

Structures and Stabilities of Carbon Chain Clusters Influenced by Atomic Antimony
 , , ,
, , ,
Abstract
:1. Introduction
2. Results and Discussion
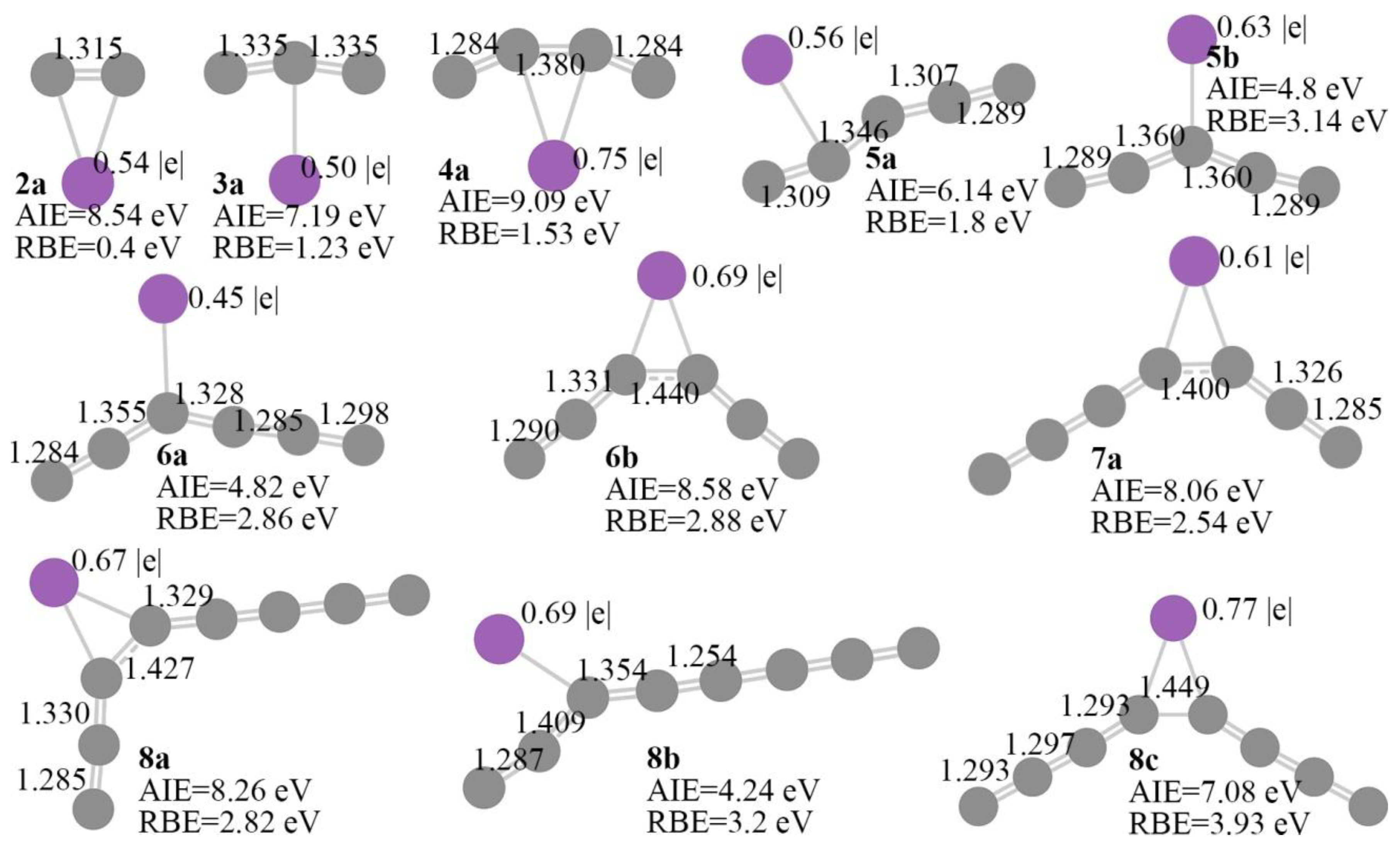
2.1. Structural Optimization
2.2. Electronic Configuration
2.3. Electronic Properties
2.4. Incremental Energies, Fragmental Energies, and Binding Sites
3. Computational Methods
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Leyva-Perez, A. Sub-nanometre metal clusters for catalytic carbon-carbon and carbon-heteroatom cross-coupling reactions. Dalton T. 2017, 46, 15987–15990. [Google Scholar] [CrossRef] [PubMed]
- de Lara-Castells, M.P.; Mitrushchenkov, A.O. Mini Review: Quantum Confinement of Atomic and Molecular Clusters in Carbon Nanotubes. Front. Chem. 2021, 9, 796890. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Song, Z.; Wang, J.; Li, Q.; An, Q. Phytic acid conversion film interfacial engineering for stabilizing zinc metal anode. Chem. Eng. J. 2022, 446, 137295. [Google Scholar] [CrossRef]
- Shao, X.; Li, L.; Huang, S.; Song, Z.; Wang, K. First-Principles Calculations of Magnetic Moment Modulation of 3D Transition Metal Atoms Encapsulated in C-60/C-70 Cages on Si(100) Surfaces: Implications for Spintronic Devices. ACS Appl. Nano Mater. 2021, 4, 12356–12364. [Google Scholar] [CrossRef]
- Chen, W.; Huang, J.; He, Z.-C.; Ji, X.; Zhang, Y.-F.; Sun, H.-L.; Wang, K.; Su, Z.-W. Accelerated photocatalytic degradation of tetracycline hydrochloride over CuAl2O4/g-C3N4 p-n heterojunctions under visible light irradiation. Sep. Purif. Technol. 2021, 277, 119461. [Google Scholar] [CrossRef]
- Liu, M.; Yu, T.; Huang, R.; Qi, W.; He, Z.; Su, R. Fabrication of nanohybrids assisted by protein-based materials for catalytic applications. Catal. Sci. Technol. 2020, 10, 3515–3531. [Google Scholar] [CrossRef]
- Pu, D.; Pan, Y. First-principles investigation of solution mechanism of C in TM-Si-C matrix as the potential high-temperature ceramics. J. Am. Ceram. Soc. 2022, 105, 2858–2868. [Google Scholar] [CrossRef]
- Liu, M.; Shan, C.; Chang, H.; Zhang, Z.; Huang, R.; Lee, D.W.; Qi, W.; He, Z.; Su, R. Nano-engineered natural sponge as a recyclable and deformable reactor for ultrafast conversion of pollutants from water. Chem. Eng. Sci. 2022, 247, 117049. [Google Scholar] [CrossRef]
- Tuktarov, A.R.; Khuzin, A.A.; Dzhemilev, U.M. Light-controlled molecular switches based on carbon clusters. Synthesis, properties and application prospects. Russ. Chem. Rev. 2017, 86, 474–509. [Google Scholar] [CrossRef]
- Hadad, C.Z.; Florez, E.; Merino, G.; Cabellos, J.L.; Ferraro, F.; Restrepo, A. Potential Energy Surfaces of WC6 Clusters in Different Spin States. J. Phys. Chem. A 2014, 118, 5762–5768. [Google Scholar] [CrossRef]
