
Prediction of a Cyclic Hydrogenated Boron Molecule as a Promising Building Block for Borophane
 , , , and
, , , and
Abstract
:1. Introduction
2. Results and Discussion
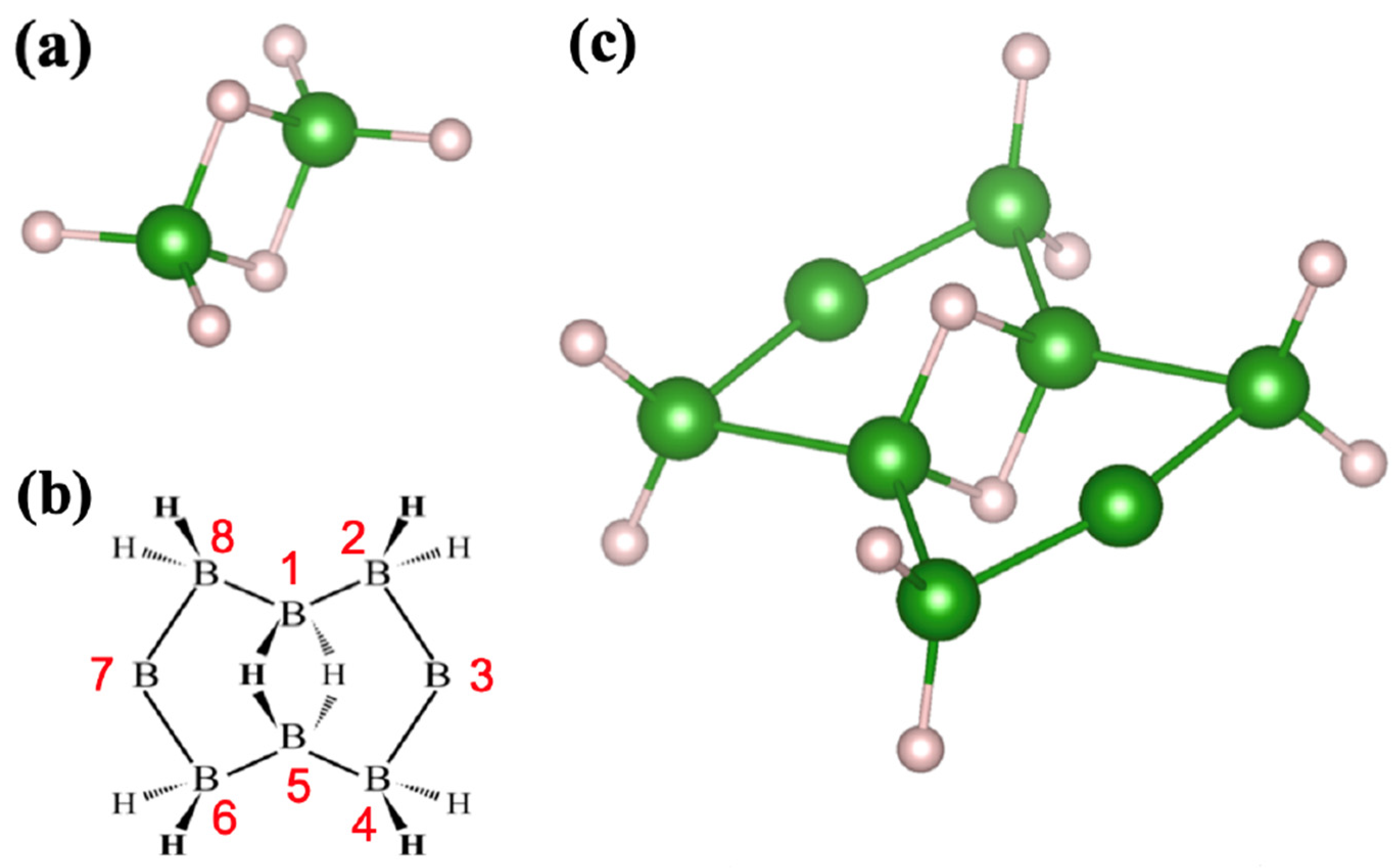
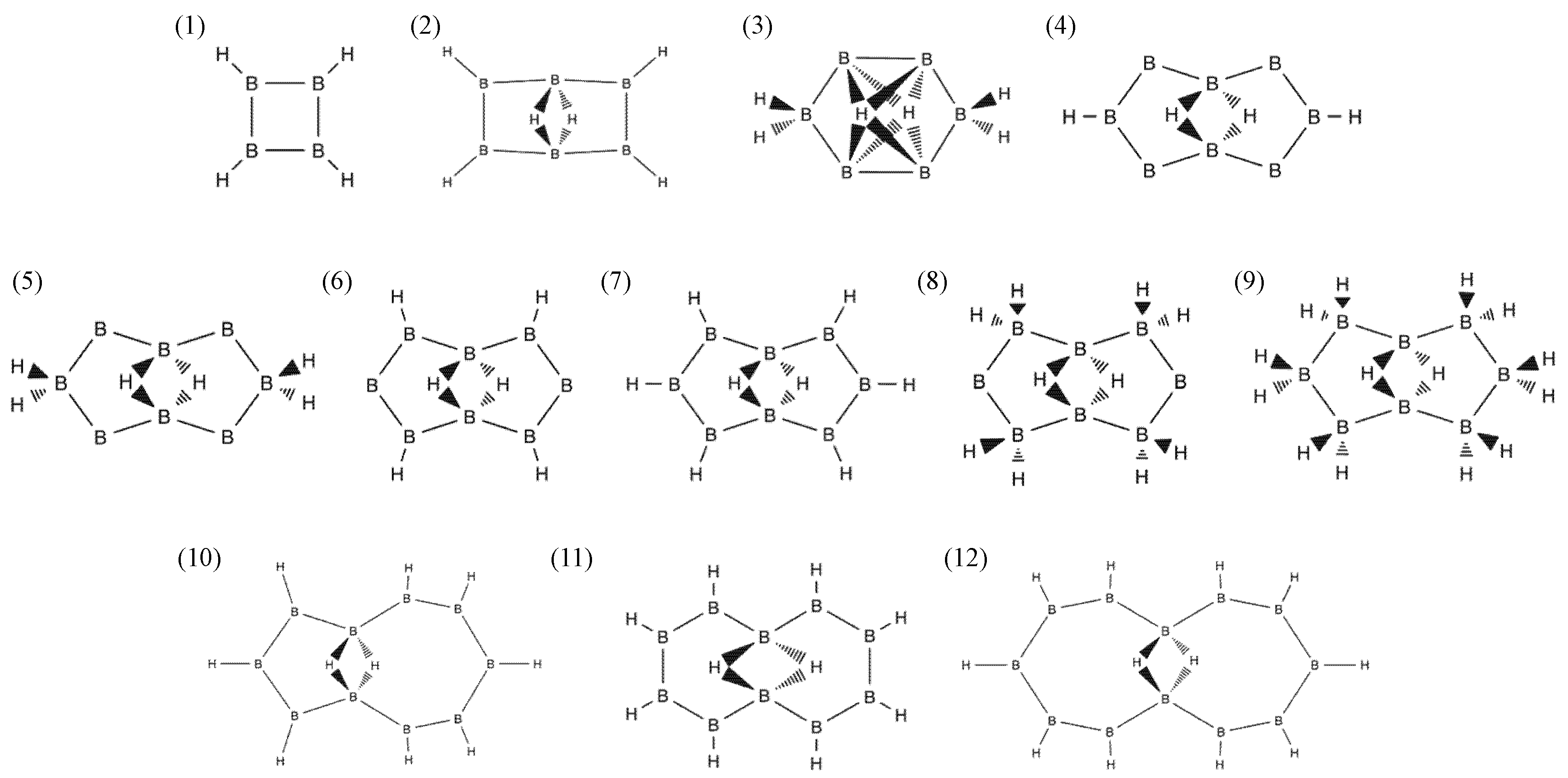
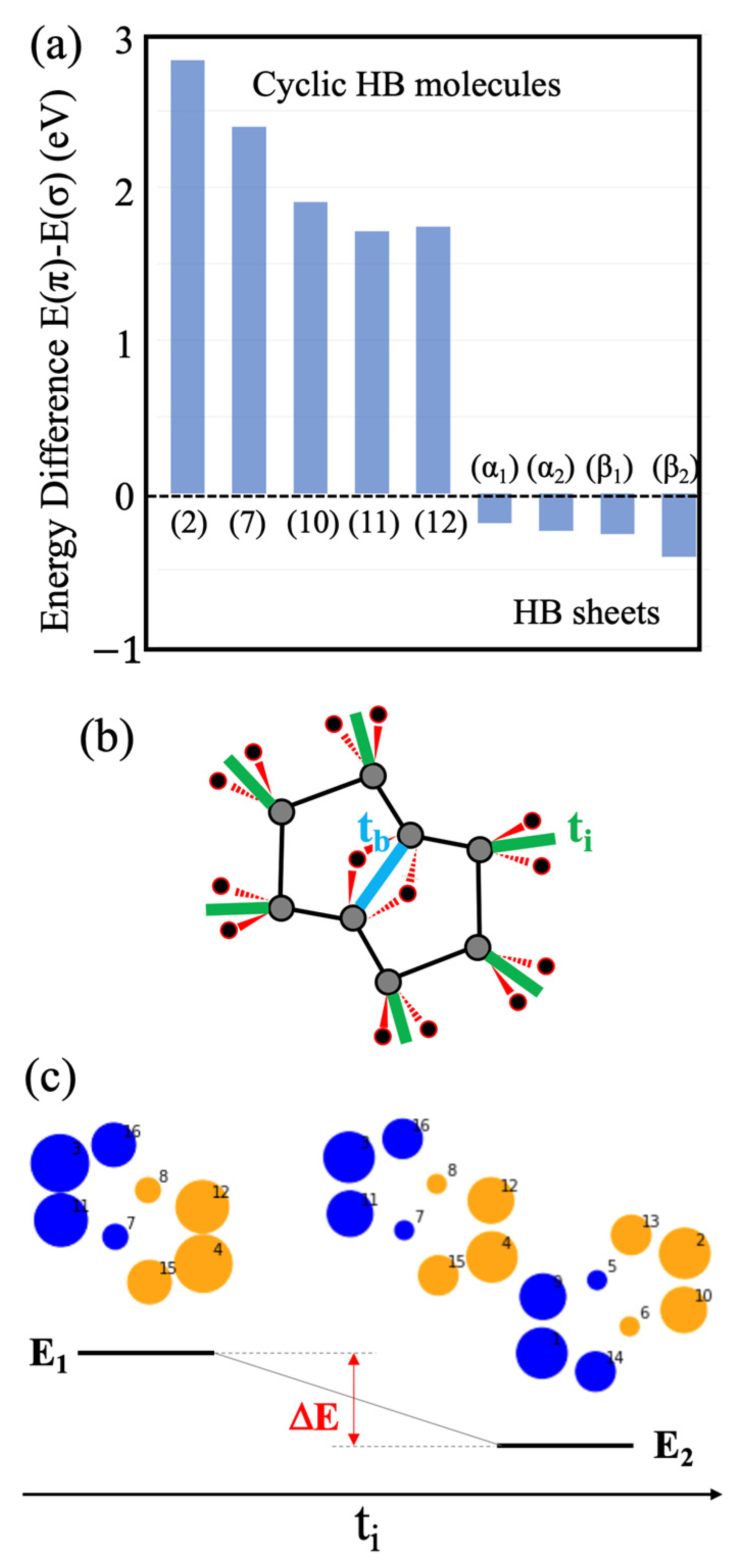
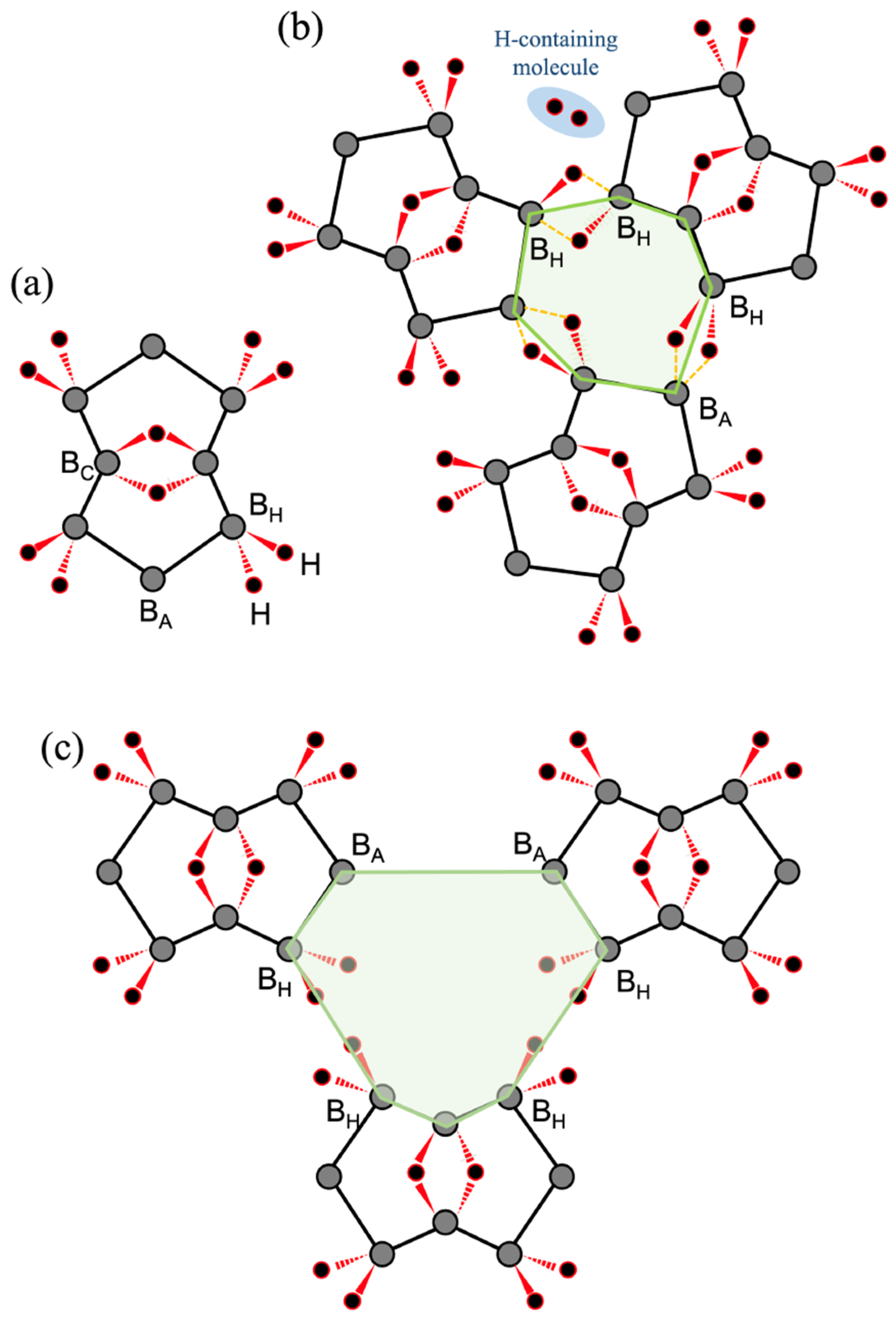
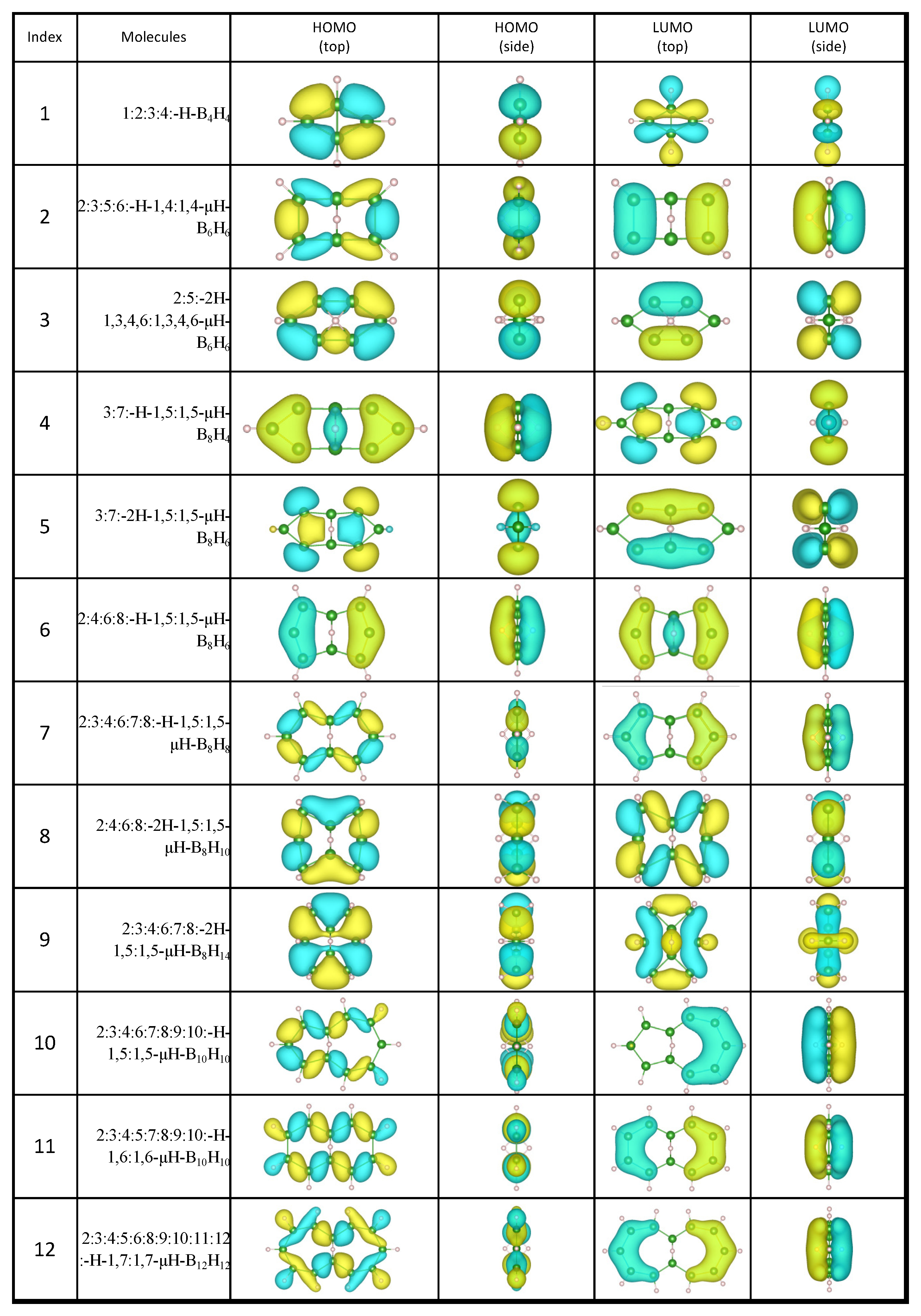
2.1. Candidates from the Cyclic Hydrogenated Boron Compounds
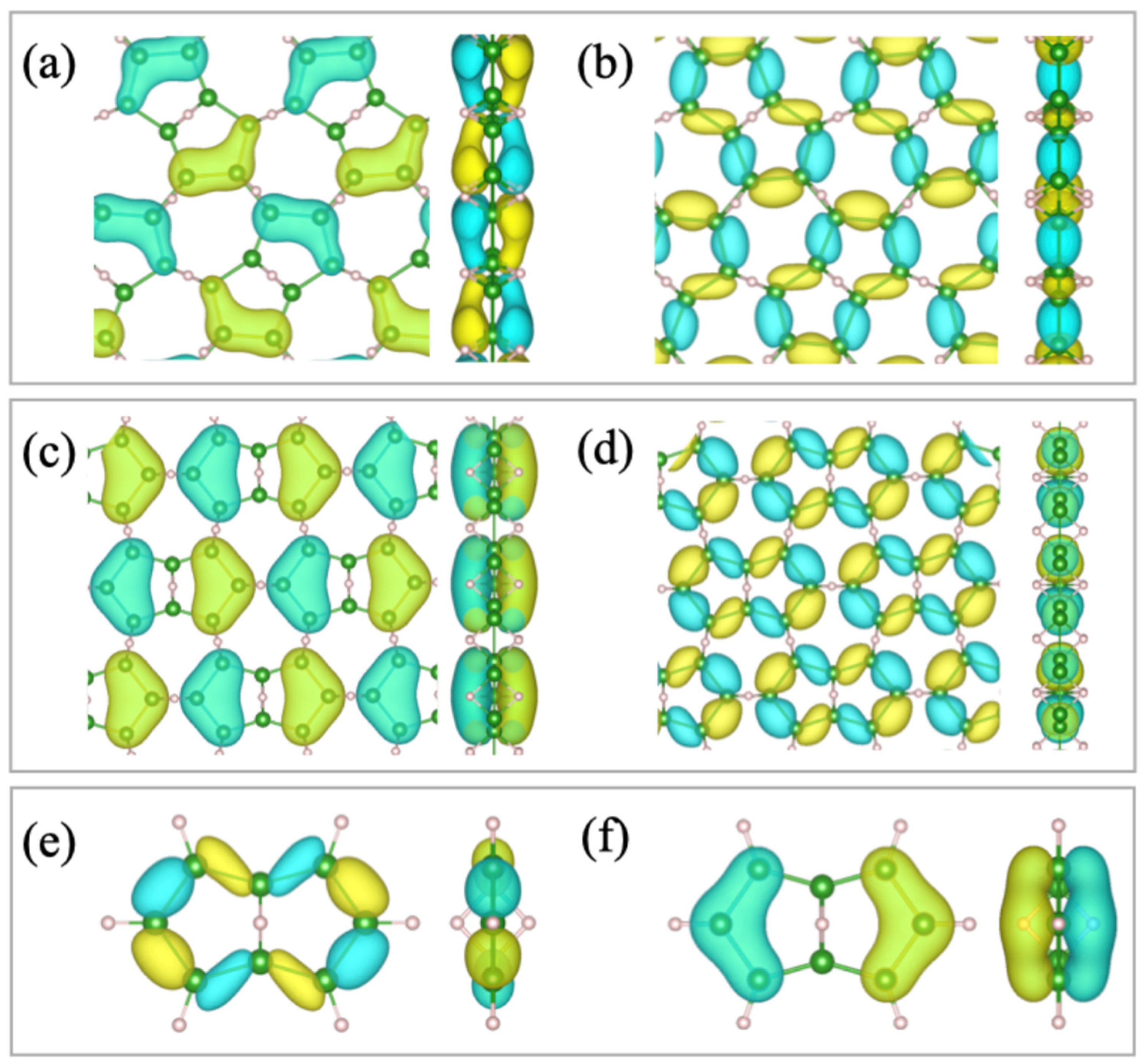
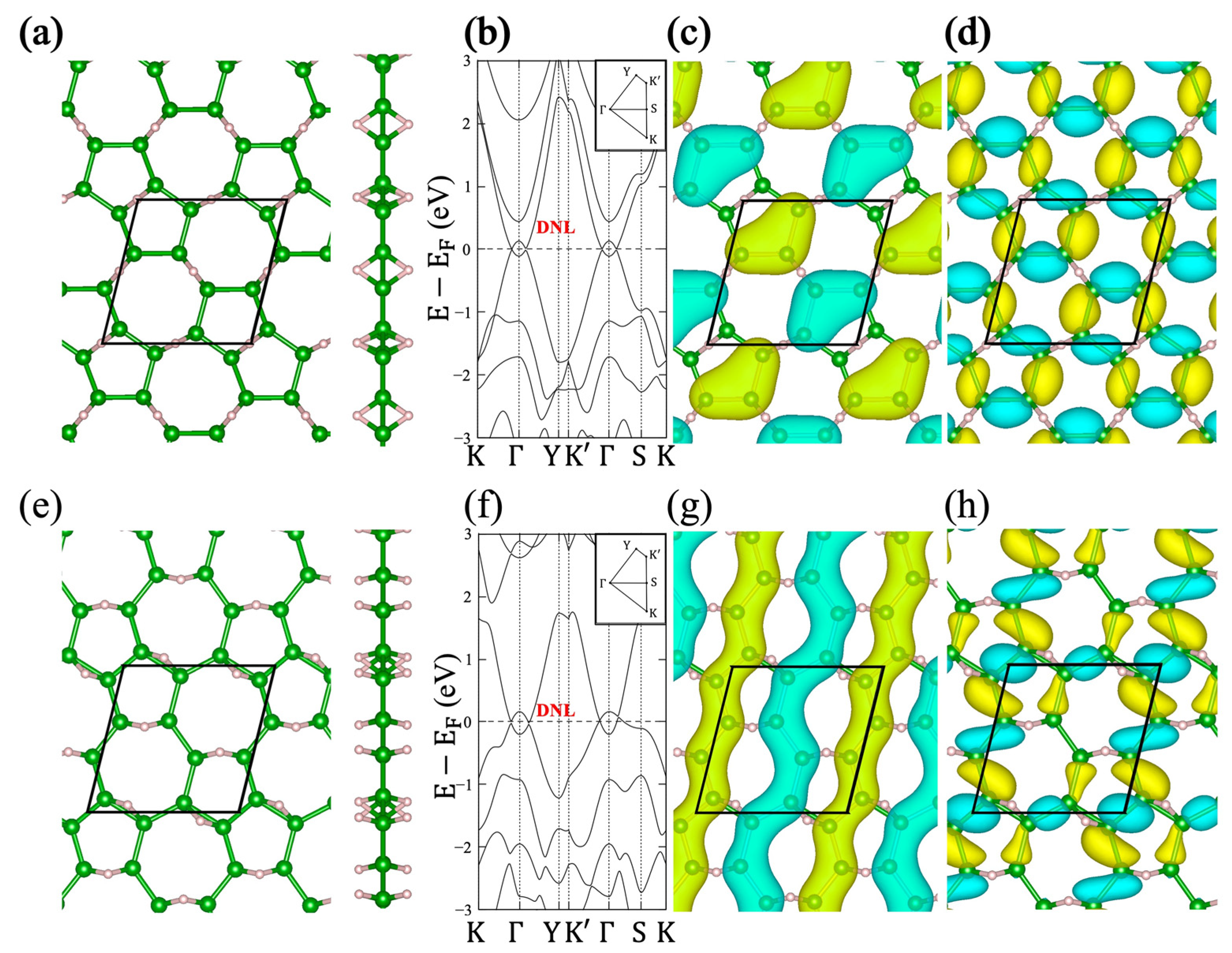
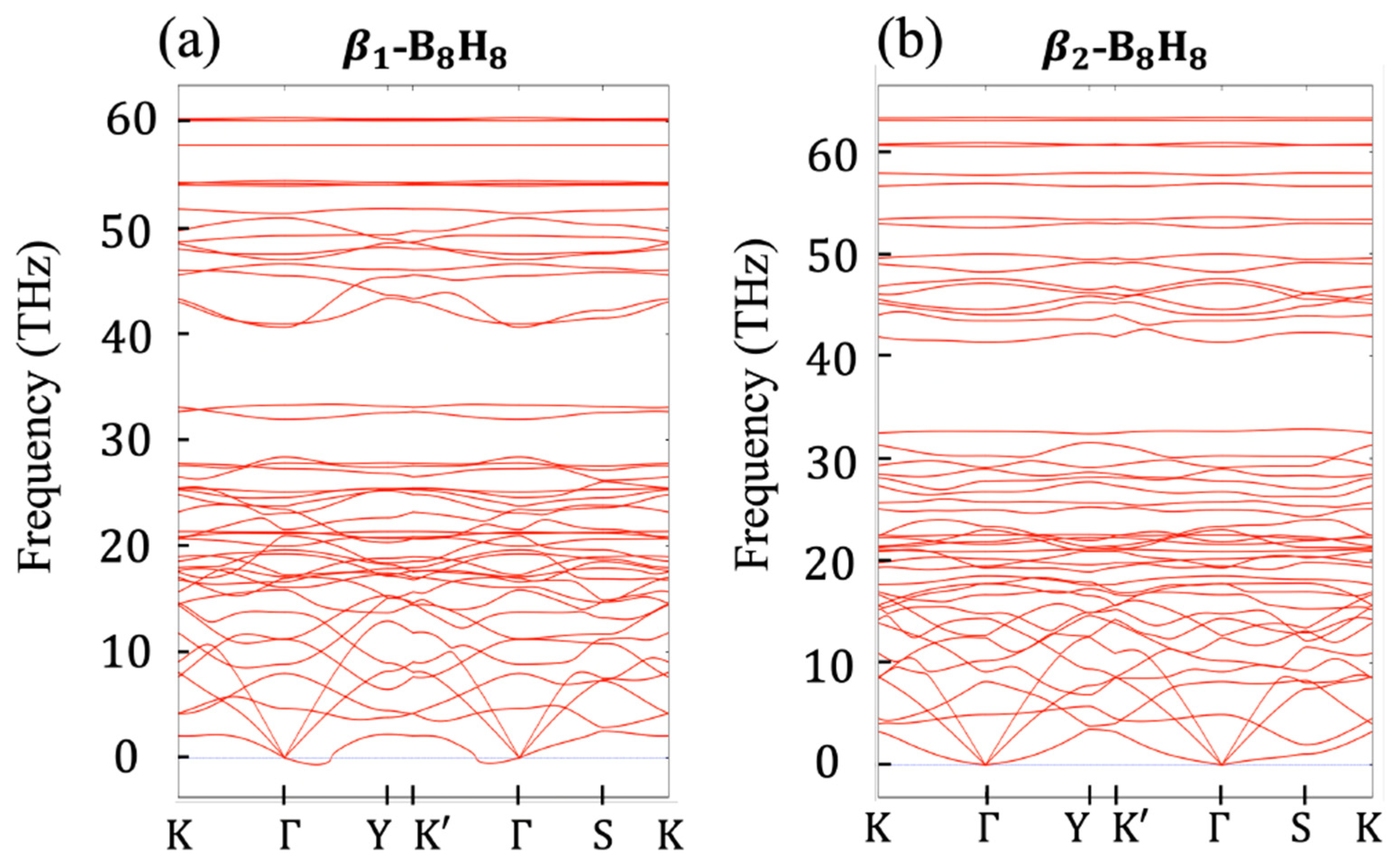
2.2. Relation to the Borophane Layer
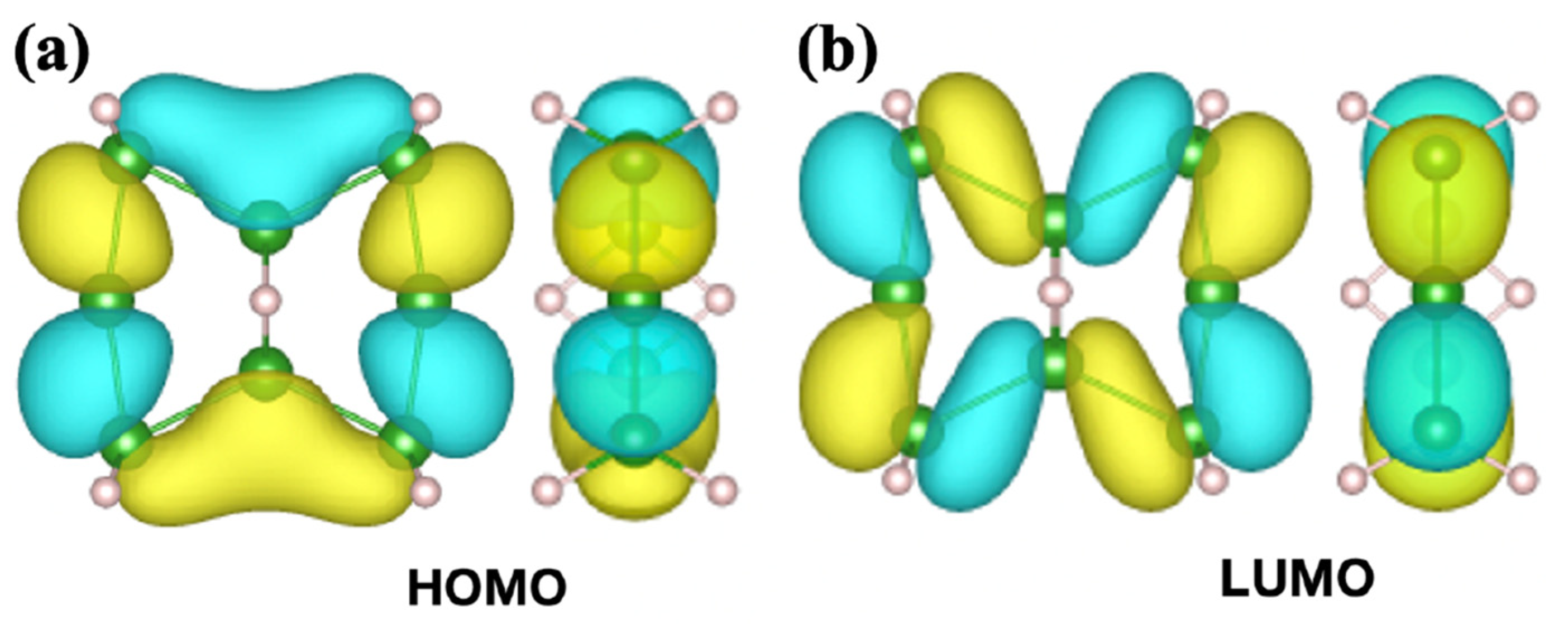
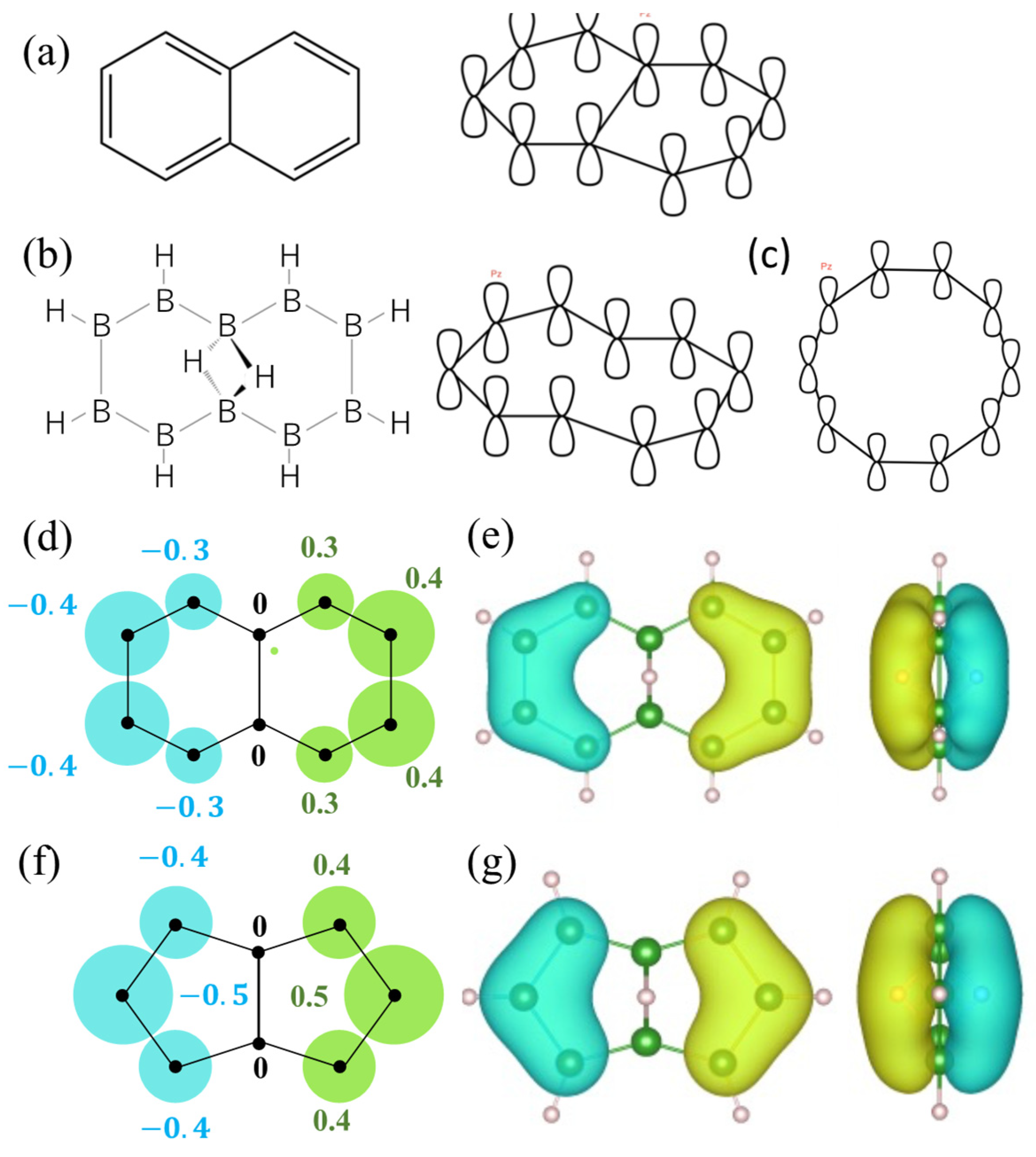
2.3. Orbitals of the Cyclic Boron Molecule in the Hückel Model
3. Calculation Methods
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

Appendix B


References
- Callister, W.D., Jr.; Rethwisch, D.G. Materials Science and Engineering: An Introduction, 10th ed.; Wiley: Hoboken, NJ, USA, 2018. [Google Scholar]
- Hagemann, H. Boron Hydrogen Compounds: Hydrogen Storage and Battery Applications. Molecules 2021, 26, 7425. [Google Scholar] [CrossRef] [PubMed]
