

Neuroprotectant Effects of Hibiscetin in 3-Nitropropionic Acid-Induced Huntington’s Disease via Subsiding Oxidative Stress and Modulating Monoamine Neurotransmitters in Rats Brain
 , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Acute Toxicity Assessment
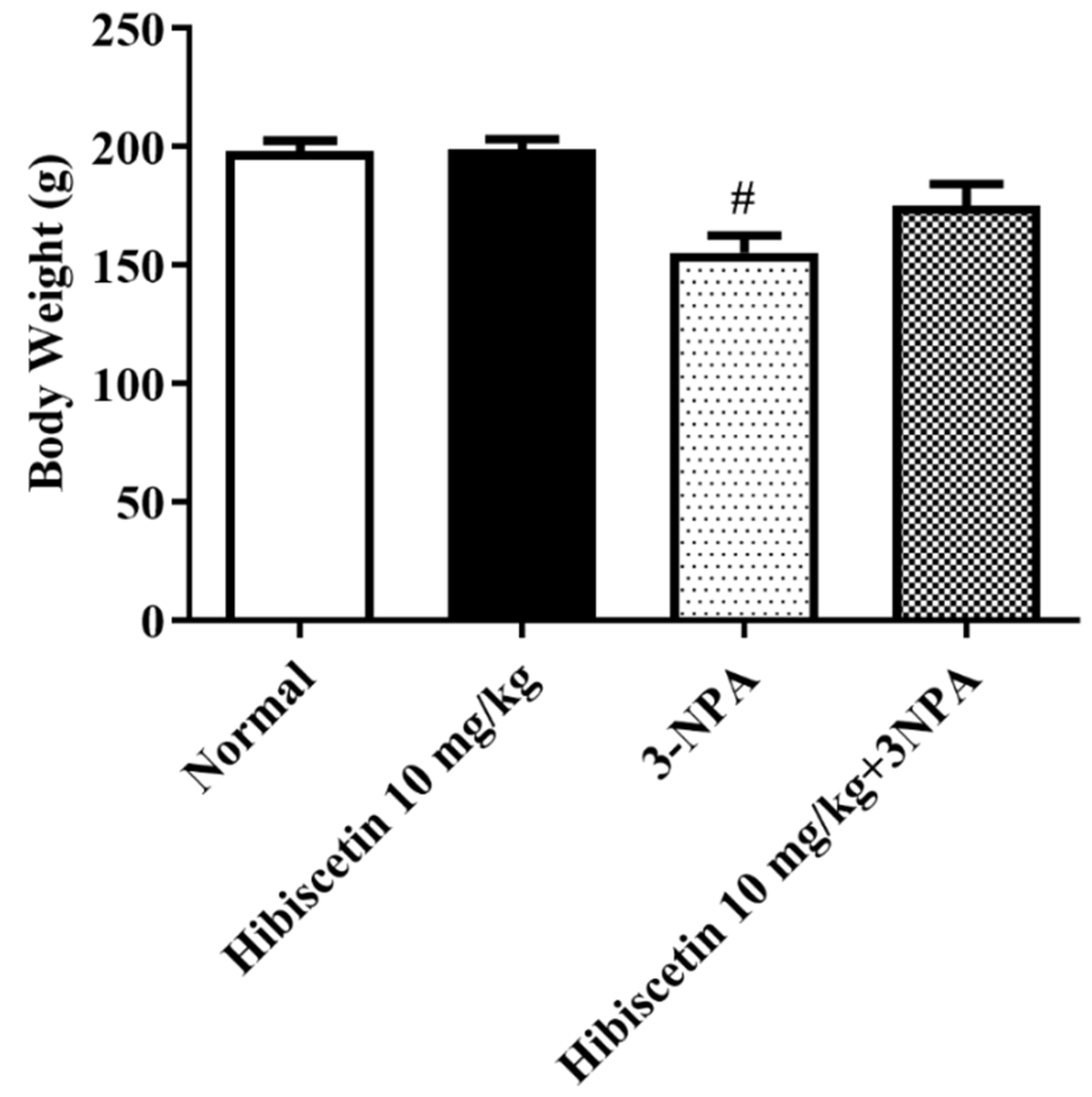
2.2. Mean Body Weight
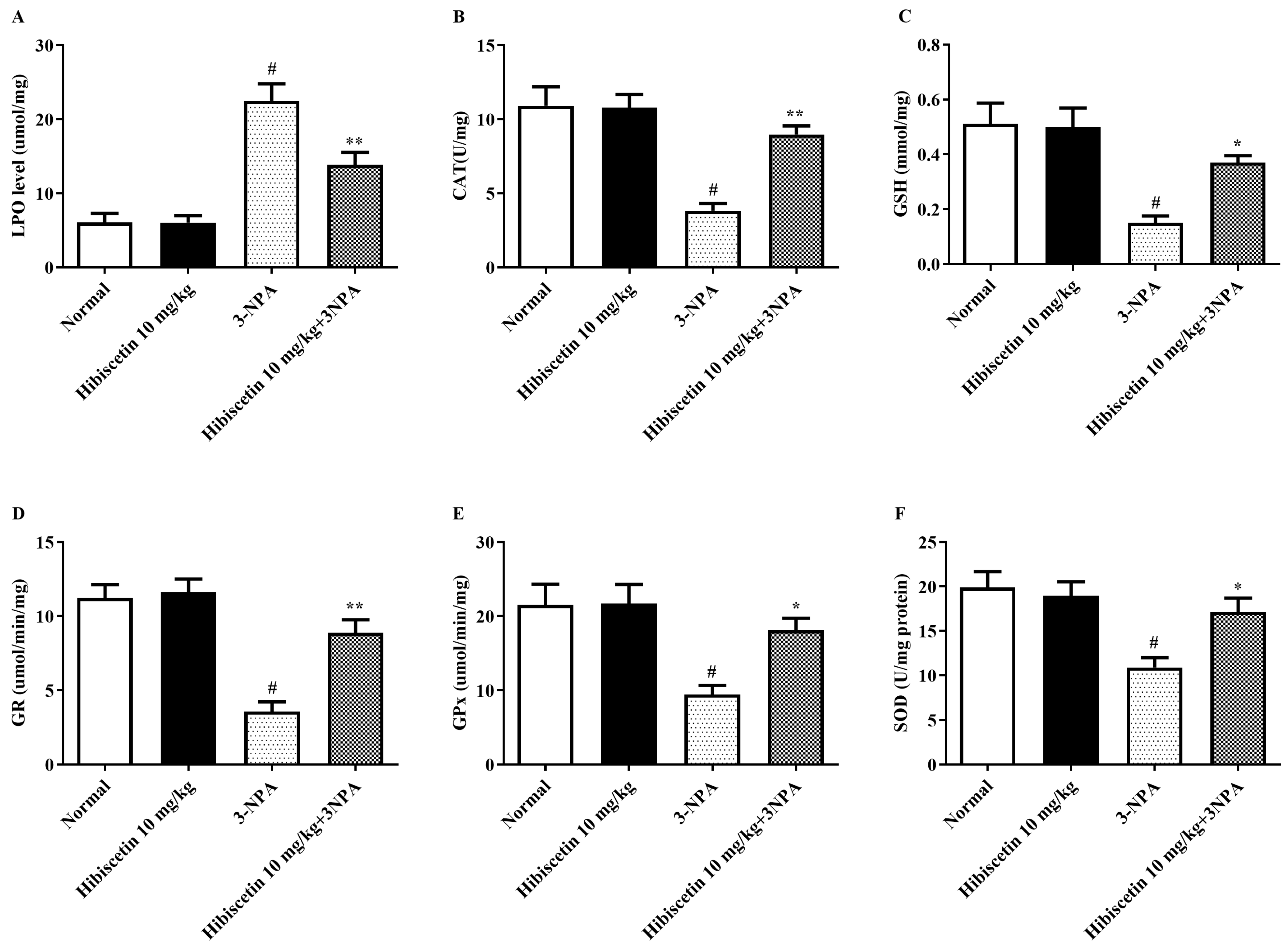
2.3. Oxidative Stress Parameters
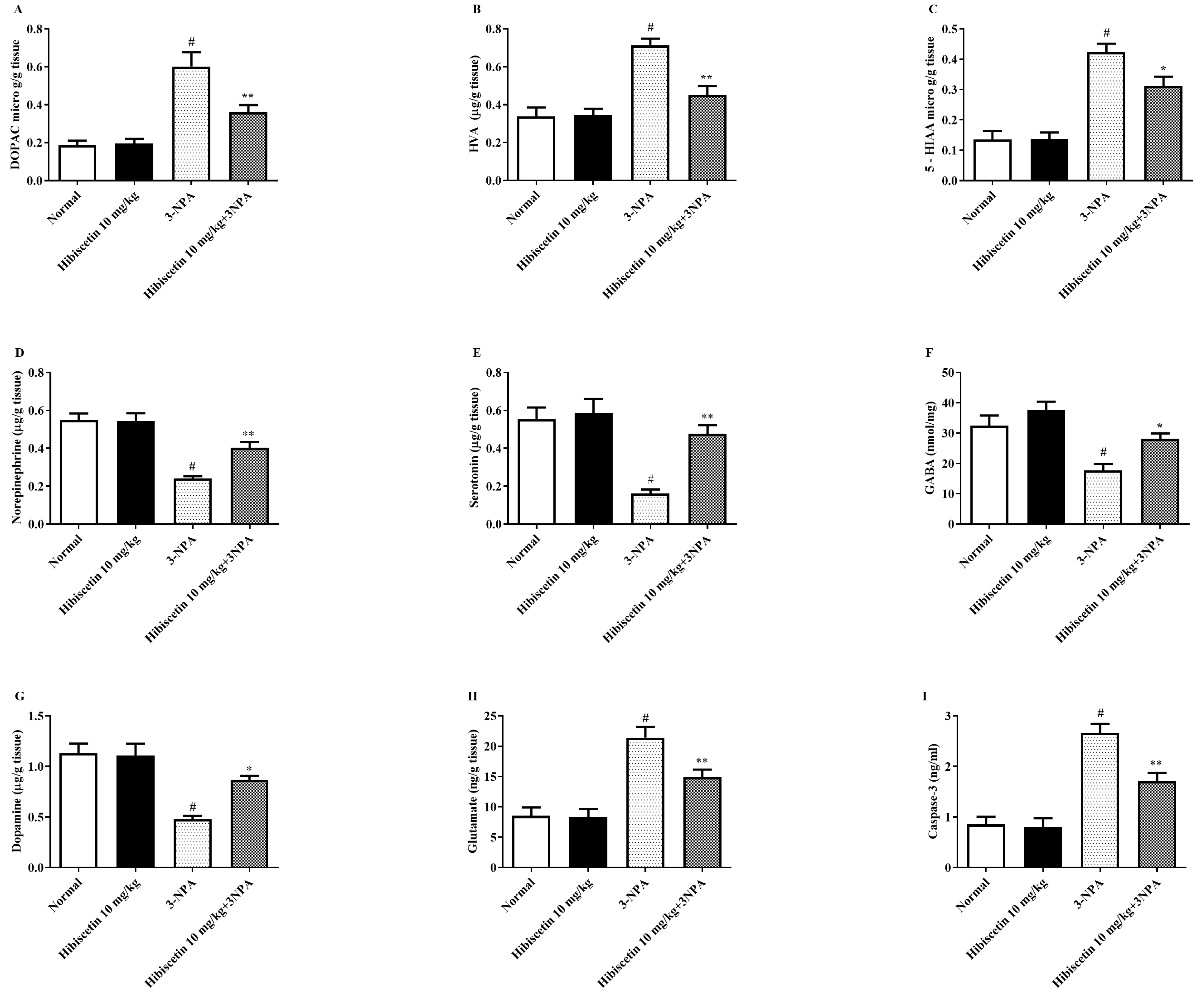
2.4. Monoamine Metabolites and Caspase 3 Biomarkers Estimation
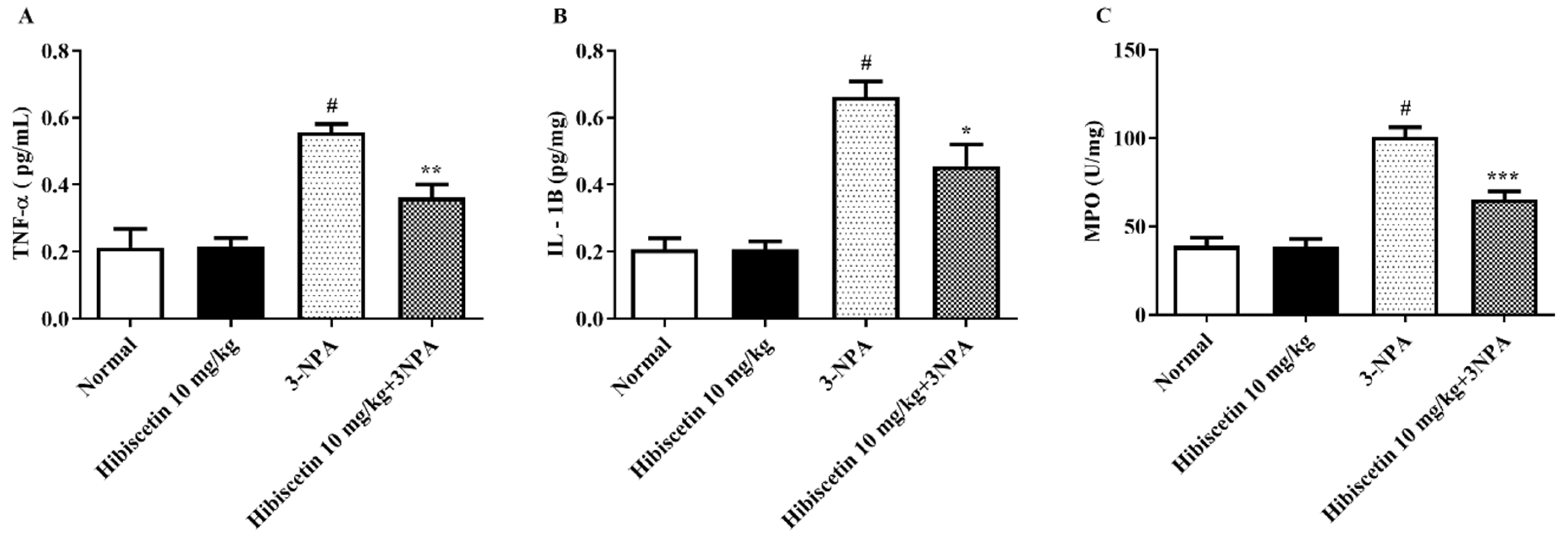
2.5. Estimation of Inflammatory Biomarkers
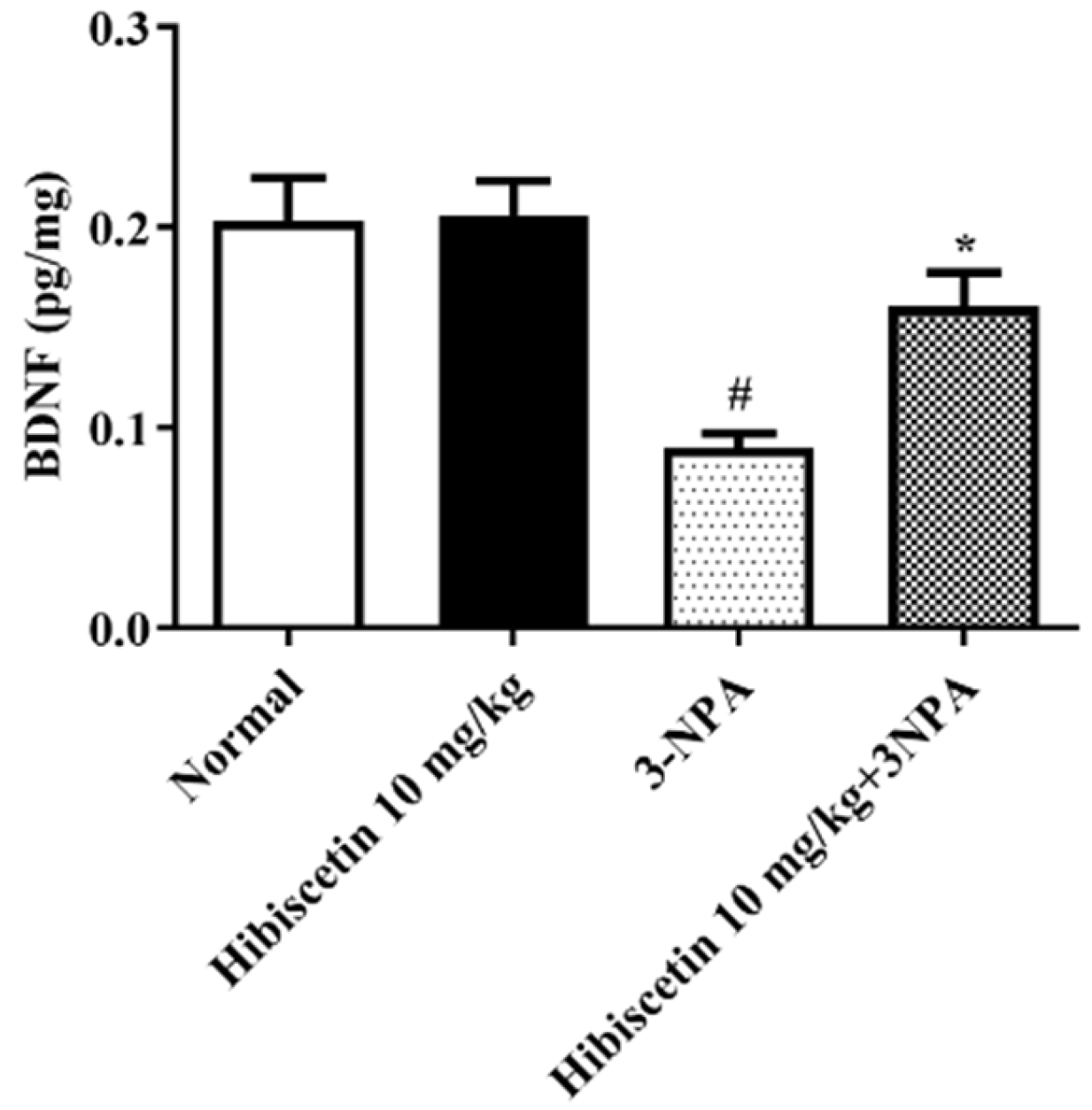
2.6. Estimation of BDNF Activity
3. Discussion
4. Methodology
4.1. Animals
4.2. Chemicals
4.3. Acute Toxicity Studies
4.4. Experimental Design
4.5. Body Weights
4.6. Biological Assessment
4.6.1. Brain Tissue Homogenate
4.6.2. Brain Biochemical Parameters
4.6.3. Estimation of Brain-Derived Neurotrophic Factor (BDNF) Activity
4.6.4. Estimation of GPx Activity
4.6.5. Assessment of Monoamines and Caspase 3 Biomarkers
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kerkis, I.; Haddad, M.S.; Valverde, C.W.; Glosman, S. Neural and mesenchymal stem cells in animal models of Huntington’s disease: Past experiences and future challenges. Stem Cell Res. Ther. 2015, 6, 232. [Google Scholar] [CrossRef]
- Komatsu, H. Innovative Therapeutic Approaches for Huntington’s Disease: From Nucleic Acids to GPCR-Targeting Small Molecules. Front. Cell. Neurosci. 2021, 15, 785703. [Google Scholar] [CrossRef] [PubMed]
- Montoya, A.; Price, B.H.; Menear, M.; Lepage, M. Brain imaging and cognitive dysfunctions in Huntington’s disease. J. Psychiatry Neurosci. 2006, 31, 21–29. [Google Scholar] [PubMed]
- Schiefer, J.; Werner, C.J.; Reetz, K. Clinical diagnosis and management in early Huntington’s disease: A review. Degener. Neurol. Neuromuscul. Dis. 2015, 5, 37. [Google Scholar] [PubMed]
- Nogueira, J.M.; Franco, A.M.; Mendes, S.; Valadas, A.; Semedo, C.; Jesus, G. Huntington’s disease in a patient misdiagnosed as conversion disorder. Case Rep. Psychiatry 2018, 2018, 3915657. [Google Scholar] [CrossRef]
- MacDonald, M.E.; Ambrose, C.M.; Duyao, M.P.; Myers, R.H.; Lin, C.; Srinidhi, L.; Barnes, G.; Taylor, S.A.; James, M.; Groot, N. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef]
- Ross, C.A.; Tabrizi, S.J. Huntington’s disease: From molecular pathogenesis to clinical treatment. Lancet Neurol. 2011, 10, 83–98. [Google Scholar] [CrossRef]