- Stein, T.; Bera, P.P.; Lee, T.J.; Head-Gordon, M. Molecular growth upon ionization of van der Waals clusters containing HCCH and HCN is a pathway to prebiotic molecules. Phys. Chem. Chem. Phys. 2020, 22, 20337–20348. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Xie, S. Structures and Progress of Carbon Clusters. Prog. Chem. 2019, 31, 50–62. [Google Scholar]
- Ticknor, B.W.; Bandyopadhyay, B.; Duncan, M.A. Photodissociation of Noble Metal-Doped Carbon Clusters. J. Phys. Chem. A 2008, 112, 12355–12366. [Google Scholar] [CrossRef] [PubMed]
- Lacovig, P.; Pozzo, M.; Alfe, D.; Vilmercati, P.; Baraldi, A.; Lizzit, S. Growth of dome-shaped carbon nanoislands on Ir(111): The intermediate between carbidic clusters and quasi-free-standing graphene. Phys. Rev. Lett. 2009, 103, 166101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niu, T.C.; Zhou, M.; Zhang, J.L.; Feng, Y.P.; Chen, W. Growth Intermediates for CVD Graphene on Cu(111): Carbon Clusters and Defective Graphene. J. Am. Chem. Soc. 2013, 135, 8409–8414. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.J. First-principle investigation on growth patterns and properties of cobalt-doped lithium nanoclusters. J. Mol. Model. 2016, 22, 133. [Google Scholar] [CrossRef]
- Song, Z.; Zhao, B.; Wang, Q.; Cheng, P. Homolytic cleavage of water on magnesia film promoted by interfacial oxide-metal nanocomposite. Appl. Surf. Sci. 2019, 487, 1222–1232. [Google Scholar] [CrossRef] [Green Version]
- Song, Z.; Shao, X.; Wang, Q.; Ma, C.; Wang, K.; Han, D. Generation of molybdenum hydride species via addition of molecular hydrogen across metal-oxygen bond at monolayer oxide/metal composite interface. Int. J. Hydrog. Energ. 2020, 45, 2975–2988. [Google Scholar] [CrossRef]
- Song, Z.; Fang, S.; Xie, P.; Zhong, A.; Ma, C.; Han, D. Unveiling the interaction profile of cisplatin with gold-supported magnesia film. Appl. Surf. Sci. 2021, 540, 148365. [Google Scholar] [CrossRef]
- Pei, C.-Y.; Li, T.; Zhang, M.; Wang, J.-W.; Chang, L.; Xiong, X.; Chen, W.; Huang, G.-B.; Han, D.-M. Synergistic effects of interface coupling and defect sites in WO3/InVO4 architectures for highly efficient nitrogen photofixation. Sep. Purif. Technol. 2022, 290, 120875. [Google Scholar] [CrossRef]
- Guo, X.-G.; Zhang, J.-L.; Zhao, Y. Ab initio Characterization of Size Dependence of Electronic Spectra for Linear Anionic Carbon Clusters Cn−(n = 4–17). J. Comput. Chem. 2012, 33, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; He, H.; Zhang, J.; Wang, L. Direct dynamics study of the hydrogen abstraction reaction of CF3CH2Cl + Cl -> CF3CHCl + HCl. Int. J. Chem. Kinet. 2012, 44, 661–667. [Google Scholar] [CrossRef]
