
PROTACs in Epigenetic Cancer Therapy: Current Status and Future Opportunities
Abstract
:1. Introduction
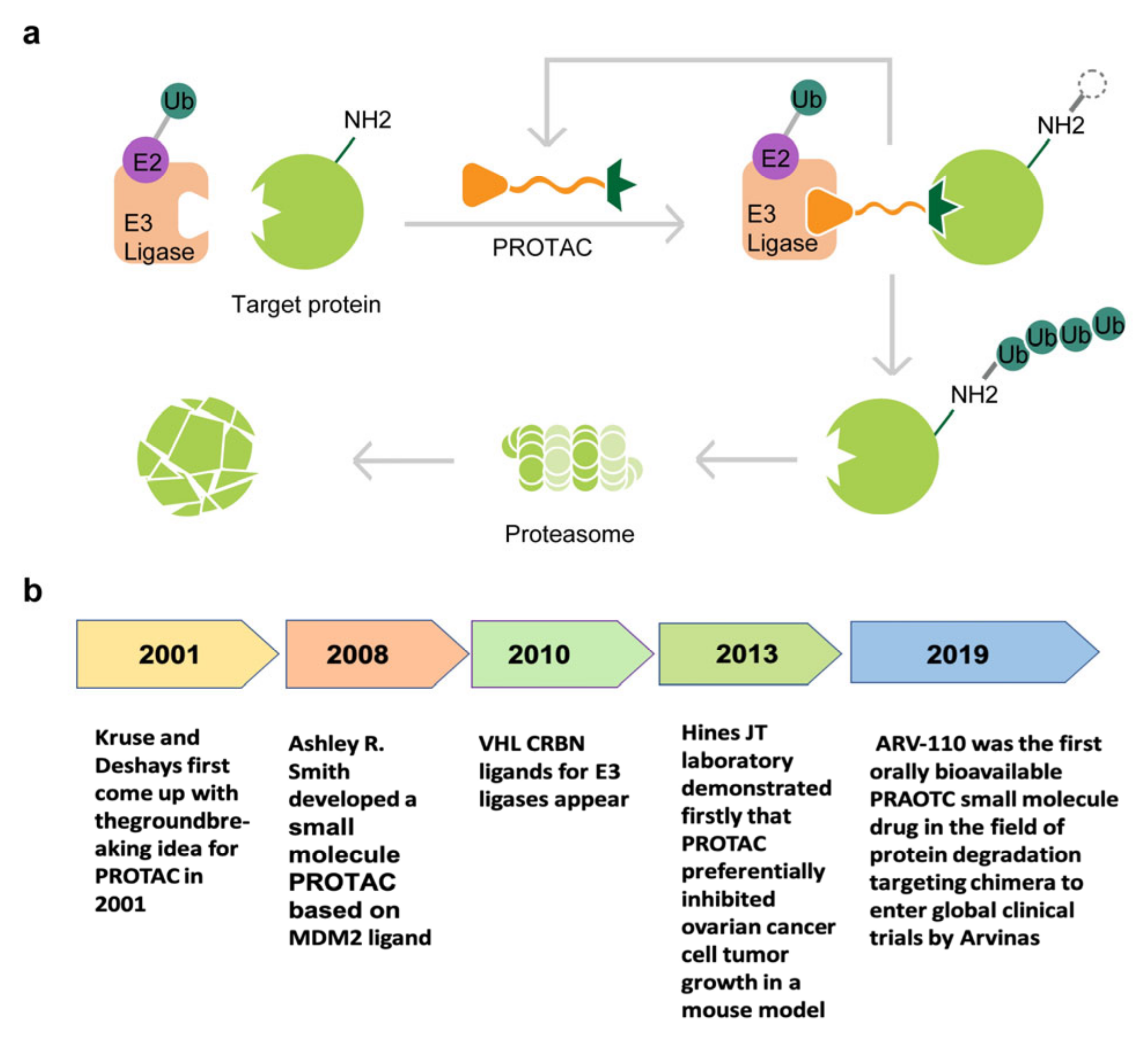
2. Development of PROTACs
3. Epigenetic Regulation in Cancer Therapy
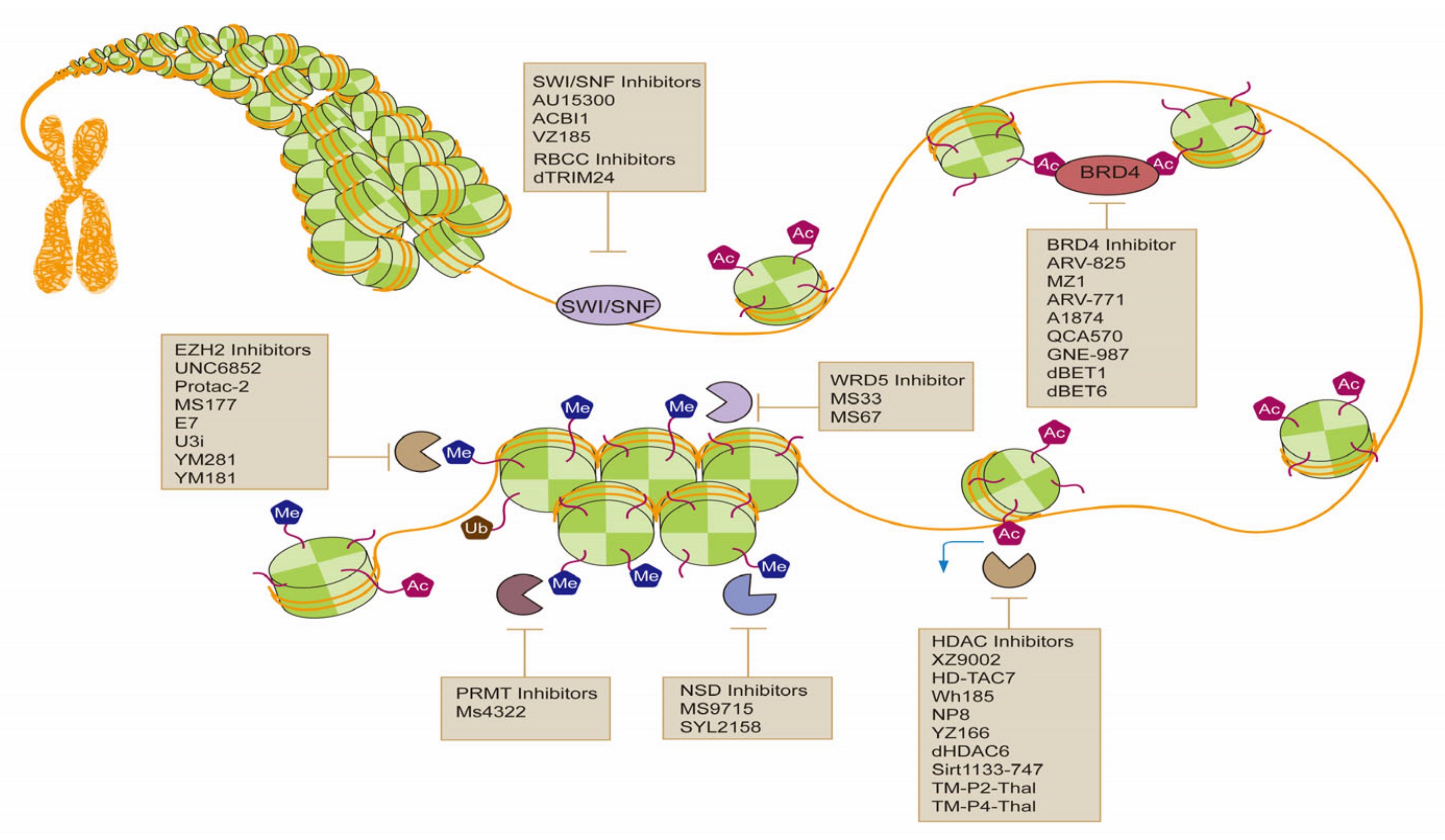
4. PROTACs for Epigenetic Targets in Cancer
4.1. Histone Acetylation
4.1.1. ARV-825
4.1.2. MZ1
4.1.3. ARV-771
4.1.4. dBET1/6
4.1.5. Other Histone Acetylation PROTACs
4.2. HDACs
4.2.1. HDAC6
4.2.2. HDAC3
4.2.3. Sirtuin 2
4.3. Histone Methylation
4.3.1. NSD3
4.3.2. PRC2
4.3.3. PRMT5
4.3.4. WRD5
4.4. Chromatin Remodeling
4.4.1. SMARCA2/SMARCA4
4.4.2. BRD9
4.4.3. TRIM24
4.4.4. ARID1A
5. Summary and Future Prospects
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Bird, A. Perceptions of epigenetics. Nature 2007, 447, 396–398. [Google Scholar] [CrossRef] [PubMed]
- Toh, T.B.; Lim, J.J.; Chow, E.K.-H. Epigenetics in cancer stem cells. Mol. Cancer 2017, 16, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nebbioso, A.; Tambaro, F.P.; Dell’Aversana, C.; Altucci, L. Cancer epigenetics: Moving forward. PLOS Genet. 2018, 14, e1007362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Zhang, H.; Gao, P. Metabolic reprogramming and epigenetic modifications on the path to cancer. Protein Cell 2021, 13, 877–919. [Google Scholar] [CrossRef]
- Zhang, L.; Lu, Q.; Chang, C. Epigenetics in Health and Disease. Adv. Exp. Med. Biol. 2020, 1253, 3–55. [Google Scholar] [CrossRef]
- Li, X.; Song, Y. Proteolysis-targeting chimera (PROTAC) for targeted protein degradation and cancer therapy. J. Hematol. Oncol. 2020, 13, 50. [Google Scholar] [CrossRef]
- Békés, M.; Langley, D.R.; Crews, C.M. PROTAC targeted protein degraders: The past is prologue. Nat. Rev. Drug Discov. 2022, 21, 181–200. [Google Scholar] [CrossRef]
- Gadd, M.S.; Testa, A.; Lucas, X.; Chan, K.H.; Chen, W.Z.; Lamont, D.J.; Zengerle, M.; Ciulli, A. Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat. Chem. Biol. 2017, 13, 514–521. [Google Scholar] [CrossRef]
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: Chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, K.M.; Kim, K.B.; Verma, R.; Ransick, A.; Stein, B.; Crews, C.M.; Deshaies, R. Development of Protacs to Target Cancer-promoting Proteins for Ubiquitination and Degradation. Mol. Cell. Proteom. 2003, 2, 1350–1358. [Google Scholar] [CrossRef]
