
μ-Raman Determination of Essential Oils’ Constituents from Distillates and Leaf Glands of Origanum Plants

 and
and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Raman Excitation/Collection Geometry
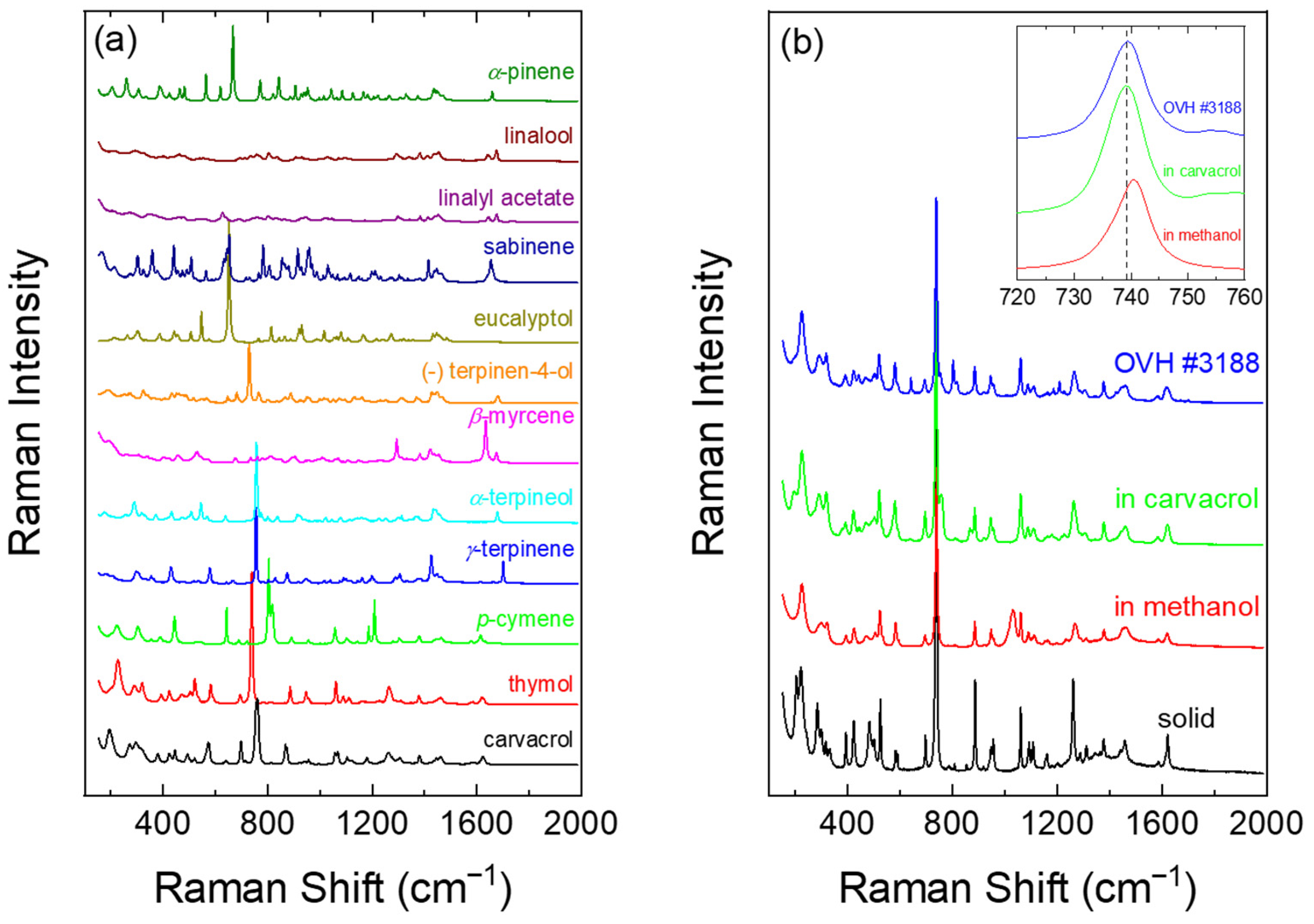
2.2. Raman Spectra of Reference Compounds
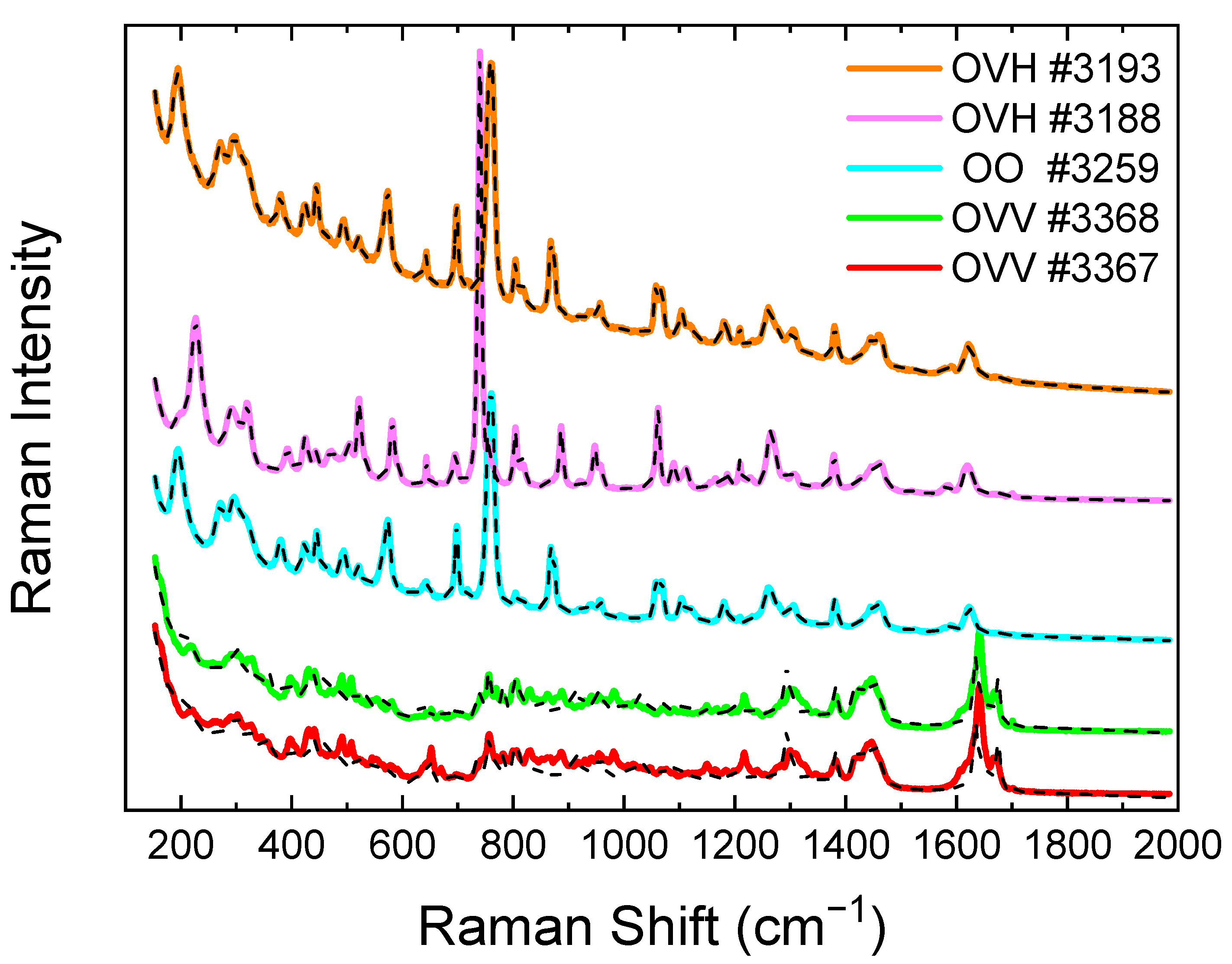
2.3. Raman Spectroscopy of the Essential Oils from the Collected Origanum Samples
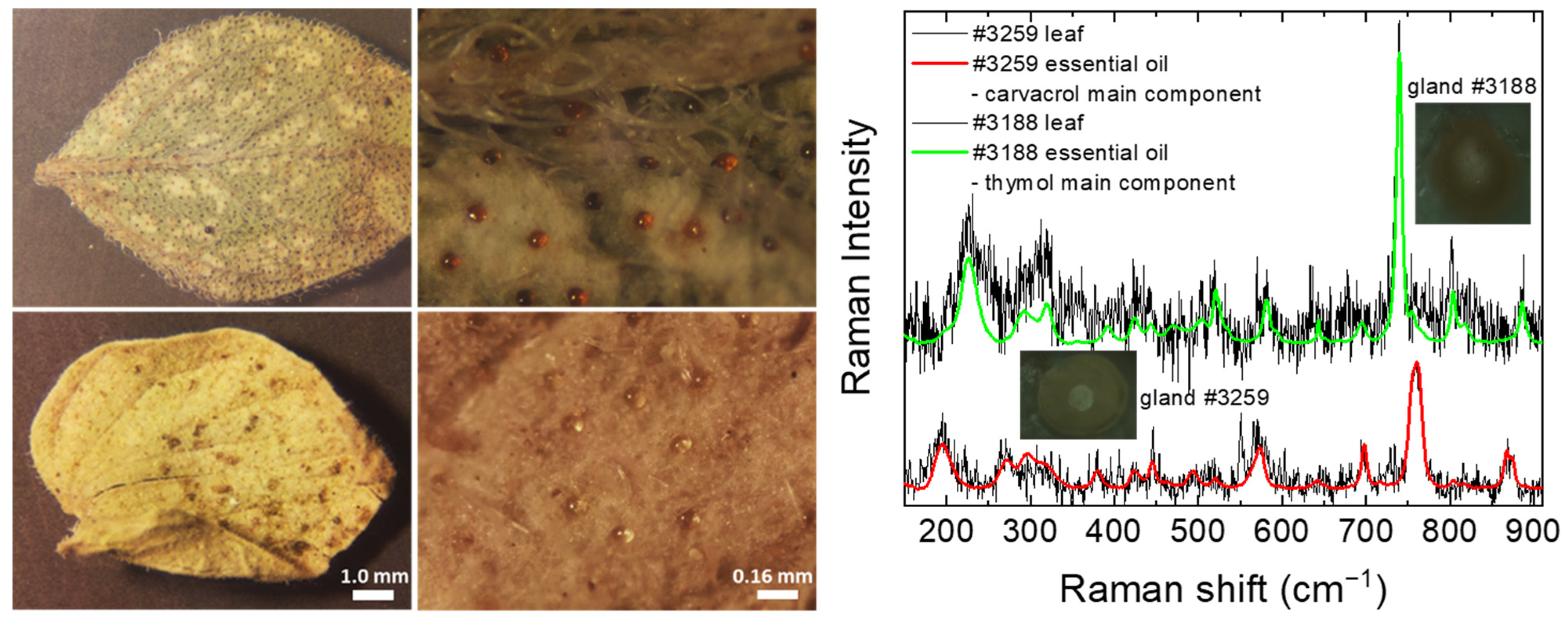
2.4. Origanum Leaves—Feasibility Investigation of In Situ Raman Spectroscopy
3. Materials and Methods
3.1. Raman Instrumentation
3.2. Quantification Method
3.3. Materials
3.3.1. Reference Compounds
3.3.2. Plant Material—Essential Oils’ Isolation
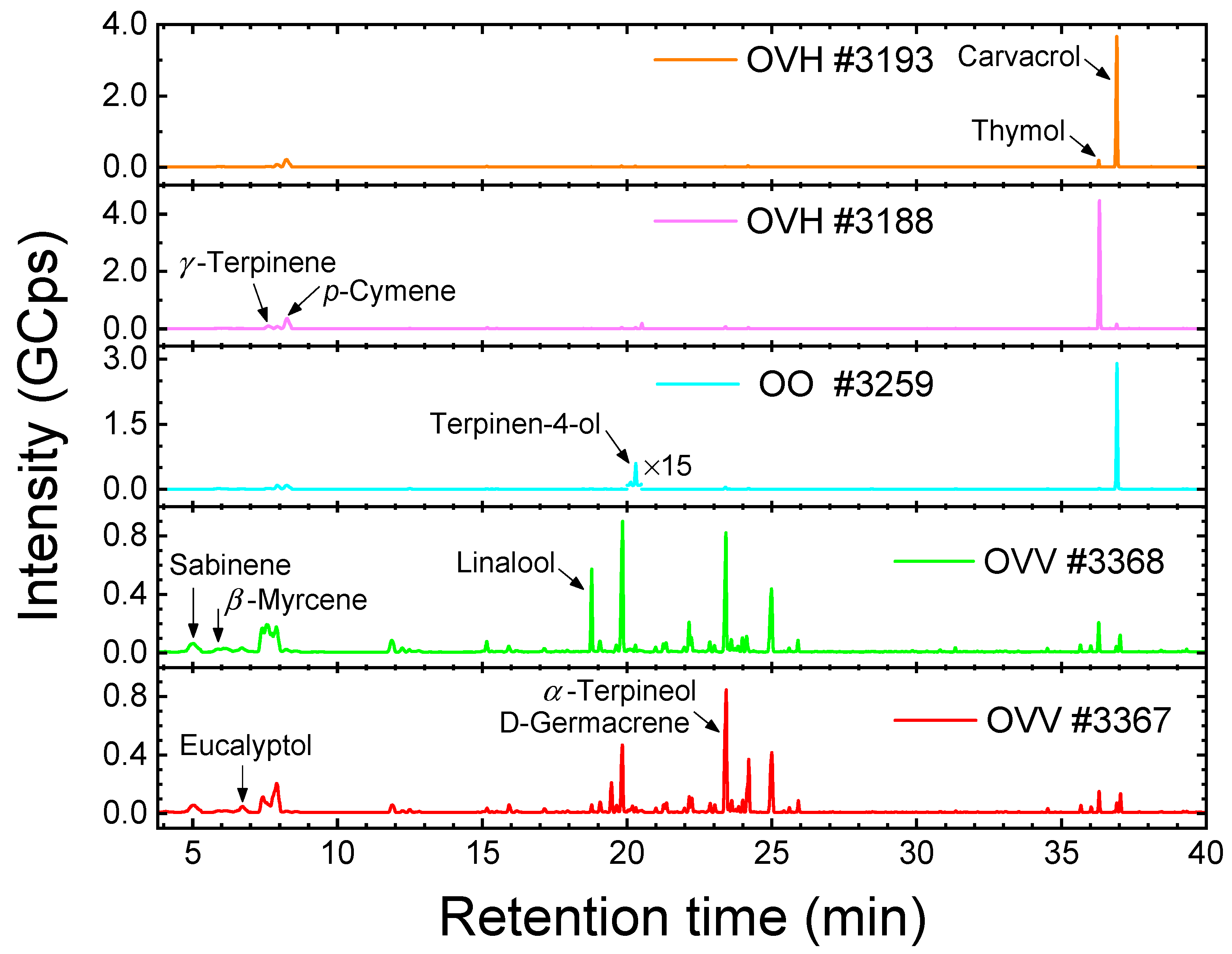
3.3.3. Headspace Gas Chromatography–Mass Spectrometry
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dhifi, W.; Bellili, S.; Jazi, S.; Bahloul, N.; Mnif, W. Essential Oils’ Chemical Characterization and Investigation of Some Biological Activities: A Critical Review. Medicines 2016, 3, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palla, F.; Bruno, M.; Mercurio, F.; Tantillo, A.; Rotolo, V. Essential Oils as Natural Biocides in Conservation of Cultural Heritage. Molecules 2020, 25, 730. [Google Scholar] [CrossRef] [Green Version]
- Guzmán, E.; Lucia, A. Essential Oils and Their Individual Components in Cosmetic Products. Cosmetics 2021, 8, 114. [Google Scholar] [CrossRef]
