum. J. Nat. Prod. 2008, 71, 81–86. [Google Scholar] [CrossRef]
- Shen, T.; Wan, W.; Yuan, H.; Kong, F.; Guo, H.; Fan, P.; Lou, H. Secondary metabolites from Commiphora opobalsamum and their antiproliferative effect on human prostate cancer cells. Phytochemistry 2007, 68, 1331–1337. [Google Scholar] [CrossRef]
- Gao, W.; Su, X.; Dong, X.; Chen, Y.; Zhou, C.; Xin, P.; Yu, C.; Wei, T. Cycloartan-24-ene-1α,2α,3β-triol, a cycloartane-type triterpenoid from the resinous exudates of Commiphora myrrha, induces apoptosis in human prostatic cancer PC-3 cells. Oncol. Rep. 2015, 33, 1107–1114. [Google Scholar] [CrossRef]
- Mills, J.S.; Werner, A.E.A. The chemistry of dammar resin. J. Chem. Soc. 1955, 3132–3140. [Google Scholar] [CrossRef]
- Asakawa, J.; Kasai, R.; Yamasaki, K.; Tanaka, O. 13C NMR study of ginseng sapogenins and their related dammarane type triterpenes. Tetrahedron 1977, 33, 1935–1939. [Google Scholar] [CrossRef]
- Freischmidt, A.; Jürgenliemk, G.; Kraus, B.; Okpanyi, S.N.; Müller, J.; Kelber, O.; Weiser, D.; Heilmann, J. Contribution of flavonoids and catechol to the reduction of ICAM-1 expression in endothelial cells by a standardised willow bark extract. Phytomedicine 2012, 19, 245–252. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Monographie Myrrhe, Myrrha. Europäisches Arzneibuch 10. Ausgabe, Grundwerk 2020: Amtliche Deutsche Ausgabe (Ph. Eur. 10.0); Deutscher Apotheker Verlag: Stuttgart, Germany, 2020; ISBN 9783769275155. [Google Scholar]
- Alqahtani, A.S.; Nasr, F.A.; Noman, O.M.; Farooq, M.; Alhawassi, T.; Qamar, W.; El-Gamal, A. Cytotoxic evaluation and anti-angiogenic effects of two furano-sesquiterpenoids from Commiphora myrrh resin. Molecules 2020, 25, 1318. [Google Scholar] [CrossRef]
- Marston, A.; Borel, C.; Hostettmann, K. Separation of natural products by centrifugal partition chromatography. J. Chromatogr. A 1988, 450, 91–99. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | 1 | 2 | 3 | |||
|---|---|---|---|---|---|---|
| δH | δC | δH | δC | δH | δC | |
| 1 | 7.02 (1H, d, 7.1) | 128.5 | 7.04 (1H, d, 7.7) | 128.6 | 5.83 (1H, dd, 11.0, 17.6) | 149.6 |
| 2 | 7.09 (1H, dd, 6.9, 8.0) | 127.3 | 7.10 (1H, dd, 7.1, 8.2) | 127.2 | 4.90 (1H, brd, 18.7) 4.93 (1H, dd, 1.4, 12.4) | 110.7 |
| 3 | 7.02 (1H, d, 7.1) | 128.5 | 7.04 (1H, d, 7.7) | 128.6 | 4.69 (1H, brs) 4.90 (1H, brs) | 112.8 |
| 4 | 136.8 | 136.8 | 146.8 | |||
| 5 | 132.7 | 134.8 | 2.57 (1H, dd, 3.0, 13.5) | 46.2 | ||