- Fan, Y.; Wen, J.; Zhao, Y.; Wang, L. Theoretical study on the hydrogen abstraction reactions of CF3CHFCF3 and CF3CF2CHF2 with X atoms (X = F, Cl, and Br). J. Fluorine Chem. 2013, 150, 39–45. [Google Scholar] [CrossRef]
- Zhao, Y.; Guo, J.; Zhang, J. A theoretical study of electronic spectra in the linear cationic chains NC2n+1N+ (n = 1–6). Chem. Phys. 2011, 386, 23–28. [Google Scholar] [CrossRef]
- Zhan, C.-G.; Iwata, S. Ab initio studies on the structures, vertical electron detachment energies, and stabilities of CnP− clusters. J. Chem. Phys. 1997, 107, 7323–7330. [Google Scholar] [CrossRef]
- Sun, S.; Cao, Y.; Sun, Z.; Tang, Z.; Zheng, L. Experimental and Theoretical Studies on Carbon−Nitrogen Clusters C2nN7−. J. Phys. Chem. A 2006, 110, 8064–8072. [Google Scholar] [CrossRef]
- Tang, Z.; BelBruno, J.J.; Huang, R.; Zheng, L. Collision-induced dissociation and density functional study of the structures and energies of cyclic C2nN5− clusters. J. Chem. Phys. 2000, 112, 9276–9281. [Google Scholar] [CrossRef]
- Li, G.; Xing, X.; Tang, Z. Structures and properties the lead-doped carbon clusters PbCn/PbCn+/PbCn− (n = 1–10). J. Chem. Phys. 2003, 118, 6884–6897. [Google Scholar] [CrossRef]
- Zhang, C.-J.; Jiang, Z.-Y.; Wang, Y.-L.; Zhu, H.-Y. Structures and stabilities of GaCn (n = 1–10) clusters. Comput. Theor. Chem. 2013, 1004, 12–17. [Google Scholar] [CrossRef]
- Chen-Jun, Z.; Yang-Li, W.; Chao-Kang, C. Density functional theory of InCn+ (n = 1–10) clusters. Acta Phys. Sin. 2018, 67, 113101. [Google Scholar] [CrossRef]
- Wang, P.; Yan, S.-T.; Xu, H.-G.; Xu, X.-L.; Zheng, W.-J. Anion photoelectron spectroscopy and density functional theory studies of AuCn−/0 (n = 3−8): Odd-even alternation in electron binding energies and structures. Chin. J. Chem. Phys. 2022, 35, 177–184. [Google Scholar] [CrossRef]
- Zhang, C.; Xu, H.; Xu, X.-L.; Zheng, W.-J. Electronic structures, chemical bonds, and stabilities of Ta4C−/0n (n = 0–4) clusters: Anion photoelectron spectroscopy and theoretical calculations. Acta Phys. Sin. 2021, 70, 023601. [Google Scholar] [CrossRef]
- Freitas, A.; Azevedo, S.; Kaschny, J.R. Structural and electronic properties of linear carbon chains encapsulated by flattened nanotubes. Phys. E-Low-Dimens. Syst. Nanostruct. 2016, 84, 444–453. [Google Scholar] [CrossRef]
- Rocha, R.A.; dos Santos, R.B.; Ribeiro Junior, L.A.; Aguiar, A.L. On the Stabilization of Carbynes Encapsulated in Penta-Graphene Nanotubes: A DFT Study. J. Mol. Model. 2021, 27, 318. [Google Scholar] [CrossRef]
- Zhang, Y.; Su, Y.; Wang, L.; Kong, E.S.-W.; Chen, X.; Zhang, Y. A one-dimensional extremely covalent material: Monatomic carbon linear chain. Nanoscale Res. Lett. 2011, 6, 577. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Li, W.; Li, Y.F.; Chen, X.M.; Zhang, H.; Xu, B.Q.; Yang, B. Structural, electronic and catalytic properties of AgnSnn (n = 2–14) clusters by density functional theory. Phys. Chem. Chem. Phys. 2022, 24, 26631–26641. [Google Scholar] [CrossRef] [PubMed]