- Napoli, E.; Giovino, A.; Carrubba, A.; How Yuen Siong, V.; Rinoldo, C.; Nina, O.; Ruberto, G. Variations of Essential Oil Constituents in Oregano (Origanum vulgare subsp. viridulum (= O. heracleoticum) over Cultivation Cycles. Plants 2020, 9, 1174. [Google Scholar] [CrossRef] [PubMed]
- Aminkhah, M.; Asgarpanah, J. GC-MS analysis of the essential oil from Artemisia aucheri Boiss. Fruits. J. Chil. Chem. Soc. 2017, 62, 3581. [Google Scholar] [CrossRef] [Green Version]
- Long, D.A. The Raman Effect: A Unified Treatment of the Theory of Raman Scattering by Molecules; John Wiley & Sons Ltd.: West Sussex, UK, 2002; ISBN 978-0-471-49028-9. [Google Scholar]
- Vandenabeele, P. Practical Raman Spectroscopy; John Wiley & Sons Ltd.: West Sussex, UK, 2013. [Google Scholar] [CrossRef]
- Jentzsch, P.V.; Ciobotă, V. Raman spectroscopy as an analytical tool for analysis of vegetable and essential oils. Flavour Fragr. J. 2014, 29, 287. [Google Scholar] [CrossRef]
- Cebi, N. Chemical Fingerprinting of the Geranium (Pelargonium graveolens) Essential Oil by Using FTIR, Raman and GC-MS Techniques. Eur. J. Sci. Technol. 2021, 25, 810. [Google Scholar] [CrossRef]
- Schulz, H.; Özkan, G.; Baranska, M.; Krüger, H.; Özcan, M. Characterisation of essential oil plants from Turkey by IR and Raman spectroscopy. Vib. Spectrosc. 2005, 39, 249. [Google Scholar] [CrossRef]
- Schulz, H.; Baranska, M. Identification and quantification of valuable plant substances by IR and Raman spectroscopy. Vib. Spectrosc. 2007, 43, 13. [Google Scholar] [CrossRef]