- Nopoulos, P.C. Huntington disease: A single-gene degenerative disorder of the striatum. Dialogues Clin. Neurosci. 2022, 18, 91–98. [Google Scholar] [CrossRef]
- Jimenez-Sanchez, M.; Licitra, F.; Underwood, B.R.; Rubinsztein, D.C. Huntington’s disease: Mechanisms of pathogenesis and therapeutic strategies. Cold Spring Harb. Perspect. Med. 2017, 7, a024240. [Google Scholar] [CrossRef]
- Gorman, A.M. Neuronal cell death in neurodegenerative diseases: Recurring themes around protein handling. J. Cell. Mol. Med. 2008, 12, 2263–2280. [Google Scholar] [CrossRef] [Green Version]
- Vonsattel, J.; DiFiglia, M. Huntington disease. J. Neuropathol. Exp. Neurol. 1997, 57, 369–384. [Google Scholar] [CrossRef]
- DiFiglia, M.; Sapp, E.; Chase, K.; Schwarz, C.; Meloni, A.; Young, C.; Martin, E.; Vonsattel, J.-P.; Carraway, R.; Reeves, S.A. Huntingtin is a cytoplasmic protein associated with vesicles in human and rat brain neurons. Neuron 1995, 14, 1075–1081. [Google Scholar] [CrossRef]
- Guedes-Dias, P.; Pinho, B.R.; Soares, T.R.; de Proença, J.; Duchen, M.R.; Oliveira, J.M. Mitochondrial dynamics and quality control in Huntington’s disease. Neurobiol. Dis. 2016, 90, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Reiner, A.; Dragatsis, I.; Dietrich, P. Genetics and neuropathology of Huntington’s disease. Int. Rev. Neurobiol. 2011, 98, 325–372. [Google Scholar]
- Tang, C.; Feigin, A. Monitoring Huntington’s disease progression through preclinical and early stages. Neurodegener. Dis. Manag. 2012, 2, 421–435. [Google Scholar] [CrossRef]
- Ferrante, R.J.; Kowall, N.W.; Richardson, E.P. Proliferative and degenerative changes in striatal spiny neurons in Huntington’s disease: A combined study using the section-Golgi method and calbindin D28k immunocytochemistry. J. Neurosci. 1991, 11, 3877–3887. [Google Scholar] [CrossRef] [PubMed]
- Graveland, G.; Williams, R.; DiFiglia, M. Evidence for degenerative and regenerative changes in neostriatal spiny neurons in Huntington’s disease. Science 1985, 227, 770–773. [Google Scholar] [CrossRef]
- Sotrel, A.; Paskevich, P.; Kiely, D.; Bird, E.; Williams, R.; Myers, R. Morphometric analysis of the prefrontal cortex in Huntington’s disease. Neurology 1991, 41, 1117. [Google Scholar] [CrossRef]