- Schneekloth, A.R.; Pucheault, M.; Tae, H.S.; Crews, C.M. Targeted intracellular protein degradation induced by a small molecule: En route to chemical proteomics. Bioorganic Med. Chem. Lett. 2008, 18, 5904–5908. [Google Scholar] [CrossRef] [Green Version]
- Itoh, Y.; Ishikawa, M.; Naito, M.; Hashimoto, Y. Protein Knockdown Using Methyl Bestatin−Ligand Hybrid Molecules: Design and Synthesis of Inducers of Ubiquitination-Mediated Degradation of Cellular Retinoic Acid-Binding Proteins. J. Am. Chem. Soc. 2010, 132, 5820–5826. [Google Scholar] [CrossRef]
- Ito, T.; Ando, H.; Suzuki, T.; Ogura, T.; Hotta, K.; Imamura, Y.; Yamaguchi, Y.; Handa, H. Identification of a Primary Target of Thalidomide Teratogenicity. Science 2010, 327, 1345–1350. [Google Scholar] [CrossRef] [Green Version]
- Buckley, D.L.; Gustafson, J.L.; Van Molle, I.; Roth, A.G.; Tae, H.S.; Gareiss, P.C.; Jorgensen, W.L.; Ciulli, A.; Crews, C.M. Small-Molecule Inhibitors of the Interaction between the E3 Ligase VHL and HIF1α. Angew. Chem. Int. Ed. 2012, 51, 11463–11467. [Google Scholar] [CrossRef] [Green Version]
- Hines, J.; Gough, J.D.; Corson, T.W.; Crews, C.M. Posttranslational protein knockdown coupled to receptor tyrosine kinase activation with phosphoPROTACs. Proc. Natl. Acad. Sci. USA 2013, 110, 8942–8947. [Google Scholar] [CrossRef] [Green Version]
- Cavalli, G.; Heard, E. Advances in epigenetics link genetics to the environment and disease. Nature 2019, 571, 489–499. [Google Scholar] [CrossRef] [Green Version]
- Jones, P.A.; Baylin, S.B. The epigenomics of cancer. Cell 2007, 128, 683–692. [Google Scholar] [CrossRef] [Green Version]
- Witte, T.; Plass, C.; Gerhauser, C. Pan-cancer patterns of DNA methylation. Genome Med. 2014, 6, 66. [Google Scholar] [CrossRef] [Green Version]
- Hoadley, K.A.; Yau, C.; Wolf, D.M.; Cherniack, A.D.; Tamborero, D.; Ng, S.; Leiserson, M.D.M.; Niu, B.; McLellan, M.D.; Uzunangelov, V.; et al. Multiplatform Analysis of 12 Cancer Types Reveals Molecular Classification within and across Tissues of Origin. Cell 2014, 158, 929–944. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Shen, S.; Kendig, D.M.; Mahavadi, S.; Murthy, K.S.; Grider, J.R.; Qiao, L.-Y. Inhibition of NMDAR Reduces Bladder Hypertrophy and Improves Bladder Function in Cyclophosphamide Induced Cystitis. J. Urol. 2015, 193, 1676–1683. [Google Scholar] [CrossRef]