- Jentzsch, P.V.; Pauker, C.S.; Pozo, P.Z.; Serra, M.S.; Camacho, G.J.; Rueda-Ayala, V.; Garrido, P.; Guerrero, L.R.; Ciobot, V. Raman spectroscopy in the detection of adulterated essential oils: The case of nonvolatile adulterants. J. Raman Spectrosc. 2021, 52, 1055. [Google Scholar] [CrossRef]
- Lafhal, S.; Vanloot, P.; Bombarda, I.; Valls, R.; Kister, J.; Dupuy, N. Raman spectroscopy for identification and quantification analysis of essential oil varieties: A multivariate approach applied to lavender and lavandin essential oils. J. Raman Spectrosc. 2015, 46, 577. [Google Scholar] [CrossRef]
- Boyaci, I.H.; Genis, H.E.; Guven, B.; Tamer, U.; Alper, N. A novel method for quantification of ethanol and methanol in distilled alcoholic beverages using Raman spectroscopy. J. Raman Spectrosc. 2012, 43, 1171. [Google Scholar] [CrossRef]
- Papaspyridakou, P.; Lykouras, M.; Kontoyannis, C.; Orkoula, M. Comparative Study of Sample Carriers for the Identification of Volatile Compounds in Biological Fluids Using Raman Spectroscopy. Molecules 2022, 27, 3279. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Uragami, N.; Miyazaki, T.; Yokoyama, N.; Inoue, H. Raman spectroscopic evaluation of human serum using metal plate and 785- and 1064-nm excitation lasers. PLoS ONE 2019, 14, e0211986. [Google Scholar] [CrossRef]
- Hanif, M.A.; Nawaz, H.; Naz, S.; Mukhtar, R.; Rashid, N.; Bhatti, I.A.; Saleem, M. Raman spectroscopy for the characterization of different fractions of hemp essential oil extracted at 130 °C using steam distillation method. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2017, 182, 16. [Google Scholar] [CrossRef]
