
Effects of Morphology and Solvent/Temperature on THz Spectra: Take Nucleosides as Example
 , ,
, ,
Abstract
:1. Introduction
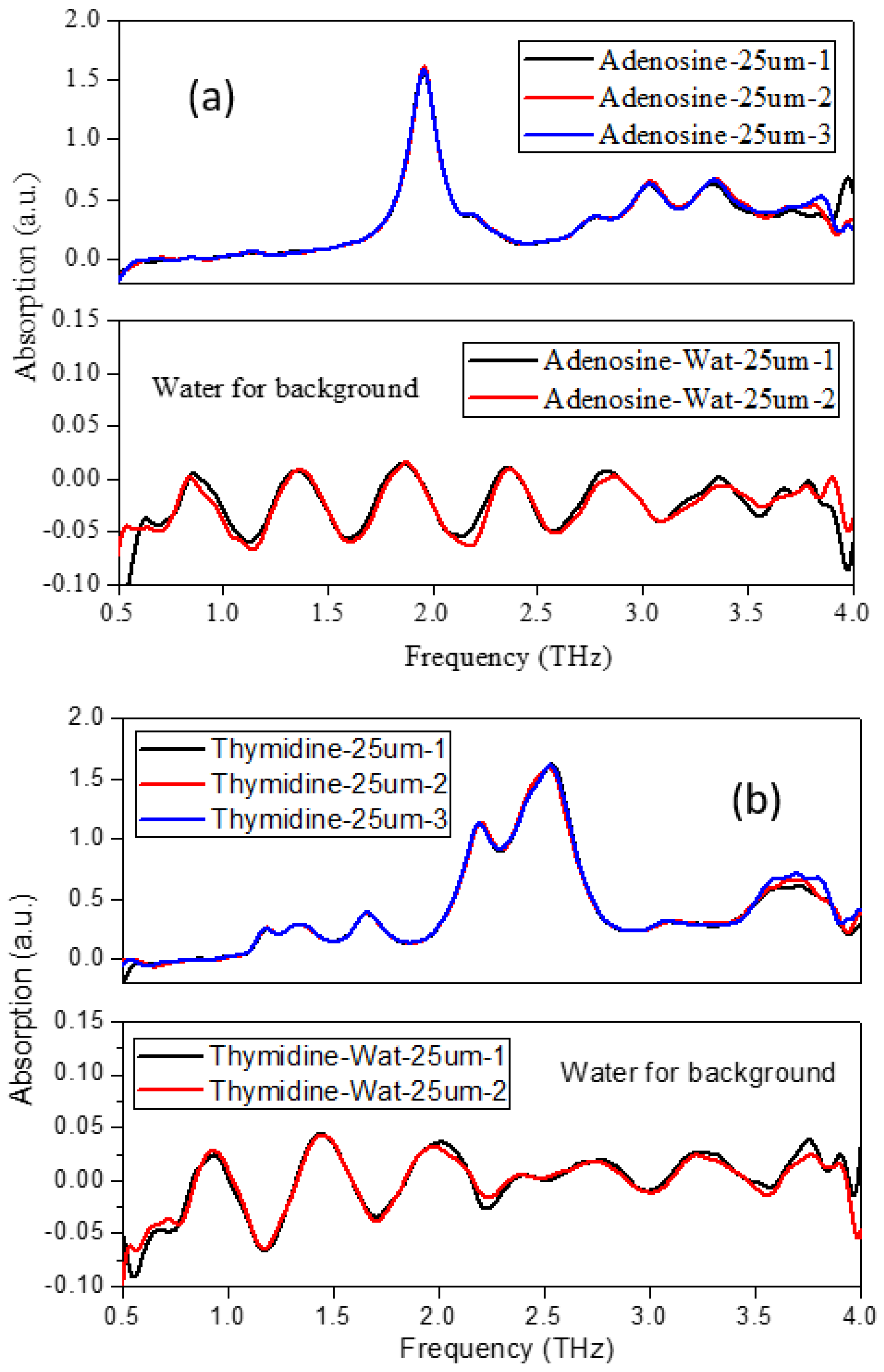
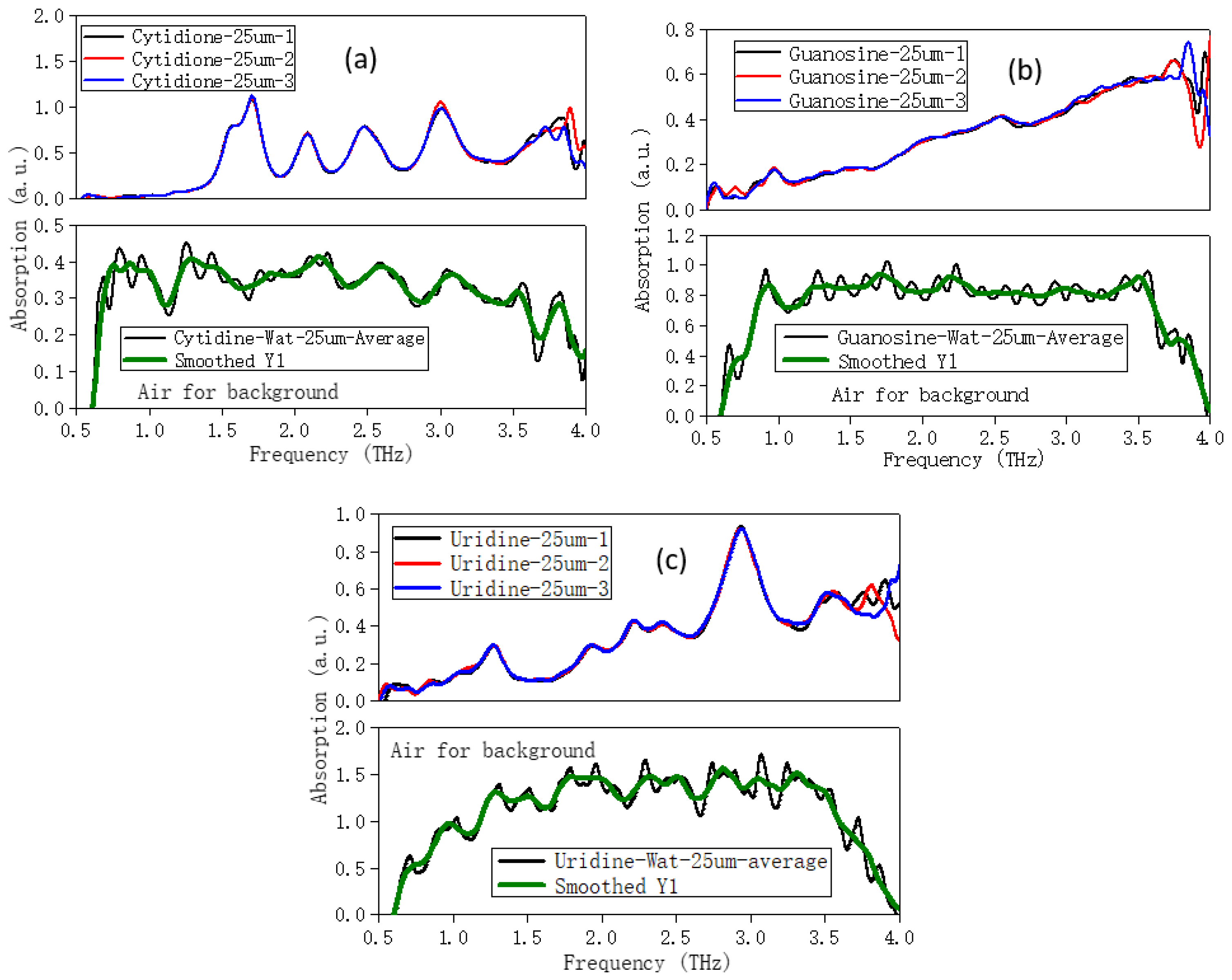
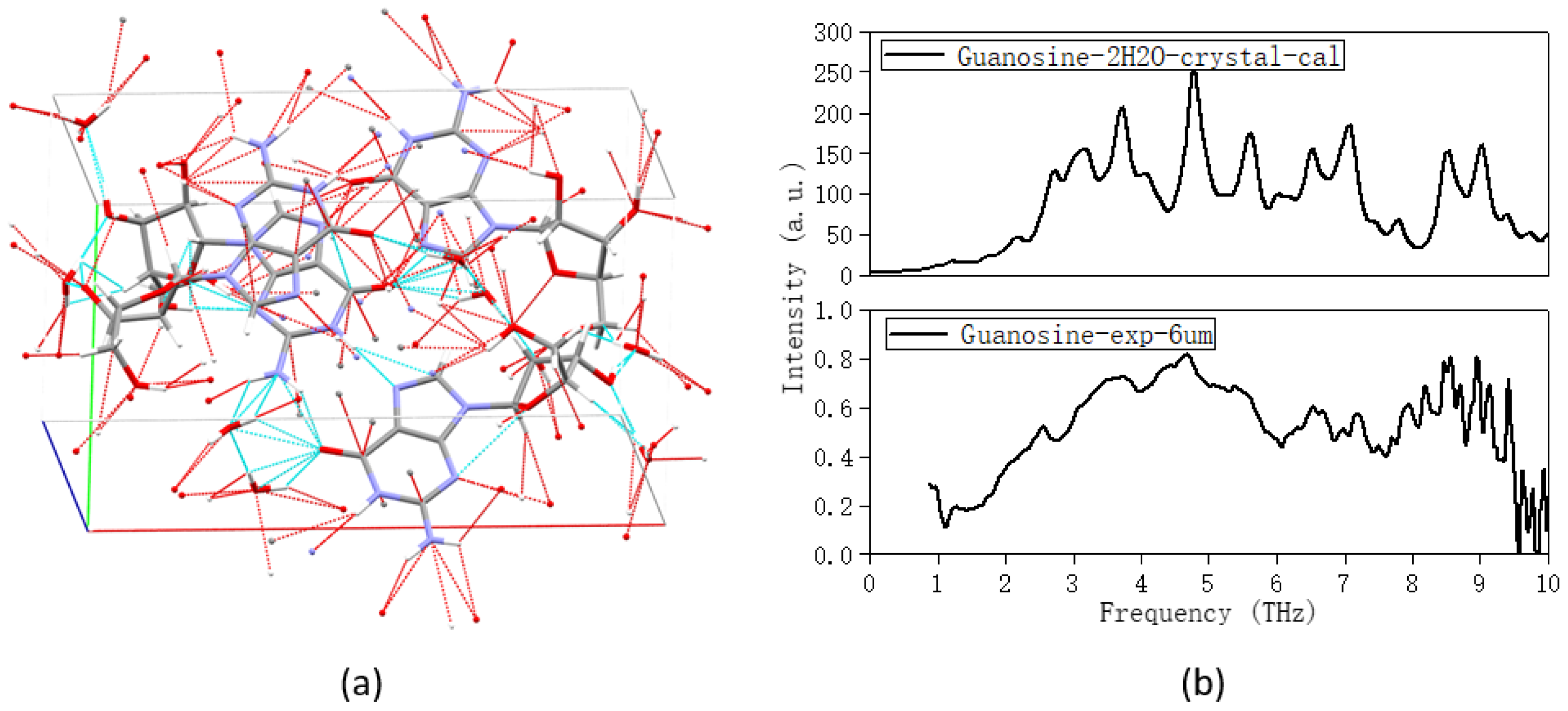
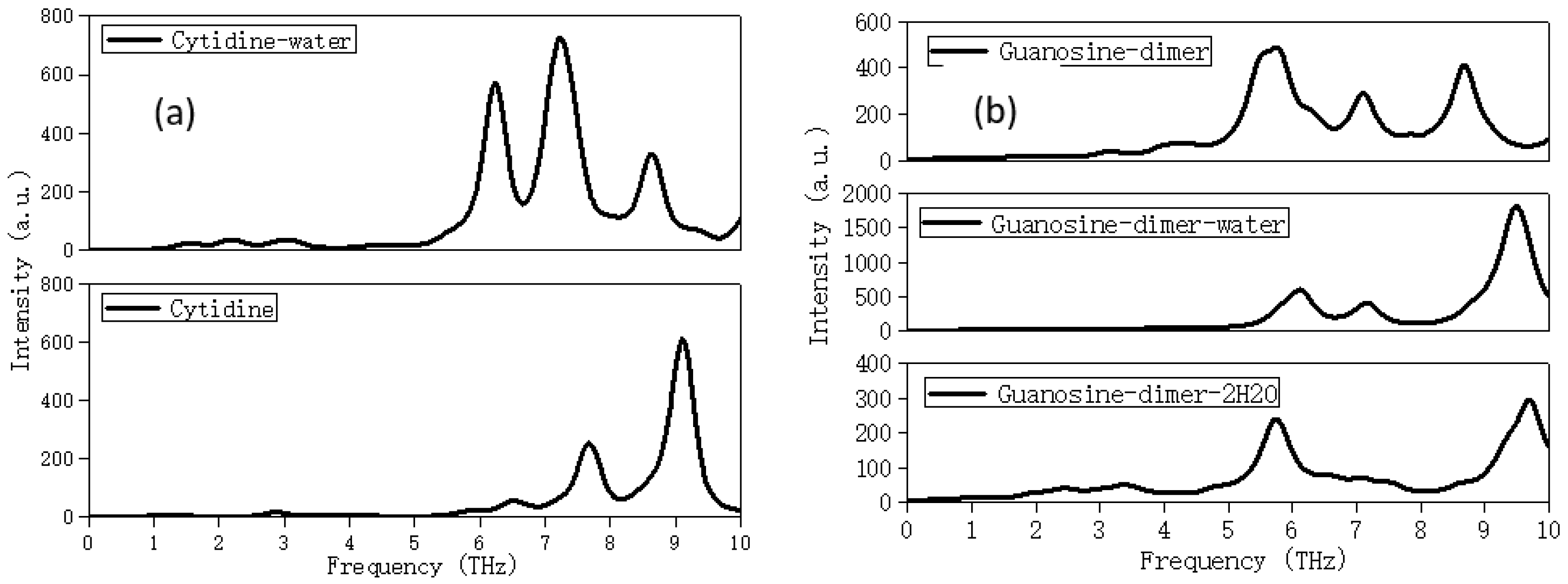
2. Results and Discussion
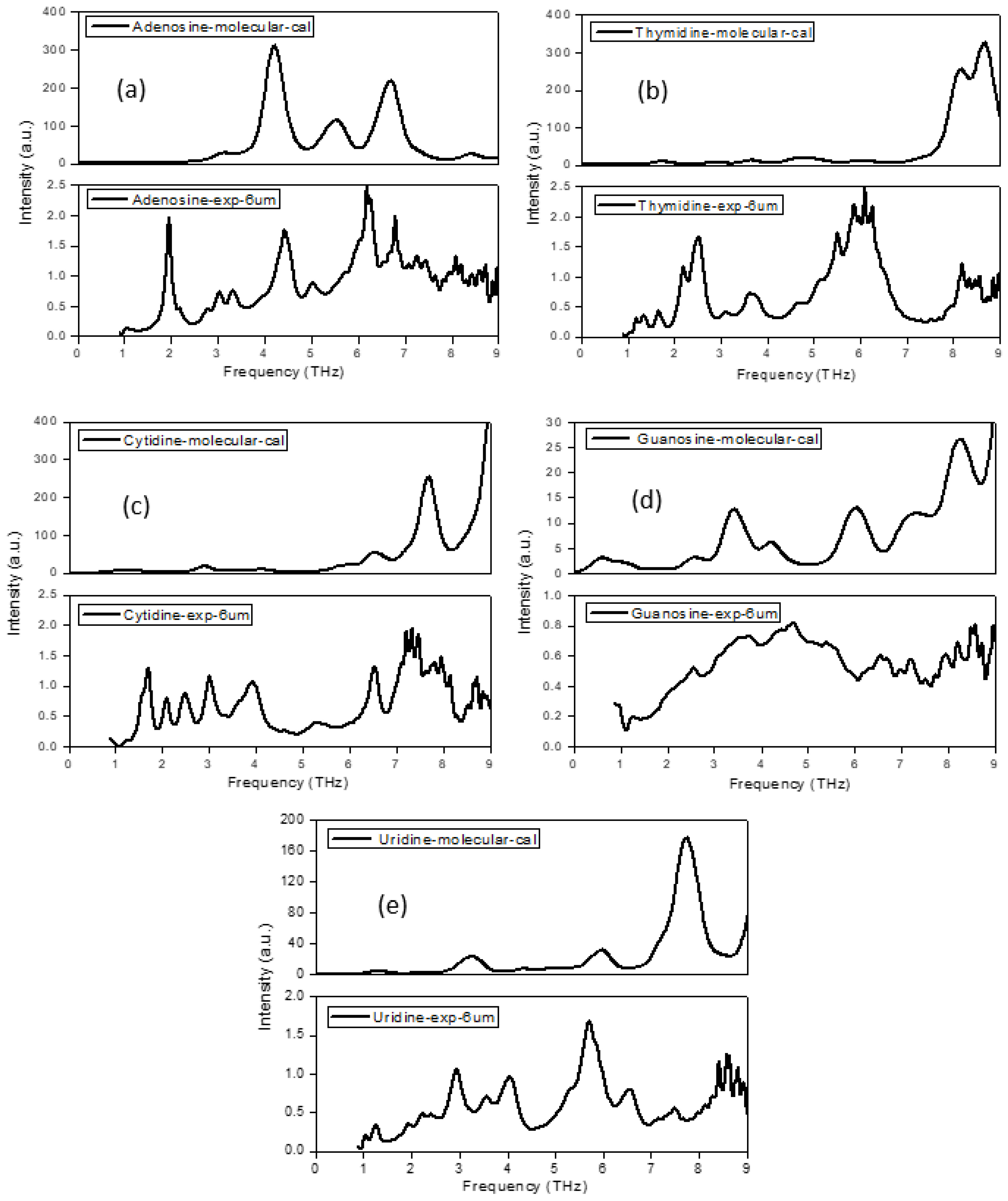
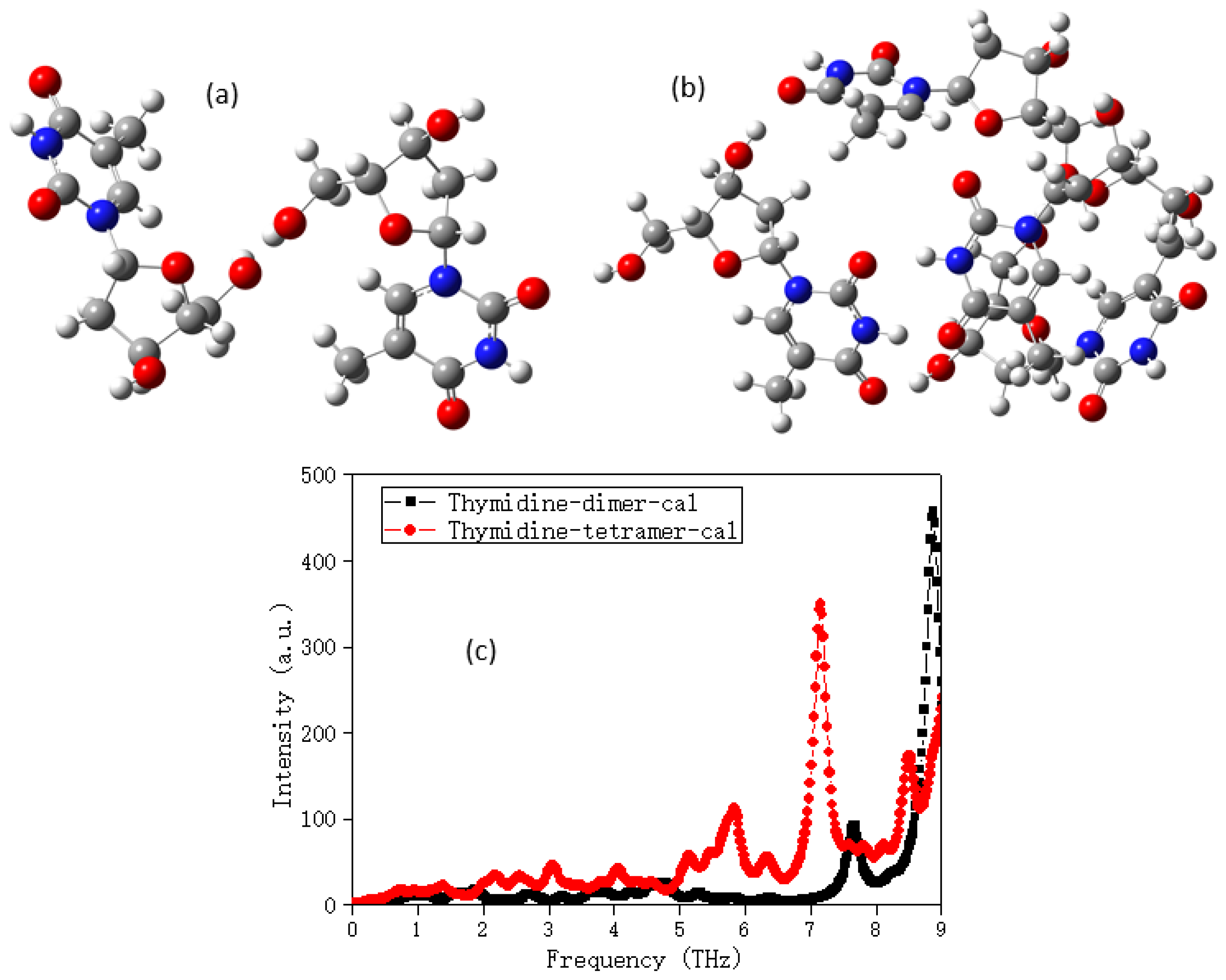
2.1. The Morphology Affects the Calculation Results
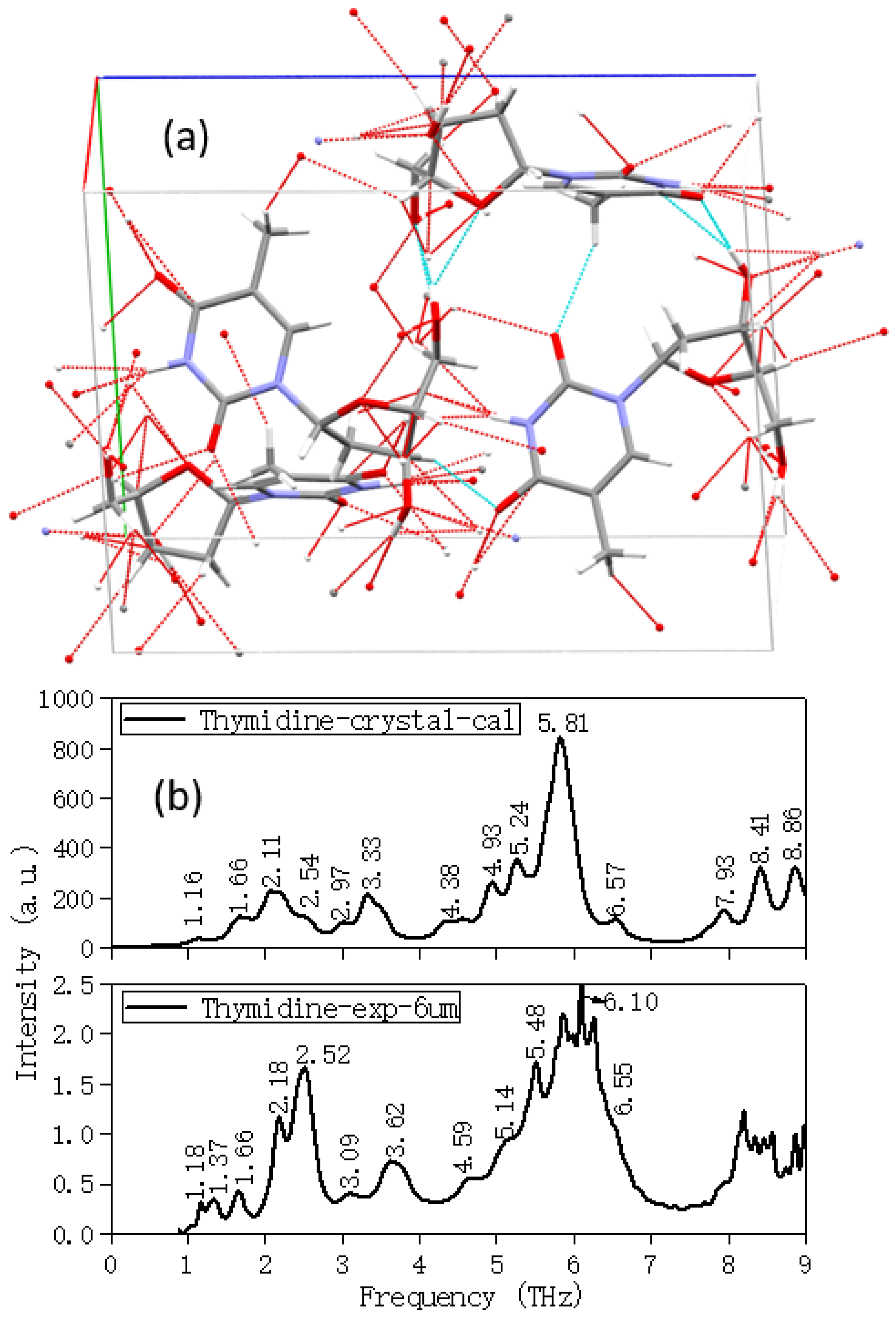
2.2. Temperature and Calculation Models Affect the Results
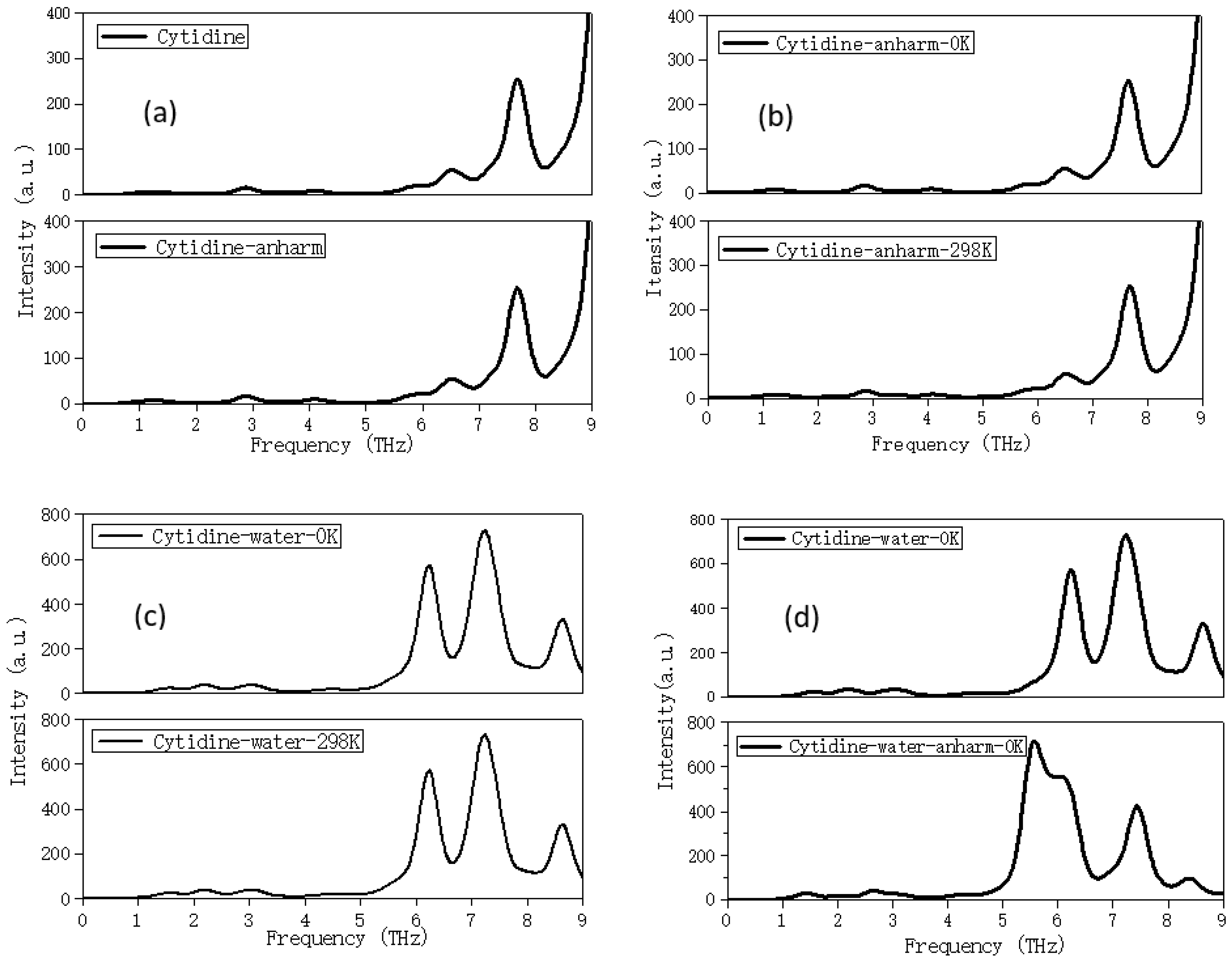
2.3. Solvation Affects the Calculation Results
3. Procedure for Experiment
4. Theoretical Method
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jordheim, L.P.; Durantel, D.; Zoulim, F.; Dumontet, C. Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat. Rev. Drug Discov. 2013, 12, 447–464. [Google Scholar] [CrossRef] [PubMed]
- Zhong, S.C. Progress in terahertz nondestructive testing: A review. Front. Mech. Eng. 2019, 14, 273–281. [Google Scholar] [CrossRef]
- Ferguson, B.; Zhang, X.C. Materials for terahertz science and technology. Nat. Mater. 2002, 1, 26–33. [Google Scholar] [CrossRef]