- Roberti, A.; Valdes, A.F.; Torrecillas, R.; Fraga, M.F.; Fernandez, A.F. Epigenetics in cancer therapy and nanomedicine. Clin. Epigenetics 2019, 11, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Topper, M.J.; Vaz, M.; Marrone, K.A.; Brahmer, J.R.; Baylin, S.B. The emerging role of epigenetic therapeutics in immuno-oncology. Nat. Rev. Clin. Oncol. 2020, 17, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Skourti, E.; Dhillon, P. Cancer epigenetics: Promises and pitfalls for cancer therapy. FEBS J. 2022, 289, 1156–1159. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.S. Epigenetics of glioblastoma multiforme: From molecular mechanisms to therapeutic approaches. Semin. Cancer Biol. 2022, 83, 100–120. [Google Scholar] [CrossRef]
- Jerónimo, C.; Bastian, P.J.; Bjartell, A.; Carbone, G.M.; Catto, J.W.F.; Clark, S.J.; Henrique, R.; Nelson, W.G.; Shariat, S.F. Epigenetics in Prostate Cancer: Biologic and Clinical Relevance. Eur. Urol. 2011, 60, 753–766. [Google Scholar] [CrossRef]
- Belkina, A.; Denis, G.V. BET domain co-regulators in obesity, inflammation and cancer. Nat. Rev. Cancer 2012, 12, 465–477. [Google Scholar] [CrossRef] [Green Version]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef] [Green Version]
- French, C.A.; Ramirez, C.L.; Kolmakova, J.; Hickman, T.T.; Cameron, M.J.; Thyne, M.E.; Kutok, J.L.; Toretsky, J.A.; Tadavarthy, A.K.; Kees, U.R.; et al. BRD–NUT oncoproteins: A family of closely related nuclear proteins that block epithelial differentiation and maintain the growth of carcinoma cells. Oncogene 2008, 27, 2237–2242. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Gao, C.; Liu, M.; Liu, Y.-C.; Kwon, J.; Qi, J.; Tian, X.; Stein, A.; Liu, Y.V.; Kong, N.R.; et al. Targeting an Inducible SALL4-Mediated Cancer Vulnerability with Sequential Therapy. Cancer Res 2021, 81, 6018–6028. [Google Scholar] [CrossRef]