- Baranska, M.; Schulz, H.; Krüger, H.; Quilitzsch, R. Chemotaxonomy of aromatic plants of the genus Origanum via vibrational spectroscopy. Anal. Bioanal. Chem. 2005, 381, 1241. [Google Scholar] [CrossRef]
- Siatis, N.G.; Kimbaris, A.C.; Pappas, C.S.; Tarantilis, P.A.; Daferera, D.J.; Polissiou, M.G. Rapid Method for Simultaneous Quantitative Determination of Four Major Essential Oil Components from Oregano (Oreganum sp.) and Thyme (Thymus sp.) Using FT-Raman Spectroscopy. J. Agric. Food Chem. 2005, 53, 202. [Google Scholar] [CrossRef]
- Jentzsch, P.V.; Ramos, L.A.; Ciobotă, V. Handheld Raman Spectroscopy for the Distinction of Essential Oils Used in the Cosmetics Industry. Cosmetics 2015, 2, 162. [Google Scholar] [CrossRef] [Green Version]
- Deidda, R.; Sacre, P.-Y.; Clavaud, M.; Coïc, L.; Avohou, H.; Hubert, P.; Ziemons, E. Vibrational spectroscopy in analysis of pharmaceuticals: Critical review of innovative portable and handheld NIR and Raman spectrophotometers. Trends Anal. Chem. 2019, 114, 251. [Google Scholar] [CrossRef]
- Jehlicka, J.; Culka, A. Critical evaluation of portable Raman spectrometers: From rock outcrops and planetary analogs to cultural heritage—A review. Anal. Chim. Acta 2022, 1209, 339027. [Google Scholar] [CrossRef]
- Farber, C.; Sanchez, L.; Kurouski, D. Confirmatory non-invasive and non-destructive identification of poison ivy using a hand-held Raman spectrometer. RSC Adv. 2020, 10, 21530. [Google Scholar] [CrossRef] [PubMed]