- Unschuld, P.G.; Joel, S.E.; Liu, X.; Shanahan, M.; Margolis, R.L.; Biglan, K.M.; Bassett, S.S.; Schretlen, D.J.; Redgrave, G.W.; van Zijl, P.C. Impaired cortico-striatal functional connectivity in prodromal Huntington’s disease. Neurosci. Lett. 2012, 514, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Unschuld, P.G.; Joel, S.E.; Pekar, J.J.; Reading, S.A.; Oishi, K.; McEntee, J.; Shanahan, M.; Bakker, A.; Margolis, R.L.; Bassett, S.S. Depressive symptoms in prodromal Huntington’s Disease correlate with Stroop-interference related functional connectivity in the ventromedial prefrontal cortex. Psychiatry Res. Neuroimaging 2012, 203, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Wolf, R.C.; Sambataro, F.; Vasic, N.; Schönfeldt-Lecuona, C.; Ecker, D.; Landwehrmeyer, B. Aberrant connectivity of lateral prefrontal networks in presymptomatic Huntington’s disease. Exp. Neurol. 2008, 213, 137–144. [Google Scholar] [CrossRef]
- Ikeda, H.; Yamaguchi, M.; Sugai, S.; Aze, Y.; Narumiya, S.; Kakizuka, A. Expanded polyglutamine in the Machado–Joseph disease protein induces cell death in vitro and in vivo. Nat. Genet. 1996, 13, 196–202. [Google Scholar] [CrossRef]
- Mangiarini, L.; Sathasivam, K.; Seller, M.; Cozens, B.; Harper, A.; Hetherington, C.; Lawton, M.; Trottier, Y.; Lehrach, H.; Davies, S.W. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 1996, 87, 493–506. [Google Scholar] [CrossRef]
- Crossman, A. Functional anatomy of movement disorders. J. Anat. 2000, 196, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Graybiel, A.M.; Aosaki, T.; Flaherty, A.W.; Kimura, M. The basal ganglia and adaptive motor control. Science 1994, 265, 1826–1831. [Google Scholar] [CrossRef] [PubMed]
- Barron, J.C.; Hurley, E.P.; Parsons, M.P. Huntingtin and the synapse. Front. Cell. Neurosci. 2021, 15, 225. [Google Scholar] [CrossRef] [PubMed]
- André, V.M.; Cepeda, C.; Venegas, A.; Gomez, Y.; Levine, M.S. Altered cortical glutamate receptor function in the R6/2 model of Huntington’s disease. J. Neurophysiol. 2006, 95, 2108–2119. [Google Scholar] [CrossRef] [PubMed]
- Cepeda, C.; Hurst, R.S.; Calvert, C.R.; Hernández-Echeagaray, E.; Nguyen, O.K.; Jocoy, E.; Christian, L.J.; Ariano, M.A.; Levine, M.S. Transient and progressive electrophysiological alterations in the corticostriatal pathway in a mouse model of Huntington’s disease. J. Neurosci. 2003, 23, 961–969. [Google Scholar] [CrossRef]
- DiProspero, N.A.; Chen, E.-Y.; Charles, V.; Plomann, M.; Kordower, J.H.; Tagle, D.A. Early changes in Huntington’s disease patient brains involve alterations in cytoskeletal and synaptic elements. J. Neurocytol. 2004, 33, 517–533. [Google Scholar] [CrossRef]