- Fischer, B.M.; Walther, M.; Jepsen, P.U. Far-infrared vibrational modes of DNA components studied by terahertz time-domain spectroscopy. Phys. Med. Biol. 2002, 47, 3807–3814. [Google Scholar] [CrossRef]
- Globus, T.; Sizov, I.; Ferrance, J.; Jazaeri, A.; Bryant, J.; Moyer, A.; Gelmont, B.; Kester, M.; Bykhovski, A. Sub-Terahertz Vibrational Spectroscopy for microRNA Based Diagnostic of Ovarian Cancer. Converg. Sci. Phys. Oncol. 2016, 2, 045001. [Google Scholar] [CrossRef]
- Dean, P.; Valavanis, A.; Keeley, J.; Bertling, K.; Lim, Y.L.; Alhathlool, R.; Burnett, A.D.; Li, L.H.; Khanna, S.P.; Indjin, D.; et al. Terahertz imaging using quantum cascade lasers-a review of systems and applications. J. Phys. D Appl. Phys. 2014, 47, 374008. [Google Scholar] [CrossRef]
- Wang, F.; Li, M.; Jiang, L.; Liu, Y.F. Terahertz spectral signature of DNA macromolecule. In Proceedings of the 1015 Asia-Pacific Microwave Conference (APMC), Nanjing, China, 6–9 December 2015; pp. 1106–1108. [Google Scholar]