- Weng, G.; Cai, X.; Cao, D.; Du, H.; Shen, C.; Deng, Y.; He, Q.; Yang, B.; Li, D.; Hou, T. PROTAC-DB 2.0: An updated database of PROTACs. Nucleic Acids Res. 2023, 51, D1367–D1372. [Google Scholar] [CrossRef]
- Weng, G.; Shen, C.; Cao, D.; Gao, J.; Dong, X.; He, Q.; Yang, B.; Li, D.; Wu, J.; Hou, T. PROTAC-DB: An online database of PROTACs. Nucleic Acids Res. 2021, 49, D1381–D1387. [Google Scholar] [CrossRef]
- Lu, J.; Qian, Y.; Altieri, M.; Dong, H.; Wang, J.; Raina, K.; Hines, J.; Winkler, J.D.; Crew, A.P.; Coleman, K.; et al. Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem. Biol. 2015, 22, 755–763. [Google Scholar] [CrossRef] [Green Version]
- Sun, B.; Fiskus, W.; Qian, Y.; Rajapakshe, K.; Raina, K.; Coleman, K.G.; Crew, A.P.; Shen, A.; Saenz, D.T.; Mill, C.P.; et al. BET protein proteolysis targeting chimera (PROTAC) exerts potent lethal activity against mantle cell lymphoma cells. Leukemia 2017, 32, 343–352. [Google Scholar] [CrossRef]
- Zhang, K.; Gao, L.; Wang, J.; Chu, X.; Zhang, Z.; Zhang, Y.; Fang, F.; Tao, Y.; Li, X.; Tian, Y.; et al. A Novel BRD Family PROTAC Inhibitor dBET1 Exerts Great Anti-Cancer Effects by Targeting c-MYC in Acute Myeloid Leukemia Cells. Pathol. Oncol. Res. 2022, 28, 1610447. [Google Scholar] [CrossRef]
- Lu, Q.; Ding, X.; Huang, T.; Zhang, S.; Li, Y.; Xu, L.; Chen, G.; Ying, Y.; Wang, Y.; Feng, Z.; et al. BRD4 degrader ARV-825 produces long-lasting loss of BRD4 protein and exhibits potent efficacy against cholangiocarcinoma cells. Am. J. Transl. Res. 2019, 11, 5728–5739. [Google Scholar]
- He, L.; Chen, C.; Gao, G.; Xu, K.; Ma, Z. ARV-825-induced BRD4 protein degradation as a therapy for thyroid carcinoma. Aging 2020, 12, 4547–4557. [Google Scholar] [CrossRef]
- Liao, X.; Qian, X.; Zhang, Z.; Tao, Y.; Li, Z.; Zhang, Q.; Liang, H.; Li, X.; Xie, Y.; Zhuo, R.; et al. ARV-825 Demonstrates Antitumor Activity in Gastric Cancer via MYC-Targets and G2M-Checkpoint Signaling Pathways. Front. Oncol. 2021, 11, 753119. [Google Scholar] [CrossRef]
- Lim, S.-L.; Xu, L.; Han, B.-C.; Shyamsunder, P.; Chng, W.-J.; Koeffler, H.P. Multiple myeloma: Combination therapy of BET proteolysis targeting chimeric molecule with CDK9 inhibitor. PLoS ONE 2020, 15, e0232068. [Google Scholar] [CrossRef]