- Graham, R.K.; Pouladi, M.A.; Joshi, P.; Lu, G.; Deng, Y.; Wu, N.P.; Figueroa, B.E.; Metzler, M.; André, V.M.; Slow, E.J.; et al. Differential susceptibility to excitotoxic stress in YAC128 mouse models of Huntington disease between initiation and progression of disease. J. Neurosci. 2009, 29, 2193–2204. [Google Scholar] [CrossRef]
- Joshi, P.R.; Wu, N.-P.; André, V.M.; Cummings, D.M.; Cepeda, C.; Joyce, J.A.; Carroll, J.B.; Leavitt, B.R.; Hayden, M.R.; Levine, M.S. Age-dependent alterations of corticostriatal activity in the YAC128 mouse model of Huntington disease. J. Neurosci. 2009, 29, 2414–2427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, F.O. Huntington’s disease. Lancet 2007, 369, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Borlongan, C.; Koutouzis, T.; Randall, T.; Freeman, T.; Cahill, D.; Sanberg, P. Systemic 3-nitropropionic acid: Behavioral deficits and striatal damage in adult rats. Brain Res. Bull. 1995, 36, 549–556. [Google Scholar] [CrossRef]
- Wijeyekoon, R.; Barker, R.A. The current status of neural grafting in the treatment of Huntington’s disease. A review. Front. Integr. Neurosci. 2011, 5, 78. [Google Scholar] [CrossRef]
- Kumar, P.; Kalonia, H.; Kumar, A. Possible GABAergic mechanism in the neuroprotective effect of gabapentin and lamotrigine against 3-nitropropionic acid induced neurotoxicity. Eur. J. Pharmacol. 2012, 674, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Dhir, A.; Akula, K.K.; Kulkarni, S. Tiagabine, a GABA uptake inhibitor, attenuates 3-nitropropionic acid-induced alterations in various behavioral and biochemical parameters in rats. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2008, 32, 835–843. [Google Scholar] [CrossRef] [PubMed]
- Weir, D.W.; Sturrock, A.; Leavitt, B.R. Development of biomarkers for Huntington’s disease. Lancet Neurol. 2011, 10, 573–590. [Google Scholar] [CrossRef] [PubMed]
- Costa, V.; Scorrano, L. Shaping the role of mitochondria in the pathogenesis of Huntington’s disease. EMBO J. 2012, 31, 1853–1864. [Google Scholar] [CrossRef]
- Pires, T.C.; Dias, M.I.; Barros, L.; Calhelha, R.C.; Alves, M.J.; Oliveira, M.B.P.; Santos-Buelga, C.; Ferreira, I.C. Edible flowers as sources of phenolic compounds with bioactive potential. Food Res. Int. 2018, 105, 580–588. [Google Scholar] [CrossRef]
- Kaisoon, O.; Konczak, I.; Siriamornpun, S. Potential health enhancing properties of edible flowers from Thailand. Food Res. Int. 2012, 46, 563–571. [Google Scholar] [CrossRef]