- Wang, F.; Zhao, D.B.; Jiang, L.; Sun, H.J.; Liu, Y.F. A comparative study on the experimental and calculated results of mid-Infrared and Raman vibrational modes of nucleic acid bases. J. Mol. Graph. Model. 2017, 74, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhao, D.B.; Jiang, L.; Liu, Y.F.; Li, S.H. Terahertz spectra of DNA nucleobase crystals: A joint experimental and computational study. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2017, 179, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhao, D.B.; Jiang, L.; Liu, Y.F. THz spectra and corresponding vibrational modes of DNA base pair cocrystals and polynucleotides. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2018, 200, 195–201. [Google Scholar] [CrossRef]
- Wang, F.; Zhao, D.B.; Jiang, L.; Liu, Y.F. THz vibrational spectroscopy for RNA basepair cocrystals and oligonucleotide sequences. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2019, 209, 49–54. [Google Scholar] [CrossRef]
- Wang, F.; Jiang, L.; Song, J.; Huang, L.; Liu, Y.F. Sub-THz spectroscopic characterization identification for Pine Wood Nematode Ribosomal DNA. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2020, 232, 118152. [Google Scholar] [CrossRef]
- Wang, F.; Sun, X.L.; Zan, J.N.; Li, M.S.; Liu, Y.F.; Chen, J.Y. Terahertz spectra and weak intermolecular interactions of nucleosides or nucleoside drugs. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2022, 265, 120344. [Google Scholar] [CrossRef]
- Wang, F.; Zhu, N.; Sun, X.L.; Liu, C.; Zhuang, Y.; Zan, J.N.; Dai, C.F.; Liu, Y.F. Characterization of degradation behaviors of PLA biodegradable plastics by infrared spectroscopy. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2022, 279, 121376. [Google Scholar] [CrossRef]
- Heinz, A.; Strachan, C.J.; Gordon, K.C.; Rades, T. Analysis of solid-state transformations of pharmaceutical compounds using vibrational spectroscopy. J. Pharm. Pharmacol. 2009, 61, 971–988. [Google Scholar] [CrossRef]
- Li, X.W.; Globus, T.; Gelmont, B.; Salay, L.C.; Bykhovski, A. Terahertz absorption of DNA decamer duplex. J. Phys. Chem. A 2008, 112, 12090–12096. [Google Scholar] [CrossRef] [PubMed]
- Young, D.W.; Tollin, P.; Wilsin, H.R. The crystal and molecular structure of thymidine. Acta Cryst. 1969, 25, 1423. [Google Scholar] [CrossRef]
- Thewalt, U.; Bugg, C.E.; Marsh, R.E. The crystal structure of guanosine dihydrate and inosine dihydrate. Acta Crystallogr. Sect. B 1970, 26, 1089–1101. [Google Scholar] [CrossRef] [PubMed]