| 6 | 3.64 (1H, d, 17.6) 3.72 (1H, d, 18.2) | 28.3 | 4.09 (2H, s) | 25.6 | 1.54 (1H, ddd, 1.8, 2.9, 14.2) 2.35 (1H, dd, 13.8, 13.8) | 33.3 |
| 7 | 160.9 | 135.9 | 73.9 | |||
| 8 | 4.72 (1H, q, 7.0) | 79.1 | 149.2 | 4.97 1 (1H, m) | 72.3 | |
| 9 | 1.29 (3H, d, 6.6) | 18.7 | 8.84 (1H, s) | 178.5 | 1.56 (1H, dd, 3.0, 15.1) 2.06 (1H, dd, 3.3, 14.9) | 38.3 |
| 10 | 136.8 | 136.8 | 39.0 | |||
| 11 | 123.4 | 123.5 | 148.5 | |||
| 12 | 174.4 | 7.37 (1H, s) | 144.8 | 4.95 (1H, brs) 5.05 (1H, brs) | 112.4 | |
| 13 | 1.47 (3H, brs) | 8.3 | 1.83 (3H, s) | 8.1 | 1.79 (3H, s) | 18.5 |
| 14 | 2.24 (3H, s) | 20.3 | 2.26 (3H, s) | 20.2 | 1.06 (3H, s) | 18.6 |
| 15 | 2.24 (3H, s) | 20.3 | 2.26 (3H, s) | 20.2 | 1.76 (3H, s) | 24.9 |
| 1′ | 169.7 | |||||
| 2′ | 2.00 (3H, s) | 21.2 | ||||
| No. | 4 | 5 | 6 | |||
|---|---|---|---|---|---|---|
| δH | δC | δH | δC | δH | δC | |
| 1 | 130.4 | 4.99 (1H, d, 10.8) | 132.1 | 4.89 (1H, d, 10.0) | 133.3 | |
| 2 | 4.33 (1H, dd, 2.8, 2.8) | 73.9 | 4.13 (1H, ddd, 5.0, 10.1, 10.5) | 78.2 | 4.16 (1H, ddd, 5.0, 10.1, 10.1) | 77.2 |
| 3 | 1.28 1 (1H, m) 2.28 1 (1H, m) | 34.0 | 1.96 (1H, dd, 11.3, 11.3) 2.55 (1H, dd, 5.0, 11.3) | 45.9 | 1.94 (1H, dd, 11.0, 11.0) 2.54 (1H, dd, 5.0, 10.1) | 45.3 |
| 4 | 2.08 (1H, m) | 22.7 | 134.8 | 133.3 | ||
| 5 | 2.24 1 (1H, m) 2.72 (1H, ddd, 1.7, 4.7, 16.5) | 36.2 | 4.96 (1H, dd, 4.4, 10.5) | 124.9 | 4.56 (1H, brd, 11.2) | 126.1 |
| 6 | 134.8 | 3.14 (1H, dd, 3.3, 18.2) 3.34 (1H, dd, 10.5, 17.9) | 27.0 | 2.88 (1H, dd, 11.2, 14.9) 3.42 (1H, brd, 14.7) | 27.7 | |
| 7 | 123.3 | 162.0 | 162.2 | |||
| 8 | 152.6 | 5.12 (1H, dd, 6.6, 6.6) | 83.3 | 4.93 (1H, dd, 4.3, 11.7) | 82.6 | |
| 9 | 6.84 (1H, s) | 110.7 | 2.23 (1H, dd, 6.6, 14.5) 3.06 (1H, dd, 6.6, 14.5) | 42.9 | 2.12 (1H, dd, 11.7, 12.4) 3.09 (1H, dd, 4.3, 12.8) | 46.8 |
| 10 | 140.1 | 135.4 | 134.5 | |||
| 11 | 3.64 1 (1H, m) | 38.2 | 125.9 | 126.3 | ||
| 12 | 178.6 | 173.5 | 173.6 | |||
| 13 | 1.55 (3H, d, 7.7) | 15.4 | 1.88 (3H, brs) | 9.3 | 1.87 (3H, brs) | 8.9 |
| 14 | 2.37 (3H, s) | 19.3 | 1.44 (3H, s) | 19.6 | 1.57 (3H, s) | 17.4 |
| 15 | 1.13 (3H, d, 6.6) | 22.1 | 1.61 (3H, s) | 18.0 | 1.67 (3H, brs) | 18.0 |
| 1′ | 3.44 (3H, s) | 56.2 | 3.35 (3H, s) | 57.4 | 3.34 (3H, s) | 57.4 |
| No. | 7 | 8 (Part I) | 8 (Part II) | ||||
|---|---|---|---|---|---|---|---|
| δH | δC | δH | δC | No. | δH | δC | |
| 1 | 5.72 (1H, d, 8.8) | 135.0 | 2.71 1 (1H, d, 8.8) | 57.5 | 1’ | 5.49 (1H, d, 16.0) | 143.4 |
| 2 | 5.86 (1H, dd, 5.0, 9.4) | 123.7 | 3.48 1 (1H, m) | 83.5 | 2’ | 5.58 (1H, dd, 9.1, 15.7) | 133.6 |