- Rice-Evans, C. Flavonoid antioxidants. Curr. Med. Chem. 2001, 8, 797–807. [Google Scholar] [CrossRef] [PubMed]
- Schroeter, H.; Boyd, C.; Spencer, J.P.; Williams, R.J.; Cadenas, E.; Rice-Evans, C. MAPK signaling in neurodegeneration: Influences of flavonoids and of nitric oxide. Neurobiol. Aging 2002, 23, 861–880. [Google Scholar] [CrossRef] [PubMed]
- Ghani, A. Medicinal Plants of Bangladesh: Chemical Constituents and Uses; Asiatic Society of Bangladesh: Nimtari, India, 1998. [Google Scholar]
- Tsuda, T.; Horio, F.; Uchida, K.; Aoki, H.; Osawa, T. Dietary cyanidin 3-O-β-D-glucoside-rich purple corn color prevents obesity and ameliorates hyperglycemia in mice. J. Nutr. 2003, 133, 2125–2130. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.-Y.; Peng, C.-H.; Chan, K.-C.; Yang, Y.-S.; Huang, C.-N.; Wang, C.-J. The hypolipidemic effect of Hibiscus sabdariffa polyphenols via inhibiting lipogenesis and promoting hepatic lipid clearance. J. Agric. Food Chem. 2010, 58, 850–859. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Moemin, A. Effect of Roselle calyces extract on the chemical and sensory properties of functional cupcakes. Food Sci. Hum. Wellness 2016, 5, 230–237. [Google Scholar] [CrossRef]
- Rengarajan, S.; Melanathuru, V.; Govindasamy, C.; Chinnadurai, V.; Elsadek, M.F. Antioxidant activity of flavonoid compounds isolated from the petals of Hibiscus rosa sinensis. J. King Saud Univ. Sci. 2020, 32, 2236–2242. [Google Scholar] [CrossRef]
- Alzarea, S.I.; Afzal, M.; Alharbi, K.S.; Alzarea, A.I.; Alenezi, S.K.; Alshammari, M.S.; Alquraini, A.; Kazmi, I. Hibiscetin attenuates oxidative, nitrative stress and neuroinflammation via suppression of TNF-α signaling in rotenone induced Parkinsonism in rats. Saudi Pharm. J. 2022, 30, 1710–1717. [Google Scholar] [CrossRef]
- Abdelfattah, M.S.; Badr, S.E.; Lotfy, S.A.; Attia, G.H.; Aref, A.M.; Abdel Moneim, A.E.; Kassab, R.B. Rutin and selenium co-administration reverse 3-nitropropionic acid-induced neurochemical and molecular impairments in a mouse model of Huntington’s disease. Neurotox. Res. 2020, 37, 77–92. [Google Scholar] [CrossRef]
- Brouillet, E.; Jacquard, C.; Bizat, N.; Blum, D. 3-Nitropropionic acid: A mitochondrial toxin to uncover physiopathological mechanisms underlying striatal degeneration in Huntington’s disease. J. Neurochem. 2005, 95, 1521–1540. [Google Scholar] [CrossRef]
- Túnez, I.; Tasset, I.; Pérez-De La Cruz, V.; Santamaría, A. 3-Nitropropionic acid as a tool to study the mechanisms involved in Huntington’s disease: Past, present and future. Molecules 2010, 15, 878–916. [Google Scholar] [CrossRef]