- Fang, T.; Jia, J.; Li, S. Vibrational spectra of Molecular Crystals with the Generalized Energy-Based Fragmentation Approach. J. Phys. Chem. A 2016, 120, 2700–2711. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Chen, C.; Zhao, D.B.; Li, S. LSQC: Low Scaling Quantum Chemistry Program. Int. J. Quantum. Chem. 2015, 115, 641–646. [Google Scholar] [CrossRef]
- Yuan, D.; Shen, X.L.; Li, W.; Li, S. Are fragment-based quantum chemistry methods applicable to medium-sized water clusters. Phys. Chem. Chem. Phys 2016, 18, 16491–16500. [Google Scholar] [CrossRef]
- Zhao, D.B.; Shen, X.L.; Cheng, Z.; Li, W.; Dong, H.; Li, S.H. Accurate and Efficient Prediction of NMR Parameters of Condensed-Phase Systems with the Generalized Energy-Based Fragmentation Method. J. Chem. Theory Comput. 2020, 16, 2995–3005. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
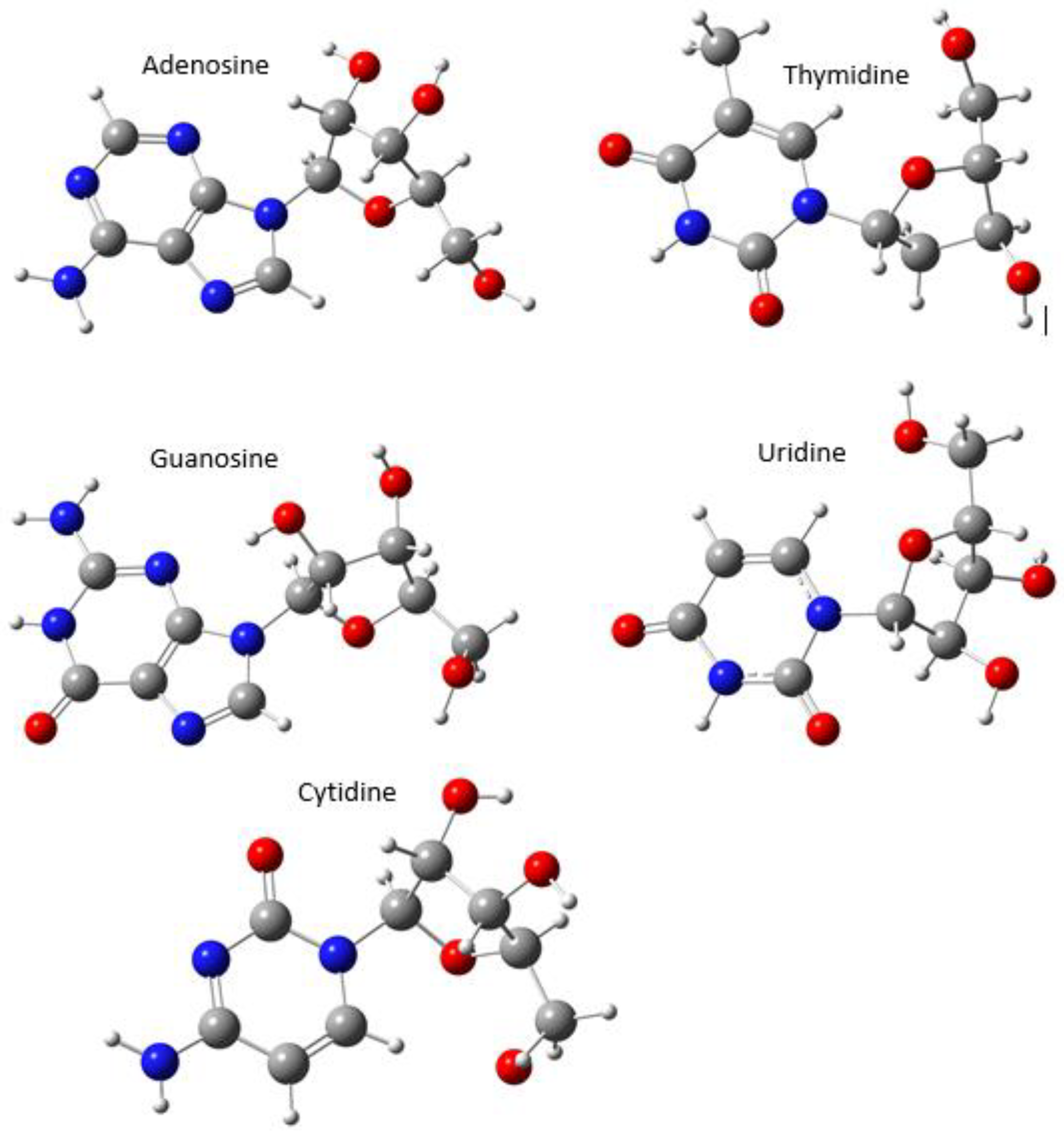{kind=link}
| Thymidine | |||
|---|---|---|---|
| Experimental | Calculated | ||
| Peaks (THz) | Main Vibrational Modes | Description | |
| 1.18 | 1.16 | Collective vibration | Overall out-of-plane swing |
| 1.37 | |||