- van’t Hoff, J.H. A suggestion looking to the extension into space of the structural formulas at present used in chemistry, and a note upon the relation between the optical activity and the chemical constitution of organic compounds. Arch. Neerl. Sci. Exactes Nat. 1874, 9, 445–454. [Google Scholar]
- Le Bel, J.A. Sur les Relations qui existent entre les Formules atomiques des Corps organiques et le Pouvoir rotatoire de leurs Dissolutions. Bell. Soc. Chim. Fr. 1874, 22, 337–347. [Google Scholar]
- Fau, S.; Frenking, G. Anti van’t Hoff/Le Bel geometries of boron compounds. A theoretical study of classical and nonclassical isomers of B2CH4, B2NH3 and B2OH2. J. Mol. Struct. THEOCHEM 1995, 338, 117–130. [Google Scholar] [CrossRef]
- Song, B.; Zhou, Y.; Yang, H.-M.; Liao, J.-H.; Yang, L.-M.; Yang, X.-B.; Ganz, E. Two-dimensional anti-Van’t Hoff/Le Bel array AlB6 with high stability, unique motif, triple dirac cones, and superconductivity. J. Am. Chem. Soc. 2019, 141, 3630–3640. [Google Scholar] [CrossRef]
- Matsuda, I.; Wu, K. 2D Boron: Boraphene, Borophene, Boronene, 1st ed.; Springer: Cham, Switzerland, 2021. [Google Scholar]
- Feng, B.; Zhang, J.; Zhong, Q.; Li, W.; Li, S.; Li, H.; Cheng, P.; Meng, S.; Chen, L.; Wu, K. Experimental Realization of Two-Dimensional Boron Sheets. Nat. Chem. 2016, 8, 563–568. [Google Scholar] [CrossRef] [Green Version]