- Gipson, T.A.; Neueder, A.; Wexler, N.S.; Bates, G.P.; Housman, D. Aberrantly spliced HTT, a new player in Huntington’s disease pathogenesis. RNA Biol. 2013, 10, 1647–1652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamwal, S.; Kumar, P. Spermidine ameliorates 3-nitropropionic acid (3-NP)-induced striatal toxicity: Possible role of oxidative stress, neuroinflammation, and neurotransmitters. Physiol. Behav. 2016, 155, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Ratan, R.R. Oxidative stress and Huntington’s disease: The good, the bad, and the ugly. J. Huntingt. Dis. 2016, 5, 217–237. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, J.; Yang, L.; Zhou, S.-m.; Guan, S.-y.; Yang, L.-k.; Shi, Q.-x.; Zhao, M.-G.; Yang, Q. Effect of Praeruptorin C on 3-nitropropionic acid induced Huntington’s disease-like symptoms in mice. Biomed. Pharmacother. 2017, 86, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, L.R.; Charrin, B.C.; Borrell-Pagès, M.; Dompierre, J.P.; Rangone, H.; Cordelières, F.P.; De Mey, J.; MacDonald, M.E.; Leßmann, V.; Humbert, S. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell 2004, 118, 127–138. [Google Scholar] [CrossRef]
- Yu, C.; Li, C.H.; Chen, S.; Yoo, H.; Qin, X.; Park, H. Decreased BDNF release in cortical neurons of a knock-in mouse model of Huntington’s disease. Sci. Rep. 2018, 8, 16976. [Google Scholar] [CrossRef]
- Smith-Dijak, A.I.; Sepers, M.D.; Raymond, L.A. Alterations in synaptic function and plasticity in Huntington disease. J. Neurochem. 2019, 150, 346–365. [Google Scholar] [CrossRef]
- Shalaby, H.N.; El-Tanbouly, D.M.; Zaki, H.F. Topiramate mitigates 3-nitropropionic acid-induced striatal neurotoxicity via modulation of AMPA receptors. Food Chem. Toxicol. 2018, 118, 227–234. [Google Scholar] [CrossRef]
- Zuccato, C.; Cattaneo, E. Role of brain-derived neurotrophic factor in Huntington’s disease. Prog. Neurobiol. 2007, 81, 294–330. [Google Scholar] [CrossRef]
- Aliaghaei, A.; Boroujeni, M.E.; Ahmadi, H.; Bayat, A.-H.; Tavirani, M.R.; Abdollahifar, M.A.; Pooyafar, M.H.; Mansouri, V. Dental pulp stem cell transplantation ameliorates motor function and prevents cerebellar atrophy in rat model of cerebellar ataxia. Cell Tissue Res. 2019, 376, 179–187. [Google Scholar] [CrossRef]