- Pascoli, G.; Lavendy, H. Theoretical Study of CnP, CnP+, CnP−(n = 1–7) Clusters. J. Phys. Chem. A 1999, 103, 3518–3524. [Google Scholar] [CrossRef]
- Pascoli, G.; Lavendy, H. Are CnN−clusters really bent? Chem. Phys. Lett. 1999, 312, 333–340. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.K.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 03 Rev. A01; Gaussian Inc.: Wallingford, CT, USA, 2004. [Google Scholar]
- Pan, Y.; Yu, E. New insight into the structural and physical properties of AlH3. Int. J. Energ. Res. 2022, 46, 19678–19685. [Google Scholar] [CrossRef]
- Song, Z.; Zhao, B.; Wang, Q.; Cheng, P. Steering reduction and decomposition of peroxide compounds by interface interactions between MgO thin film and transition-metal support. Appl. Surf. Sci. 2018, 459, 812–821. [Google Scholar] [CrossRef]
- Chen, S.; Pan, Y. Enhancing catalytic properties of noble metal@MoS2/WS2 heterojunction for the hydrogen evolution reaction. Appl. Surf. Sci. 2022, 591, 153168. [Google Scholar] [CrossRef]
- Pan, Y. First-principles investigation of the effect of noble metals on the electronic and optical properties of GaN nitride. Mat. Sci. Semicon. Proc. 2022, 151, 107051. [Google Scholar] [CrossRef]
- Zhou, Y.; Shao, X.J.; Lam, K.H.; Zheng, Y.; Zhao, L.Z.; Wang, K.D.; Zhao, J.Z.; Chen, F.M.; Hou, X.H. Symmetric Sodium-Ion Battery Based on Dual-Electron Reactions of NASICON-Structured Na3MnTi(PO4)3 Material. ACS Appl. Mater. Inter. 2020, 12, 30328–30335. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.P.; Zhao, J.Z.; Xu, H. Adsorption Induced Indirect-to-Direct Band Gap Transition in Monolayer Blue Phosphorus. J. Phys. Chem. C 2018, 122, 15792–15798. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.Y.; Yan, H.; Zhong, A.G.; Chen, H.; Jin, Y.X.; Dai, G.L. Theoretical and conceptual DFT study of pnicogen-and halogen-bonded complexes of PH2X—BrCl. J. Mol. Model. 2019, 25, 28. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.Y.; Wu, J.Y.; Dai, G.L. Study on the halogen bond and pi-pi stacking interaction between fluoro substituted iodobenzene and pyrazine. J. Mol. Model. 2020, 26, 333. [Google Scholar] [CrossRef]
- Wu, J.Y.; Yan, H.; Chen, H.; Jin, Y.X.; Zhong, A.G.; Wang, Z.X.; Dai, G.L. Three types of noncovalent interactions studied between pyrazine and XF. J. Mol. Model. 2022, 28, 15. [Google Scholar] [CrossRef]
- Wu, J. Investigations into the nature of halogen-and hydrogen-bonding interactions of some heteroaromatic rings with dichlorine monoxide. J. Mol. Model. 2014, 20, 2424. [Google Scholar] [CrossRef]