- Feng, B.; Sugino, O.; Liu, R.-Y.; Zhang, J.; Yukawa, R.; Kawamura, M.; Iimori, T.; Kim, H.; Hasegawa, Y.; Li, H.; et al. Dirac Fermions in Borophene. Phys. Rev. Lett. 2017, 118, 096401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, B.; Zhang, J.; Ito, S.; Arita, M.; Cheng, C.; Chen, L.; Wu, K.; Komori, F.; Sugino, O.; Miyamoto, K.; et al. Discovery of 2D anisotropic Dirac cones. Adv. Mater. 2018, 30, 1704025. [Google Scholar] [CrossRef]
- Nishino, H.; Fujita, T.; Cuong, N.T.; Tominaka, S.; Miyauchi, M.; Iimura, S.; Hirata, A.; Umezawa, N.; Okada, S.; Nishibori, E.; et al. Formation and Characterization of Hydrogen Boride Sheets Derived from MgB2 by Cation Exchange. J. Am. Chem. Soc. 2017, 139, 13761–13769. [Google Scholar] [CrossRef]
- Niibe, M.; Cameau, M.; Cuong, N.T.; Sunday, O.I.; Zhang, X.; Tsujikawa, Y.; Okada, S.; Yubuta, K.; Kondo, T.; Matsuda, I. Electronic structure of a borophene layer in rare-earth aluminum/chromium boride and its hydrogenated derivative borophane. Phys. Rev. Mater. 2021, 5, 084007. [Google Scholar] [CrossRef]
- Zhang, X.; Tsujikawa, Y.; Tateishi, I.; Niibe, M.; Wada, T.; Horio, M.; Hikichi, M.; Ando, Y.; Yubuta, K.; Kondo, T.; et al. Electronic Topological Transition of 2D Boron by the Ion Exchange Reaction. J. Phys. Chem. C 2022, 126, 12802–12808. [Google Scholar] [CrossRef]