| 1.66 | 1.66 | Collective vibration | Overall out-of-plane bending |
| 2.18 | 2.11 | Collective vibration | Overall out-of-plane swing and bending |
| 2.52 | 2.54 | Collective vibration | Overall out-of-plane swing and bending |
| 3.09 | 2.97 | Collective vibration | Overall out-of-plane swing |
| 3.62 | 3.33 | Collective vibration | Overall out-of-plane swing |
| 4.59 | 4.38 | Collective vibration | Out-of-plane swing and bending |
| 5.14 | 4.93 | Collective vibration | Out-of-plane swing and bending |
| 5.48 | 5.24 | Collective vibration | Out-of-plane swing and bending |
| 6.10 | 5.81 | Partial vibration | Partial out-of-plane bending |
| 6.55 | 6.57 | Partial vibration | Pentose bases out-of-plane swing and bending |
| 7.93 | Partial vibration | Purine ring expanding, pentose bases out-of-plane swing | |
| 8.41 | Partial vibration | Partial weak out-of-plane bending | |
| 8.86 | Partial vibration | CH3 on pentose group in-plane swing | |
| Adenosine | Thymidine | Cytidine | Guanosine | Uridine | |||||
|---|---|---|---|---|---|---|---|---|---|
| Solid | Liquid | Solid | Liquid | Solid | Liquid | Solid | Liquid | Solid | Liquid |