- Pascoli, G.; Lavendy, H. Geometrical structures of the phosphorus-doped carbon cluster cations CnP+ (n = 1–20). Int. J. Mass Spectrom. 1999, 189, 125–132. [Google Scholar] [CrossRef]
- Pascoli, G.; Lavendy, H. Structures and energies of CnSi+ (4 ≤ n ≤ 15) silicon carbide clusters. Int. J. Mass Spectrom. 1998, 173, 41–54. [Google Scholar] [CrossRef]




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CSb | 1.888 | |||||||||||||||
| C2Sb | 1.300 | 1.941 | ||||||||||||||
| C3Sb | 1.301 | 1.296 | 1.949 | |||||||||||||
| C4Sb | 1.291 | 1.304 | 1.277 | 1.941 | ||||||||||||
| C5Sb | 1.294 | 1.297 | 1.280 | 1.288 | 1.946 | |||||||||||
| C6Sb | 1.291 | 1.301 | 1.271 | 1.295 | 1.277 | 1.949 | ||||||||||
| C7Sb | 1.292 | 1.299 | 1.274 | 1.288 | 1.282 | 1.286 | 1.945 | |||||||||
| C8Sb | 1.291 | 1.301 | 1.271 | 1.293 | 1.272 | 1.296 | 1.276 | 1.956 | ||||||||
| C9Sb | 1.291 | 1.300 | 1.272 | 1.289 | 1.276 | 1.285 | 1.284 | 1.285 | 1.947 | |||||||
| C10Sb | 1.290 | 1.301 | 1.270 | 1.293 | 1.272 | 1.292 | 1.272 | 1.297 | 1.275 | 1.960 | ||||||
| C11Sb | 1.291 | 1.300 | 1.272 | 1.290 | 1.275 | 1.287 | 1.278 | 1.284 | 1.286 | 1.284 | 1.949 | |||||
| C12Sb | 1.290 | 1.301 | 1.270 | 1.293 | 1.272 | 1.292 | 1.272 | 1.293 | 1.271 | 1.298 | 1.274 | 1.964 | ||||
| C13Sb | 1.291 | 1.300 | 1.271 | 1.291 | 1.274 | 1.288 | 1.277 | 1.286 | 1.280 | 1.283 | 1.287 | 1.283 | 1.951 | |||
| C14Sb | 1.290 | 1.302 | 1.270 | 1.293 | 1.271 | 1.293 | 1.272 | 1.293 | 1.272 | 1.294 | 1.271 | 1.300 | 1.273 | 1.966 | ||
| C15Sb | 1.290 | 1.301 | 1.271 | 1.291 | 1.274 | 1.289 | 1.276 | 1.287 | 1.278 | 1.285 | 1.281 | 1.282 | 1.288 | 1.282 | 1.952 | |
| C16Sb | 1.290 | 1.302 | 1.270 | 1.294 | 1.271 | 1.293 | 1.271 | 1.293 | 1.272 | 1.294 | 1.271 | 1.295 | 1.270 | 1.300 | 1.273 | 1.968 |
| CSb | 1.963 | |||||||||||||||
| C2Sb | 1.326 | 1.889 | ||||||||||||||
| C3Sb | 1.334 | 1.270 | 2.009 | |||||||||||||
| C4Sb | 1.315 | 1.277 | 1.300 | 1.891 | ||||||||||||
| C5Sb | 1.321 | 1.272 | 1.302 | 1.264 | 1.995 | |||||||||||
| C6Sb | 1.311 | 1.276 | 1.291 | 1.268 | 1.301 | 1.894 | ||||||||||
| C7Sb | 1.315 | 1.276 | 1.293 | 1.264 | 1.303 | 1.265 | 1.985 | |||||||||
| C8Sb | 1.308 | 1.278 | 1.290 | 1.266 | 1.295 | 1.266 | 1.303 | 1.896 | ||||||||
| C9Sb | 1.311 | 1.278 | 1.290 | 1.268 | 1.295 | 1.264 | 1.302 | 1.267 | 1.979 | |||||||
| C10Sb | 1.306 | 1.279 | 1.289 | 1.267 | 1.295 | 1.263 | 1.298 | 1.265 | 1.304 | 1.897 | ||||||
| C11Sb | 1.309 | 1.280 | 1.288 | 1.270 | 1.292 | 1.267 | 1.296 | 1.265 | 1.302 | 1.268 | 1.976 | |||||
| C12Sb | 1.304 | 1.281 | 1.287 | 1.268 | 1.294 | 1.264 | 1.298 | 1.262 | 1.300 | 1.264 | 1.304 | 1.898 | ||||