- Kawamura, R.; Cuong, N.T.; Fujita, T.; Ishibiki, R.; Hirabayashi, T.; Yamaguchi, A.; Matsuda, I.; Okada, S.; Kondo, T.; Miyauchi, M. Photoinduced hydrogen release from hydrogen boride sheets. Nat. Commun. 2019, 10, 4880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuong, N.T.; Tateishi, I.; Cameau, M.; Niibe, M.; Umezawa, N.; Slater, B.; Yubuta, K.; Kondo, T.; Ogata, M.; Okada, S.; et al. Topological Dirac Nodal Loops in Nonsymmorphic Hydrogenated Monolayer Boron. Phys. Rev. B 2020, 101, 195412. [Google Scholar] [CrossRef]
- Tateishi, I.; Cuong, N.T.; Moura, C.A.S.; Cameau, M.; Ishibiki, R.; Fujino, A.; Okada, S.; Yamamoto, A.; Araki, M.; Ito, S.; et al. Semimetallicity of Free-Standing Hydrogenated Monolayer Boron from MgB2. Phys. Rev. Mater. 2019, 3, 024004. [Google Scholar] [CrossRef]
- Tateishi, I.; Zhang, X.; Matsuda, I. Electronic Structures of Polymorphic Layers of Borophane. Molecules 2022, 27, 1808. [Google Scholar] [CrossRef]
- Boron Hydride Chemistry; Muetterties, E.L. (Ed.) Academic Press: New York, NY, USA, 2012. [Google Scholar]