- Li, Z.; Lim, S.L.; Tao, Y.; Li, X.; Xie, Y.; Yang, C.; Zhang, Z.; Jiang, Y.; Zhang, X.; Cao, X.; et al. PROTAC Bromodomain Inhibitor ARV-825 Displays Anti-Tumor Activity in Neuroblastoma by Repressing Expression of MYCN or c-Myc. Front. Oncol. 2020, 10, 574525. [Google Scholar] [CrossRef]
- Noblejas-López, M.D.M.; Jiménez, C.N.; Burgos, M.; Gómez-Juárez, M.; Montero, J.C.; Esparís-Ogando, A.; Pandiella, A.; Galán-Moya, E.M.; Ocaña, A. Activity of BET-proteolysis targeting chimeric (PROTAC) compounds in triple negative breast cancer. J. Exp. Clin. Cancer Res. 2019, 38, 383. [Google Scholar] [CrossRef] [Green Version]
- Qiao, L.; Shen, S.; Liu, M.; Xia, C.; Kay, J.; Zhang, Q. Inflammation and activity augment brain-derived neurotrophic factor peripheral release. Neuroscience 2016, 318, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Z.; Xia, C.; Shen, S.; Corwin, F.D.; Liu, M.; Guan, R.; Grider, J.R.; Qiao, L.-Y. Suppression of the PI3K Pathway In Vivo Reduces Cystitis-Induced Bladder Hypertrophy and Restores Bladder Capacity Examined by Magnetic Resonance Imaging. PLoS ONE 2014, 9, e114536. [Google Scholar] [CrossRef] [PubMed]
- Rathod, D.; Fu, Y.; Patel, K. BRD4 PROTAC as a novel therapeutic approach for the treatment of vemurafenib resistant melanoma: Preformulation studies, formulation development and in vitro evaluation. Eur. J. Pharm. Sci. 2019, 138, 105039. [Google Scholar] [CrossRef] [PubMed]
- Saraswat, A.; Patki, M.; Fu, Y.; Barot, S.; Dukhande, V.V.; Patel, K. Nanoformulation of PROteolysis TArgeting Chimera targeting ‘undruggable’ c-Myc for the treatment of pancreatic cancer. Nanomedicine 2020, 15, 1761–1777. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Hu, Y.; Miao, J.; Chen, J.; Liu, J.; Cheng, Y.; Gao, X. A BRD4 PROTAC nanodrug for glioma therapy via the intervention of tumor cells proliferation, apoptosis and M2 macrophages polarization. Acta Pharm. Sin. B 2022, 12, 2658–2671. [Google Scholar] [CrossRef]
- Minko, T. Nanoformulation of BRD4-Degrading PROTAC: Improving Druggability to Target the ‘Undruggable’ MYC in Pancreatic Cancer. Trends Pharmacol. Sci. 2020, 41, 684–686. [Google Scholar] [CrossRef]