- Payne, W.Z.; Kurouski, D. Raman spectroscopy enables phenotyping and assessment of nutrition values of plants: A review. Plant Methods 2021, 17, 78. [Google Scholar] [CrossRef] [PubMed]
- Butler, H.J.; McAinsh, M.R.; Adams, S.; Martin, F.L. Application of vibrational spectroscopy techniques to non-destructively monitor plant health and development. Anal. Methods 2015, 7, 4059. [Google Scholar] [CrossRef] [Green Version]
- Daferera, D.; Pappas, C.; Tarantilis, P.A.; Polissiou, M. Quantitative analysis of α-pinene and β-myrcene in mastic gum oil using FT-Raman spectroscopy. Food Chem. 2002, 7, 511. [Google Scholar] [CrossRef]
- Khoshroo, H.; Khadem, H.; Bahreini, M.; Tavassoli, S.H.; Hadian, J. Quantitative analysis of essential oils of Thymus daenensis using laser-induced fluorescence and Raman spectroscopy. Appl. Opt. 2015, 54, 9533. [Google Scholar] [CrossRef] [PubMed]
- Mertzanidis, D.; Nakas, A.; Assimopoulou, A.N.; Kokkini, S. Unravelling the Chemotaxonomic Identity of “White” and “Black” Oregano (Origanum vulgare) in Northern Greece. Planta Med. 2022. [Google Scholar] [CrossRef] [PubMed]
- Ietswaart, J.H. A Taxonomic Revision of the Genus Origanum (Labiatae); Leiden University Press: The Hague, The Netherlands; Boston, MA, USA; London, UK, 1980; pp. 85–87, 106–123. [Google Scholar]