- Connelly, N.G.; Damhus, T.; Hartshorn, R.M.; Hutton, A.T. Nomenclature of Inorganic Chemistry: IUPAC Recommendations 2005; Royal Society of Chemistry: Cambridge, UK, 2005. [Google Scholar]
- Beckett, M.A.; Brellochs, B.; Chizhevsky, I.T.; Damhus, T.; Hellwich, K.-H.; Kennedy, J.D.; Laitinen, R.; Powella, W.H.; Rabinovich, D.; Viñas, C.; et al. Nomenclature for boranes and related species (IUPAC Recommendations 2019). Pure Appl. Chem. 2020, 92, 355–381. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.H.; Yun, S.J.; Won, Y.S.; Oh, C.S.; Kim, S.M.; Kim, K.K.; Lee, Y.H. Large-scale synthesis of graphene and other 2D materials towards industrialization. Nat. Commun. 2022, 13, 1484. [Google Scholar] [CrossRef]
- Solà, M. Aromaticity rules. Nat. Chem. 2022, 14, 585–590. [Google Scholar] [CrossRef]
- Mutai, K. Quantum Chemistry, Ryoshi Kagaku; SHOKODO Co. Ltd.: Tokyo, Japan, 1991. (In Japanese) [Google Scholar]
- Hückel, E. Quantum-theoretical contributions to the benzene problem. I. The electron configuration of benzene and related compounds. Z. Phys. 1931, 70, 204–286. [Google Scholar] [CrossRef]
- Krygowski, T.M.; Cyrañski, M.K.; Czarnocki, Z.; Häfelinger, G.; Katritzky, A.R. Aromaticity: A Theoretical Concept of Immense Practical Importance. Tetrahedron 2000, 56, 1783–1796. [Google Scholar] [CrossRef]