| 0.85 | 1.18 | 0.93 | 0.86 | 0.98 | 0.93 | 1.27 | 1.27 | ||
| 1.36 | 1.35 | 1.45 | 1.56 | 1.28 | 2.05 (weak) | 1.71 (weak) | 1.49 | ||
| 1.95 | 1.87 | 1.66 | 1.98 | 1.67 | 1.47 | 2.55 (weak) | 2.2 (weak) | 1.92 | 1.88 |
| 2.75 | 2.37 | 2.19 | 2.38 | 2.11 | 2.17 | 3.74 | 3.51 | 2.21 | 2.31 |
| 3.04 | 2.82 | 2.52 | 2.76 | 2.46 | 2.59 | 2.42 | 2.49 | ||
| 3.35 | 3.41 | 3.10 | 3.26 | 3.01 | 3.06 | 2.93 | 2.82 | ||
| 3.82 | 3.80 | 3.67 | 3.75 | 3.93 | 3.53 | 3.05 | |||
| 3.79 | 3.55 | 3.32 | |||||||
| Guanosine | Guanosine Dihydrate | ||
|---|---|---|---|
| Experimental | Calculated | ||
| Peaks (THz) | Main Vibrational Modes | Description | |
| 2.57 | 2.19 | Collective vibration | Overall out-of-plane distortion, H2O include |
| 3.65 | 2.69 | Collective vibration | Overall in-plane distortion, H2O include |
| 3.19 | Collective vibration | Overall out-of-plane swing, H2O include | |
| 3.69 | Collective vibration | Weak in-plane swing, H2O strong distortion | |
| 4.65 | 4.77 | Collective vibration | Weak out-of-plane distortion, H2O strong distortion |
| 5.38 | 5.56 | Collective vibration | Out-of-plane distortion, H2O weak swing |