- Fernandes, R.; Heywood, V.H. Origanum L. In Flora Europaea; Tutin, T.G., Heywood, V.H., Burges, N.A., Moore, D.M., Valentine, D.H., Walters, S.M., Webb, D.A., Eds.; Cambridge University Press: Cambridge, UK, 1972; Volume 3, pp. 171–172. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Locality | Taxon | Geographical Coordinates | Altitude (m) | Essential Oil Content | Collection Code, Date | |
|---|---|---|---|---|---|---|
| N (Latitude) | E (Longitude) | |||||
| Kassandra Peninsula | O. vulgare subsp. hirtum (Link) Ietsw. | 40.003745 | 23.419388 | 75 | 5.46 | #3188, 17 August 2015 |
| Mount Pelion | O. vulgare subsp.hirtum | 39.392600 | 22.981425 | 277 | 6.59 | #3193, 10 August 2015 |
| Island of Kos | O. onites L. | 36.842372 | 27.206357 | 380 | 4.40 | #3259, 28 July 2018 |
| Mount Belles | O. vulgare L. subsp. vulgare | 41.339199 | 23.232680 | 1299 | 0.60 | #3367, 13 July 2021 |
| Mount Belles | O. vulgare L. subsp. vulgare | 41.342830 | 23.237160 | 1177 | 0.50 | #3368, 13 July 2021 |
| HS GC-MS % of All Identified Compounds | HS GC-MS % of the Available Raman Reference Compounds | Raman % | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Constituent | Sample | ||||||||
| #3188 | #3193 | #3259 | #3188 | #3193 | #3259 | #3188 | #3193 | #3259 | |
| Carvacrol | 2.2 | 72.2 | 80.7 | 2.4 | 77.1 | 85.9 | 4.4 | 79.7 | 89.0 |
| Thymol | 64.5 | 3.3 | 0.4 | 71.7 | 3.5 | 0.4 | 69.8 | 1.4 | 0.4 |
| p-Cymene | 16.6 | 14.7 | 9.0 | 18.5 | 15.7 | 9.6 | 12.9 | 10.6 | 3.5 |
| γ-Terpinene | 4.9 | 1.6 | 1.7 | 5.5 | 1.7 | 1.8 | 3.7 | 0.0 | 0.0 |
| β-Myrcene | 0.8 | 0.9 | 1.0 | 0.9 | 1.0 | 1.1 | 1.3 | 3.0 | 2.0 |