- Hess, B.A., Jr.; Schaad, L.J. Hueckel molecular orbital .pi. resonance energies. Benzenoid hydrocarbons. J. Am. Chem. Soc. 1971, 93, 2413–2416. [Google Scholar] [CrossRef]
- Schaad, L.J.; Hess, B.A., Jr. Huckel theory and aromatically. J. Chem. Educ. 1974, 51, 640. [Google Scholar] [CrossRef]
- Smith, D.G.A.; Burns, L.A.; Simmonett, A.C.; Parrish, R.M.; Schieber, M.C.; Galvelis, R.; Kraus, P.; Kruse, H.; Remigio, R.D.; Alenaizan, A.; et al. PSI4 1.4: Open-source software for high-throughput quantum chemistry. J. Chem. Phys. 2020, 152, 184108. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785. [Google Scholar] [CrossRef] [Green Version]
- Giannozzi, P.; Andreussi, O.; Brumme, T.; Bunau, O.; Nardelli, M.B.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Cococcioni, M.; et al. Advanced capabilities for materials modelling with Quantum ESPRESSO. J. Phys. Condens. Matter 2017, 29, 465901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [Green Version]
- Prandini, G.; Marrazzo, A.; Castelli, I.E.; Mounet, N.; Marzari, N. Precision and efficiency in solid-state pseudopotential calculations. npj Comput. Mater. 2018, 4, 72. [Google Scholar] [CrossRef] [Green Version]