| C13Sb | 1.307 | 1.282 | 1.286 | 1.271 | 1.290 | 1.269 | 1.293 | 1.267 | 1.296 | 1.266 | 1.301 | 1.269 | 1.973 | |||
| C14Sb | 1.303 | 1.283 | 1.286 | 1.270 | 1.292 | 1.265 | 1.297 | 1.262 | 1.300 | 1.260 | 1.301 | 1.264 | 1.305 | 1.897 | ||
| C15Sb | 1.305 | 1.283 | 1.285 | 1.272 | 1.289 | 1.270 | 1.292 | 1.269 | 1.293 | 1.268 | 1.295 | 1.266 | 1.300 | 1.270 | 1.971 | |
| C16Sb | 1.302 | 1.284 | 1.285 | 1.271 | 1.291 | 1.266 | 1.296 | 1.263 | 1.300 | 1.260 | 1.302 | 1.259 | 1.303 | 1.263 | 1.306 | 1.898 |
| CSb | 1.952 | |||||||||||||||
| C2Sb | 1.280 | 2.014 | ||||||||||||||
| C3Sb | 1.278 | 1.326 | 1.924 | |||||||||||||
| C4Sb | 1.273 | 1.336 | 1.260 | 2.000 | ||||||||||||
| C5Sb | 1.273 | 1.326 | 1.262 | 1.314 | 1.921 | |||||||||||
| C6Sb | 1.273 | 1.329 | 1.256 | 1.323 | 1.261 | 1.997 | ||||||||||
| C7Sb | 1.272 | 1.326 | 1.256 | 1.315 | 1.263 | 1.311 | 1.919 | |||||||||
| C8Sb | 1.273 | 1.326 | 1.257 | 1.317 | 1.258 | 1.319 | 1.262 | 1.994 | ||||||||
| C9Sb | 1.273 | 1.325 | 1.256 | 1.316 | 1.257 | 1.313 | 1.264 | 1.310 | 1.917 | |||||||
| C10Sb | 1.274 | 1.324 | 1.257 | 1.314 | 1.259 | 1.314 | 1.259 | 1.317 | 1.263 | 1.992 | ||||||
| C11Sb | 1.273 | 1.324 | 1.256 | 1.315 | 1.256 | 1.314 | 1.257 | 1.312 | 1.263 | 1.310 | 1.914 | |||||
| C12Sb | 1.275 | 1.322 | 1.258 | 1.312 | 1.259 | 1.312 | 1.260 | 1.311 | 1.260 | 1.315 | 1.264 | 1.990 | ||||
| C13Sb | 1.274 | 1.322 | 1.256 | 1.314 | 1.256 | 1.314 | 1.256 | 1.314 | 1.257 | 1.312 | 1.263 | 1.310 | 1.913 | |||
| C14Sb | 1.276 | 1.321 | 1.258 | 1.312 | 1.259 | 1.311 | 1.260 | 1.310 | 1.261 | 1.310 | 1.261 | 1.314 | 1.265 | 1.987 | ||
| C15Sb | 1.275 | 1.321 | 1.257 | 1.313 | 1.257 | 1.314 | 1.256 | 1.315 | 1.255 | 1.315 | 1.256 | 1.312 | 1.262 | 1.310 | 1.912 | |
| C16Sb | 1.276 | 1.320 | 1.258 | 1.311 | 1.260 | 1.310 | 1.260 | 1.310 | 1.261 | 1.309 | 1.261 | 1.309 | 1.262 | 1.312 | 1.266 | 1.985 |
| CnSb | En (a. u.) | ΔEn (a. u.) | BE (a. u.) | ΔEI (kcal/mol) | IE (kcal/mol) | EA (kcal/mol) | μ (debye) | RC (MHz) |
|---|---|---|---|---|---|---|---|---|
| 1 | −43.35455 | 0.10241 | 212.4 | 69.2 | 3.13 | 12990 | ||
| 2 | −81.48162 | −38.12707 | 0.37201 | 169.2 | 196.3 | 62.6 | 5.57 | 3497 |
| 3 | −119.56561 | −38.08399 | 0.59853 | 142.1 | 196.1 | 72.1 | 5.36 | 1519 |
| 4 | −157.65877 | −38.09316 | 0.83422 | 147.9 | 187.7 | 72.9 | 7.46 | 821 |
| 5 | −195.74409 | −38.08532 | 1.06207 | 143.0 | 186.3 | 78.7 | 7.10 | 496 |
| 6 | −233.83823 | −38.09414 | 1.29874 | 148.5 | 183.9 | 76.7 | 8.98 | 326 |
| 7 | −271.91968 | −38.08145 | 1.52272 | 140.5 | 179.1 | 83.0 | 8.75 | 227 |
| 8 | −310.00802 | −38.08834 | 1.75359 | 144.9 | 176.1 | 84.1 | 10.39 | 165 |
| 9 | −348.09409 | −38.08607 | 1.98219 | 143.4 | 173.8 | 86.2 | 10.31 | 125 |