- Khan, A.; Jamwal, S.; Bijjem, K.; Prakash, A.; Kumar, P. Neuroprotective effect of hemeoxygenase-1/glycogen synthase kinase-3β modulators in 3-nitropropionic acid-induced neurotoxicity in rats. Neuroscience 2015, 287, 66–77. [Google Scholar] [CrossRef]
- Palpagama, T.H.; Waldvogel, H.J.; Faull, R.L.; Kwakowsky, A. The role of microglia and astrocytes in Huntington’s disease. Front. Mol. Neurosci. 2019, 12, 258. [Google Scholar] [CrossRef] [PubMed]
- Gilani, S.J.; Bin-Jumah, M.N.; Al-Abbasi, F.A.; Albohairy, F.M.; Nadeem, M.S.; Ahmed, M.M.; Alzarea, S.I.; Kazmi, I. The Ameliorative Role of Hibiscetin against High-Fat Diets and Streptozotocin-Induced Diabetes in Rodents via Inhibiting Tumor Necrosis Factor-α, Interleukin-1β, and Malondialdehyde Level. Processes 2022, 10, 1396. [Google Scholar] [CrossRef]
- Afzal, M.; Sayyed, N.; Alharbi, K.S.; Alzarea, S.I.; Alshammari, M.S.; Alomar, F.A.; Alenezi, S.K.; Quazi, A.M.; Alzarea, A.I.; Kazmi, I. Anti-Huntington’s Effect of Rosiridin via Oxidative Stress/AchE Inhibition and Modulation of Succinate Dehydrogenase, Nitrite, and BDNF Levels against 3-Nitropropionic Acid in Rodents. Biomolecules 2022, 12, 1023. [Google Scholar] [CrossRef] [PubMed]
- Alshehri, S.; Al-Abbasi, F.A.; Ghoneim, M.M.; Imam, S.S.; Afzal, M.; Alharbi, K.S.; Nadeem, M.S.; Sayyed, N.; Kazmi, I. Anti-Huntington’s Effect of Butin in 3-Nitropropionic Acid-Treated Rats: Possible Mechanism of Action. Neurotox. Res. 2022, 40, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Mehan, S.; Monga, V.; Rani, M.; Dudi, R.; Ghimire, K. Neuroprotective effect of solanesol against 3-nitropropionic acid-induced Huntington’s disease-like behavioral, biochemical, and cellular alterations: Restoration of coenzyme-Q10-mediated mitochondrial dysfunction. Indian J. Pharmacol. 2018, 50, 309. [Google Scholar] [CrossRef]
- Durg, S.; Kumar, N.; Vandal, R.; Dhadde, S.B.; Thippeswamy, B.; Veerapur, V.P.; Badami, S. Antipsychotic activity of embelin isolated from Embelia ribes: A preliminary study. Biomed. Pharmacother. 2017, 90, 328–331. [Google Scholar] [CrossRef]
- Shaikh, A.; Dhadde, S.B.; Durg, S.; Veerapur, V.; Badami, S.; Thippeswamy, B.; Patil, J.S. Effect of Embelin Against Lipopolysaccharide-induced Sickness Behaviour in Mice. Phytother. Res. 2016, 30, 815–822. [Google Scholar] [CrossRef]
- Ellman, G. Tissue sulfhvdrvl sroups. Arch. Biochem. Biophvs 1959, 82, 72–77. [Google Scholar] [CrossRef]
- Misra, H.P.; Fridovich, I. The role of superoxide anion in the autoxidation of epinephrine and a simple assay for superoxide dismutase. J. Biol. Chem. 1972, 247, 3170–3175. [Google Scholar] [CrossRef]