- Togo, A.; Tanaka, I. First principles phonon calculations in materials science. Scr. Mater. 2015, 108, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Momma, K.; Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Cryst. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Index | Molecule Candidate | Total Energy (eV) | HOMO Level (eV) | LUMO Level (eV) | Stability |
|---|---|---|---|---|---|
| 1 | 1:2:3:4:-H-B4H4 | −2768.32 | −6.64 | −2.50 | FALSE |
| 2 | 2:3:5:6:-H-1,4:1,4-μH-B6H6 | −4148.76 | −6.56 | −3.73 | FALSE |
| 3 | 2:5:-2H-1,3,4,6:1,3,4,6-μH-B6H6 | −4149.17 | −7.65 | −4.22 | FALSE |
| 4 | 3:7:-H-1,5:1,5-μH-B8H4 | −5466.93 | −5.93 | −4.30 | FALSE |
| 5 | 3:7:-2H-1,5:1,5-μH-B8H6 | −5499.07 | −5.20 | −4.54 | FALSE |
| 6 | 2:4:6:8:-H-1,5:1,5-μH-B8H6 | −5501.27 | −5.93 | −4.16 | FALSE |
| 7 | 2:3:4:6:7:8:-H-1,5:1,5-μH-B8H8 | −5533.90 | −6.64 | −4.24 | FALSE |
| 8 | 2:4:6:8:-2H-1,5:1,5-μH-B8H10 | −5568.84 | −8.14 | −4.05 | TRUE |
| 9 | 2:3:4:6:7:8:-2H-1,5:1,5-μH-B8H14 | −5635.07 | −7.10 | −3.70 | FALSE |
| 10 | 2:3:4:6:7:8:9:10:-H-1,5:1,5-μH-B10H10 | −6915.86 | −6.18 | −4.24 | FALSE |
| 11 | 2:3:4:5:7:8:9:10:-H-1,6:1,6-μH-B10H10 | −6917.17 | −6.18 | −4.46 | FALSE |
| 12 | 2:3:4:5:6:8:9:10:11:12:-H-1,7:1,7-μH-B12H12 | −8299.69 | −6.31 | −4.57 | FALSE |