| 10 | −386.18172 | −38.08763 | 2.21235 | 144.4 | 172.1 | 87.5 | 11.74 | 97 |
| 11 | −424.26787 | −38.08615 | 2.44103 | 143.5 | 169.5 | 88.6 | 11.78 | 77 |
| 12 | −462.35529 | −38.08742 | 2.67098 | 144.3 | 169.2 | 89.5 | 13.04 | 62 |
| 13 | −500.44149 | −38.08620 | 2.89971 | 143.5 | 166.2 | 90.5 | 13.19 | 51 |
| 14 | −538.52879 | −38.0873 | 3.12954 | 144.2 | 166.8 | 92.4 | 14.30 | 42 |
| 15 | −576.61521 | −38.08642 | 3.35849 | 143.7 | 163.5 | 92.0 | 14.53 | 36 |
| 16 | −614.70241 | −38.08720 | 3.58822 | 144.2 | 164.8 | 94.2 | 15.51 | 30 |
| CnSb+ | En (a. u.) | ΔEn (a. u.) | BE (a. u.) | ΔEI (kcal/mol) | μ(Debye) | RC (MHz) |
|---|---|---|---|---|---|---|
| 1 | −43.01612 | −0.23602 | 1.08 | 12013 | ||
| 2 | −81.16886 | −38.15274 | 0.05925 | 0.29527 | 2.68 | 3584 |
| 3 | −119.25339 | −38.08453 | 0.28631 | 0.22705 | 0.85 | 1474 |
| 4 | −157.35996 | −38.10657 | 0.53541 | 0.24911 | 2.26 | 835 |
| 5 | −195.44758 | −38.08762 | 0.76556 | 0.23014 | 0.52 | 489 |
| 6 | −233.54567 | −38.09809 | 1.00618 | 0.24063 | 1.97 | 331 |
| 7 | −271.63483 | −38.08916 | 1.23787 | 0.23169 | 0.43 | 226 |
| 8 | −309.72820 | −38.09337 | 1.47377 | 0.2359 | 1.89 | 167 |
| 9 | −347.81792 | −38.08972 | 1.70602 | 0.23225 | 0.48 | 124 |
| 10 | −385.90836 | −38.09044 | 1.93899 | 0.23297 | 0.83 | 98 |
| 11 | −423.99872 | −38.09036 | 2.17188 | 0.2329 | 0.65 | 77 |
| 12 | −462.08689 | −38.08817 | 2.40258 | 0.23069 | 1.99 | 62 |
| 13 | −500.17791 | −38.09102 | 2.63613 | 0.23355 | 0.87 | 51 |
| 14 | −538.26446 | −38.08655 | 2.86521 | 0.22908 | 2.06 | 43 |
| 15 | −576.35619 | −38.09173 | 3.09947 | 0.23426 | 1.11 | 36 |
| 16 | −614.44148 | −38.08529 | 3.32729 | 0.22782 | 2.10 | 31 |
| CnSb− | En (a. u.) | ΔEn (a. u.) | BE (a. u.) | ΔEI (kcal/mol) | μ (Debye) | RC (MHz) |
|---|---|---|---|---|---|---|
| 1 | −43.46493 | 0.21279 | 5.50 | 12153 | ||
| 2 | −81.58147 | −38.11654 | 0.47186 | 0.25908 | 8.89 | 3348 |
| 3 | −119.68068 | −38.09921 | 0.7136 | 0.24174 | 9.78 | 1528 |
| 4 | −157.77531 | −38.09463 | 0.95076 | 0.23715 | 13.03 | 800 |
| 5 | −195.86996 | −38.09465 | 1.18794 | 0.23718 | 13.52 | 496 |
| 6 | −233.96096 | −38.09100 | 1.42147 | 0.23353 | 16.53 | 321 |
| 7 | −272.05265 | −38.09169 | 1.65569 | 0.23422 | 16.93 | 227 |
| 8 | −310.14274 | −38.09009 | 1.88831 | 0.23262 | 19.74 | 163 |
| 9 | −348.23223 | −38.08949 | 2.12033 | 0.23202 | 20.16 | 125 |
| 10 | −386.32214 | −38.08991 | 2.35277 | 0.23243 | 22.76 | 96 |
| 11 | −424.41010 | −38.08796 | 2.58326 | 0.23049 | 23.26 | 77 |
| 12 | −462.49915 | −38.08905 | 2.81484 | 0.23158 | 25.04 | 62 |
| 13 | −500.58704 | −38.08789 | 3.04526 | 0.23042 | 26.30 | 51 |
| 14 | −538.67748 | −38.09044 | 3.27823 | 0.23296 | 28.52 | 42 |
| 15 | −576.76336 | −38.08588 | 3.50664 | 0.22841 | 29.33 | 36 |