- Fu, Y.; Saraswat, A.; Wei, Z.; Agrawal, M.; Dukhande, V.; Reznik, S.; Patel, K. Development of Dual ARV-825 and Nintedanib-Loaded PEGylated Nano-Liposomes for Synergistic Efficacy in Vemurafnib-Resistant Melanoma. Pharmaceutics 2021, 13, 1005. [Google Scholar] [CrossRef]
- Vartak, R.; Saraswat, A.; Yang, Y.; Chen, Z.-S.; Patel, K. Susceptibility of Lung Carcinoma Cells to Nanostructured Lipid Carrier of ARV-825, a BRD4 Degrading Proteolysis Targeting Chimera. Pharm. Res. 2022, 39, 2745–2759. [Google Scholar] [CrossRef]
- Saraswat, A.; Vemana, H.P.; Dukhande, V.V.; Patel, K. Galactose-decorated liver tumor-specific nanoliposomes incorporating selective BRD4-targeted PROTAC for hepatocellular carcinoma therapy. Heliyon 2022, 8, e08702. [Google Scholar] [CrossRef]
- Zengerle, M.; Chan, K.-H.; Ciulli, A. Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem. Biol. 2015, 10, 1770–1777. [Google Scholar] [CrossRef] [Green Version]
- Tarantelli, C.; Cannas, E.; Ekeh, H.; Moscatello, C.; Gaudio, E.; Cascione, L.; Napoli, S.; Rech, C.; Testa, A.; Maniaci, C.; et al. The bromodomain and extra-terminal domain degrader MZ1 exhibits preclinical anti-tumoral activity in diffuse large B-cell lymphoma of the activated B cell-like type. Explor. Target. Anti-Tumor Ther. 2021, 2, 586–601. [Google Scholar] [CrossRef]
- Zhang, X.; Guo, X.; Zhuo, R.; Tao, Y.; Liang, W.; Yang, R.; Chen, Y.; Cao, H.; Jia, S.; Yu, J.; et al. BRD4 inhibitor MZ1 exerts anti-cancer effects by targeting MYCN and MAPK signaling in neuroblastoma. Biochem. Biophys. Res. Commun. 2022, 604, 63–69. [Google Scholar] [CrossRef]
- Otto, C.; Schmidt, S.; Kastner, C.; Denk, S.; Kettler, J.; Müller, N.; Germer, C.; Wolf, E.; Gallant, P.; Wiegering, A. Targeting bromodomain-containing protein 4 (BRD4) inhibits MYC expression in colorectal cancer cells. Neoplasia 2019, 21, 1110–1120. [Google Scholar] [CrossRef]
- Noblejas-López, M.D.M.; Nieto-Jiménez, C.; Galán-Moya, E.M.; Tebar-García, D.; Montero, J.C.; Pandiella, A.; Burgos, M.; Ocaña, A. MZ1 co-operates with trastuzumab in HER2 positive breast cancer. J. Exp. Clin. Cancer Res. 2021, 40, 106. [Google Scholar] [CrossRef]