| System | Total Energy E (Hartree) | E − 4E(B2H6) (Hartree) | E − 4E(B2H6) (kcal/mol) |
|---|---|---|---|
| 4 B2H6 | −213.218 | 0.000 | 0.000 |
| 8 BH3 | −212.969 | 0.249 | 156.250 |
| B8H10 + 7H2 | −212.908 | 0.310 | 194.528 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ando, Y.; Nakashima, T.; Yin, H.; Tateishi, I.; Zhang, X.; Tsujikawa, Y.; Horio, M.; Cuong, N.T.; Okada, S.; Kondo, T.; et al. Prediction of a Cyclic Hydrogenated Boron Molecule as a Promising Building Block for Borophane. Molecules 2023, 28, 1225. https://doi.org/10.3390/molecules28031225
Ando Y, Nakashima T, Yin H, Tateishi I, Zhang X, Tsujikawa Y, Horio M, Cuong NT, Okada S, Kondo T, et al. Prediction of a Cyclic Hydrogenated Boron Molecule as a Promising Building Block for Borophane. Molecules. 2023; 28(3):1225. https://doi.org/10.3390/molecules28031225
Chicago/Turabian StyleAndo, Yasunobu, Takeru Nakashima, Heming Yin, Ikuma Tateishi, Xiaoni Zhang, Yuki Tsujikawa, Masafumi Horio, Nguyen Thanh Cuong, Susumu Okada, Takahiro Kondo, and et al. 2023. "Prediction of a Cyclic Hydrogenated Boron Molecule as a Promising Building Block for Borophane" Molecules 28, no. 3: 1225. https://doi.org/10.3390/molecules28031225