| 16 | −614.85418 | −38.09082 | 3.73999 | 0.23335 | 31.33 | 30 |
| C4Sb | C5Sb | C6Sb | C11Sb | C12Sb | |
|---|---|---|---|---|---|
| C-C | 1.277 1.267 | 1.288 1.281 | 1.277 1.267 | 1.284 1.275 | 1.274 1.263 |
| C-Sb | 1.941 1.941 | 1.946 1.941 | 1.949 1.949 | 1.949 1.949 | 1.964 1.966 |
| C-Sb Stretching frequency | 378.3 384.5 | 331.3 335.2 | 291.7 294.9 | 204.8 204.9 | 190.4 189.6 |
| HOMO-LUMO gap | 1.27 1.26 | 1.25 1.27 | 1.17 1.16 | 1.03 1.04 | 1.01 0.99 |
| n orbital configuration |
|---|
| 1 (core) 1σ22σ21π43σ1 2 (core)1σ22σ23σ24σ21π42π1 3 (core)1σ22σ23σ24σ21π45σ22π3 4 (core)1σ22σ23σ24σ25σ21π42π46σ23π1 5 (core)1σ22σ23σ24σ25σ21π46σ22π47σ23π3 6 (core)1σ22σ23σ24σ25σ26σ21π47σ22π43π48σ24π1 7 (core)1σ22σ23σ24σ25σ26σ27σ21π48σ22π43π49σ24π3 8 (core)1σ22σ23σ24σ25σ26σ27σ28σ21π49σ22π43π44π410σ25π1 9 (core)1σ22σ23σ24σ25σ26σ27σ28σ29σ21π42π410σ23π44π411σ25π3 10 (core)1σ22σ23σ24σ25σ26σ27σ28σ29σ210σ21π42π411σ23π44π45π412σ26π1 11 (core)1σ22σ23σ24σ25σ26σ27σ28σ29σ210σ211σ21π42π412σ23π44π45π413σ26π3 12 (core)1σ22σ23σ24σ25σ26σ27σ28σ29σ210σ211σ212σ21π42π413σ23π44π45π46π414σ27π1 13 (core)1σ22σ23σ24σ25σ26σ27σ28σ29σ210σ211σ212σ213σ21π42π414σ23π44π45π46π415σ27π3 14 (core)1σ22σ23σ24σ25σ26σ27σ28σ29σ210σ211σ212σ213σ214σ21π42π43π415σ24π45π46π47π416σ28π1 15 (core)1σ22σ23σ24σ25σ26σ27σ28σ29σ210σ211σ212σ213σ214σ215σ21π42π43π416σ24π45π46π47π417σ28π3 16 (core)1σ22σ23σ24σ25σ26σ27σ28σ29σ210σ211σ212σ213σ214σ215σ216σ21π42π43π417σ24π45π46π47π48π418σ29π1 |
| Species | C1 | C2 | Species | C1 | C2 | ||
|---|---|---|---|---|---|---|---|
| C1Sb | −0.33 | C9Sb | −0.76 | −0.14 | 0.63 | ||
| C2Sb | −0.47 | −0.05 | 0.42 | C10Sb | −0.80 | −0.14 | 0.66 |
| C3Sb | −0.45 | −0.14 | 0.31 | C11Sb | −0.77 | −0.13 | 0.64 |
| C4Sb | −0.73 | −0.22 | 0.51 | C12Sb | −0.80 | −0.13 | 0.67 |
| C5Sb | −0.72 | −0.19 | 0.53 | C13Sb | −0.77 | −0.13 | 0.65 |
| C6Sb | −0.80 | −0.18 | 0.62 | C14Sb | −0.80 | −0.13 | 0.67 |
| C7Sb | −0.75 | −0.15 | 0.60 | C15Sb | −0.78 | −0.13 | 0.65 |
| C8Sb | −0.80 | −0.15 | 0.65 | C16Sb | −0.80 | −0.13 | 0.67 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, Z.; Shao, X.; Wu, W.; Liu, Z.; Yang, M.; Liu, M.; Wang, H. Structures and Stabilities of Carbon Chain Clusters Influenced by Atomic Antimony. Molecules 2023, 28, 1358. https://doi.org/10.3390/molecules28031358
Song Z, Shao X, Wu W, Liu Z, Yang M, Liu M, Wang H. Structures and Stabilities of Carbon Chain Clusters Influenced by Atomic Antimony. Molecules. 2023; 28(3):1358. https://doi.org/10.3390/molecules28031358
Chicago/Turabian StyleSong, Zhenjun, Xiji Shao, Wei Wu, Zhenzhong Liu, Meiding Yang, Mingyue Liu, and Hai Wang. 2023. "Structures and Stabilities of Carbon Chain Clusters Influenced by Atomic Antimony" Molecules 28, no. 3: 1358. https://doi.org/10.3390/molecules28031358