| 3 | 5.79 (1H, m) | 120.9 | 1.14 1 (1H, m) 2.01 1 (1H, m) | 39.8 | 3’ | 3.03 (1H, dd, 9.1, 9.1) | 88.9 |
| 4 | 135.8 | 2.70 1 (1H, m) | 33.1 | 4’ | 2.57 1 (1H, m) | 37.4 | |
| 5 | 2.87 (1H, brd, 14.3) | 44.3 | 2.29 (1H, dd, 9.6, 9.6) | 59.2 | 5’ | 2.39 1 (1H, m) 2.46 (1H, dd, 11.8, 14.6) | 48.5 |
| 6 | 2.54 (1H, ddd, 2.0, 2.0, 16.2) 2.96 (1H, dd, 4.4, 17.1) | 21.3 | 196.5 | 6’ | 202.4 | ||
| 7 | 148.5 | 122.5 | 7’ | 117.6 | |||
| 8 | 147.9 | 159.0 | 8’ | 153.5 | |||
| 9 | 5.77 (1H, s) | 117.7 | 3.50 (1H, s) | 54.7 | 9’ | 2.59 (1H, d, 14.3) 2.96 (1H, d, 14.3) | 35.1 |
| 10 | 37.8 | 36.1 | 10’ | - | 42.0 | ||
| 11 | 121.1 | 121.1 | 11’ | - | 128.6 | ||
| 12 | 171.2 | 7.16 (1H, s) | 139.2 | 12’ | 6.92 (1H, s) | 138.3 | |
| 13 | 1.93 (3H, brs) | 8.6 | 2.21 (3H, s) | 10.2 | 13’ | 2.01 (3H, s) | 9.4 |
| 14 | 1.02 (3H, s) | 17.9 | 1.18 (3H, s) | 25.6 | 14’ | 1.84 (1H, d, 11.6) 2.39 1 (1H, m) | 42.9 |
| 15 | 1.90 (3H, brs) | 20.0 | 1.12 (3H, d, 6.6) | 19.4 | 15’ | 1.15 1 (3H, d, 6.1) | 18.5 |
| 16 | 3.44 (3H, s) | 56.2 | 3.33 (3H, s) | 56.3 | 16’ | 3.29 (3H, s) | 56.6 |
| No. | 23 | 24 | ||
|---|---|---|---|---|
| δH | δC | δH | δC | |
| 1 | 3.94 (1H, brs) | 75.1 | 3.22 (1H, d, 3.9) | 59.6 |
| 2 | 3.75 (1H, dd, 2.9, 9.8) | 72.3 | 3.12 (1H, d, 3.9) | 57.5 |
| 3 | 4.77 (1H, dd, 9.9, 9.9) | 80.0 | 3.47 1 (1H, m) | 72.8 |
| 4 | 1.63 1 (1H, m) | 34.8 | 1.29 1 (1H, m) | 36.1 |
| 5 | 1.62 1 (1H, m) | 39.3 | 1.21 1 (1H, m) | 35.6 |
| 6 | 1.32 1 (1H, m) 1.80 1 (1H, m) | 20.1 | 1.33 1 (1H, m) 1.74 1 (1H, m) | 19.2 |
| 7 | 2.04 1 (1H, m) 2.10 1 (1H, m) | 25.8 | 2.03 1 (2H, m) | 24.5 |
| 8 | 138.6 | 136.5 | ||
| 9 | 130.0 | 130.3 | ||
| 10 | 41.8 | 37.6 | ||
| 11 | 2.12 1 (1H, m) 2.22 1 (1H, m) | 21.5 | 2.23 (1H, m) 2.29 (1H, m) | 22.3 |
| 12 | 1.75 1 (1H, m) 1.82 1 (1H, m) | 30.8 | 1.76 1 (1H, m) 1.82 1(1H, m) | 30.7 |
| 13 | 44.6 | 44.5 | ||
| 14 | 50.1 | 49.9 | ||
| 15 | 1.21 1 (1H, m) 1.60 1 (1H, m) | 30.8 | 1.18 1 (1H, m) 1.57 1 (1H, m) | 30.6 |
| 16 | 1.33 1 (1H, m) 1.93 1 (1H, m) | 28.1 | 1.33 1 (1H, m) 1.93 (1H, m) | 28.1 |
| 17 | 1.52 1 (1H, m) | 50.3 | 1.52 1 (1H, m) | 50.2 |
| 18 | 0.72 (3H, s) | 15.7 | 0.74 1 (3H, s) | 15.7 |
| 19 | 1.00 1 (3H, s) | 18.2 | 1.06 (3H, s) | 17.9 |
| 20 | 1.40 1 (1H, m) | 36.3 | 1.40 1 (1H, m) | 36.3 |
| 21 | 0.93 1 (3H, m) | 18.7 | 0.93 (3H, d, 6.6) | 18.7 |
| 22 | 1.05 1 (1H, m) 1.45 1 (1H, m) | 36.3 | 1.05 1 (1H, m) 1.45 1 (1H, m) | 36.3 |
| 23 | 1.86 1 (1H, m) 2.04 1 (1H, m) | 25.0 | 1.87 1 (1H, m) 2.04 1 (1H, m) | 24.9 |