- Aebi, H.; Wyss, S.R.; Scherz, B.; Skvaril, F. Heterogeneity of erythrocyte catalase II: Isolation and characterization of normal and variant erythrocyte catalase and their subunits. Eur. J. Biochem. 1974, 48, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Ohkawa, H.; Ohishi, N.; Yagi, K. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal. Biochem. 1979, 95, 351–358. [Google Scholar] [CrossRef]
- Farias, J.G.; Puebla, M.; Acevedo, A.; Tapia, P.J.; Gutierrez, E.; Zepeda, A.; Calaf, G.; Juantok, C.; Reyes, J.G. Oxidative stress in rat testis and epididymis under intermittent hypobaric hypoxia: Protective role of ascorbate supplementation. J. Androl. 2010, 31, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, R.A.; Burk, R.F. Glutathione peroxidase activity in selenium-deficient rat liver. Biochem. Biophys. Res. Commun. 1976, 71, 952–958. [Google Scholar] [CrossRef] [PubMed]
- Pagel, P.; Blome, J.; Wolf, H.U. High-performance liquid chromatographic separation and measurement of various biogenic compounds possibly involved in the pathomechanism of Parkinson’s disease. J. Chromatogr. B Biomed. Sci. Appl. 2000, 746, 297–304. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahdi, W.A.; AlGhamdi, S.A.; Alghamdi, A.M.; Imam, S.S.; Alshehri, S.; Almaniea, M.A.; Hajjar, B.M.; Al-Abbasi, F.A.; Sayyed, N.; Kazmi, I. Neuroprotectant Effects of Hibiscetin in 3-Nitropropionic Acid-Induced Huntington’s Disease via Subsiding Oxidative Stress and Modulating Monoamine Neurotransmitters in Rats Brain. Molecules 2023, 28, 1402. https://doi.org/10.3390/molecules28031402
Mahdi WA, AlGhamdi SA, Alghamdi AM, Imam SS, Alshehri S, Almaniea MA, Hajjar BM, Al-Abbasi FA, Sayyed N, Kazmi I. Neuroprotectant Effects of Hibiscetin in 3-Nitropropionic Acid-Induced Huntington’s Disease via Subsiding Oxidative Stress and Modulating Monoamine Neurotransmitters in Rats Brain. Molecules. 2023; 28(3):1402. https://doi.org/10.3390/molecules28031402
Chicago/Turabian StyleMahdi, Wael A., Shareefa A. AlGhamdi, Amira M. Alghamdi, Syed Sarim Imam, Sultan Alshehri, Mohammad A. Almaniea, Baraa Mohammed Hajjar, Fahad A. Al-Abbasi, Nadeem Sayyed, and Imran Kazmi. 2023. "Neuroprotectant Effects of Hibiscetin in 3-Nitropropionic Acid-Induced Huntington’s Disease via Subsiding Oxidative Stress and Modulating Monoamine Neurotransmitters in Rats Brain" Molecules 28, no. 3: 1402. https://doi.org/10.3390/molecules28031402