| 6.53 | 6.49 | Partial vibration | Out-of-plane distortion of pentose groups, H2O weak distortion |
| 7.17 | 7.05 | Partial vibration | Out-of-plane distortion of purine ring, H2O weak distortion |
| 7.82 | Partial vibration | H2O strong distortion | |
| 8.52 | Partial vibration | Out-of-plane distortion of pentose groups, H2O weak distortion | |
| 9.05 | Partial vibration | Out-of-plane distortion of pentose groups, H2O weak distortion | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, F.; Lin, H.; Tong, J.; Tai, J.; Wu, J.; Yao, Y.; Liu, Y. Effects of Morphology and Solvent/Temperature on THz Spectra: Take Nucleosides as Example. Molecules 2023, 28, 1528. https://doi.org/10.3390/molecules28041528
Wang F, Lin H, Tong J, Tai J, Wu J, Yao Y, Liu Y. Effects of Morphology and Solvent/Temperature on THz Spectra: Take Nucleosides as Example. Molecules. 2023; 28(4):1528. https://doi.org/10.3390/molecules28041528
Chicago/Turabian StyleWang, Fang, Haifeng Lin, Jiawen Tong, Junbin Tai, Jiaen Wu, Yaru Yao, and Yunfei Liu. 2023. "Effects of Morphology and Solvent/Temperature on THz Spectra: Take Nucleosides as Example" Molecules 28, no. 4: 1528. https://doi.org/10.3390/molecules28041528