| 24 | 5.10 (1H, dd, 7.2, 7.2) | 125.0 | 5.10 (1H, dd, 7.2, 7.2) | 125.2 |
| 25 | 130.9 | 131.0 | ||
| 26 | 1.61 (3H, s) | 17.6 | 1.61 (3H, s) | 17.6 |
| 27 | 1.69 (3H, s) | 25.6 | 1.69 (3H, s) | 25.7 |
| 28 | 0.93 (3H, s) | 24.9 | 0.91 (3H, s) | 24.3 |
| 29 | 0.91 (3H, d, 6.1) | 15.0 | 0.97 (3H, d, 6.6) | 16.3 |
| 30 | ||||
| 1′ | 175.2 | |||
| 2′ | 2.27 1 (2H, m) | 43.7 | ||
| 3′ | 2.16 1 (1H, m) | 25.7 | ||
| 4′ | 0.99 (3H, d, 6.8) | 22.4 | ||
| 5′ | 0.99 (3H, d, 6.8) | 22.5 | ||
| Fraction | Gradient | Retention Time [min], Isolate | Fraction | Gradient | Retention Time [min], Isolate | ||
|---|---|---|---|---|---|---|---|
| Time [min] | ACN [%] | Time [min] | ACN [%] | ||||
| F5C2 | 0 6 10 11 16 | 65 79 88 100 100 | 8.8, 26 | F7C7F2 | 0 19 20 25 | 30 50 100 100 | 16.1, 6 15.4, 12 |
| F5C5 | 0 16 17 22 | 45 53 100 100 | 16.0, 9 11.9, 15 | F7C7F3 | 0 19 20 25 | 25 45 100 100 | 18.1, 5 |
| F6C3 | 0 15 20 | 70 100 100 | 11.6, 23 | F9C2 | 0 15 16 21 | 60 80 100 100 | 14.7, 31 |
| F6C4 | 0 20 21 26 | 60 80 100 100 | 17.3, 24 | F9C3 | 0 15 16 21 | 60 80 100 100 | 13.6, 28 11.8, 29 |
| F6C5 | 0 13 14 19 | 60 68 100 100 | 11.1, 34 | F9C4 | 0 12 13 16 | 60 76 100 100 | 9.7, 33 |
| F6C7F3 | 0 19 23 30 35 | 30 50 80 100 100 | 21.6, 16 11.6, 17 26.8, 25 | M1.2C3 | 0 15 25 26 31 | 40 50 60 95 95 | 20.7, 13 and 22 |
| F7C2 | 0 26 36 41 | 60 70 100 100 | 30.2, 30 33.8, 32 | M1.2C6 | 0 20 21 26 | 50 60 90 90 | 14.0, 1 17.4, 19 and 21 18.8, 20 20.6, 4 21.2, 3 27.2, impure 8 |
| F7C3 | 0 10 11 17 | 60 80 100 100 | 9.9, 27 | 27.2, from M1.2C6 | 0 16 17 26 | 57 68 90 90 | 15.2, 8 |
| F7C7F1 | 0 13 14 19 | 40 54 100 100 | 11.3, 10 12.4, 11 | M1.2C7 | 0 20 21 31 | 55 65 100 100 | 11.8, 14 13.1, 2 16.2, 7 16.9, 18 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuck, K.; Unterholzner, A.; Lipowicz, B.; Schwindl, S.; Jürgenliemk, G.; Schmidt, T.J.; Heilmann, J. Terpenoids from Myrrh and Their Cytotoxic Activity against HeLa Cells. Molecules 2023, 28, 1637. https://doi.org/10.3390/molecules28041637
Kuck K, Unterholzner A, Lipowicz B, Schwindl S, Jürgenliemk G, Schmidt TJ, Heilmann J. Terpenoids from Myrrh and Their Cytotoxic Activity against HeLa Cells. Molecules. 2023; 28(4):1637. https://doi.org/10.3390/molecules28041637
Chicago/Turabian StyleKuck, Katrin, Anna Unterholzner, Bartosz Lipowicz, Sebastian Schwindl, Guido Jürgenliemk, Thomas J. Schmidt, and Jörg Heilmann. 2023. "Terpenoids from Myrrh and Their Cytotoxic Activity against HeLa Cells" Molecules 28, no. 4: 1637. https://doi.org/10.3390/molecules28041637