| Sabinene | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 1.2 | 0.7 | 0.6 |
| α-Terpineol | 0.1 | 0.1 | 0.1 | 0.1 | 0.1 | 0.1 | 0.2 | 0.0 | 0.0 |
| Eucalyptol | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.5 | 0.1 | 0.5 |
| Linalool | 0.1 | 0.2 | 0.0 | 0.1 | 0.2 | 0.0 | 4.2 | 1.9 | 1.6 |
| Terpinen-4-ol | 0.7 | 0.7 | 1.1 | 0.8 | 0.7 | 1.2 | 1.1 | 1.9 | 1.7 |
| α-Pinene | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.7 | 0.9 | 0.6 |
| Linalyl acetate | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| Sum | 89.9 | 93.7 | 94.0 | 100.0 | 100.0 | 100.1 | 100.0 | 100.2 | 99.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kampasakali, E.; Nakas, A.; Mertzanidis, D.; Kokkini, S.; Assimopoulou, A.N.; Christofilos, D. μ-Raman Determination of Essential Oils’ Constituents from Distillates and Leaf Glands of Origanum Plants. Molecules 2023, 28, 1221. https://doi.org/10.3390/molecules28031221
Kampasakali E, Nakas A, Mertzanidis D, Kokkini S, Assimopoulou AN, Christofilos D. μ-Raman Determination of Essential Oils’ Constituents from Distillates and Leaf Glands of Origanum Plants. Molecules. 2023; 28(3):1221. https://doi.org/10.3390/molecules28031221
Chicago/Turabian StyleKampasakali, Elli, Alexandros Nakas, Dimitrios Mertzanidis, Stella Kokkini, Andreana N. Assimopoulou, and Dimitrios Christofilos. 2023. "μ-Raman Determination of Essential Oils’ Constituents from Distillates and Leaf Glands of Origanum Plants" Molecules 28, no. 3: 1221. https://doi.org/10.3390/molecules28031221