- Cimas, F.; Niza, E.; Juan, A.; Noblejas-López, M.; Bravo, I.; Lara-Sanchez, A.; Alonso-Moreno, C.; Ocaña, A. Controlled Delivery of BET-PROTACs: In Vitro Evaluation of MZ1-Loaded Polymeric Antibody Conjugated Nanoparticles in Breast Cancer. Pharmaceutics 2020, 12, 986. [Google Scholar] [CrossRef]
- Raina, K.; Lu, J.; Qian, Y.; Altieri, M.; Gordon, D.; Rossi, A.M.K.; Wang, J.; Chen, X.; Dong, H.; Siu, K.; et al. PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 7124–7129. [Google Scholar] [CrossRef] [Green Version]
- Kitamura, T.; Suzuki, M.; Nishimatsu, H.; Kurosaki, T.; Enomoto, Y.; Fukuhara, H.; Kume, H.; Takeuchi, T.; Miao, L.; Jiangang, H.; et al. Final Report on Low-Dose Estramustine Phosphate (EMP) Monotherapy and Very Low-Dose EMP Therapy combined with LH-RH Agonist for Previously Untreated Advanced Prostate Cancer. Aktuel Urol. 2010, 41, S34–S40. [Google Scholar] [CrossRef]
- Deng, Y.; Yu, C.; Chen, L.; Zhang, X.; Lei, Q.; Liu, Q.; Cai, G.; Liu, F. ARV-771 Acts as an Inducer of Cell Cycle Arrest and Apoptosis to Suppress Hepatocellular Carcinoma Progression. Front. Pharmacol. 2022, 13, 858901. [Google Scholar] [CrossRef]
- Wang, J. BRD4-IRF1 axis regulates chemoradiotherapy-induced PD-L1 expression and immune evasion in non-small cell lung cancer. Clin. Transl. Med. 2022, 12, e718. [Google Scholar] [CrossRef]
- Jiang, Y.Y. TP63, SOX2, and KLF5 Establish a Core Regulatory Circuitry That Controls Epigenetic and Transcription Patterns in Esophageal Squamous Cell Carcinoma Cell Lines. Gastroenterology 2020, 159, 1311–1327.e19. [Google Scholar] [CrossRef]
- Liu, J.; Chen, H.; Liu, Y.; Shen, Y.; Meng, F.; Kaniskan, H.; Jin, J.; Wei, W. Cancer Selective Target Degradation by Folate-Caged PROTACs. J. Am. Chem. Soc. 2021, 143, 7380–7387. [Google Scholar] [CrossRef] [PubMed]
- Winter, G.E.; Buckley, D.L.; Paulk, J.; Roberts, J.M.; Souza, A.; Dhe-Paganon, S.; Bradner, J.E. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015, 348, 1376–1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, K.; Berger, D.; Zielinski, C.C.; Valent, P.; Grunt, T.W. Hitting two oncogenic machineries in cancer cells: Cooperative effects of the multi-kinase inhibitor ponatinib and the BET bromodomain blockers JQ1 or dBET1 on human carcinoma cells. Oncotarget 2018, 9, 26491–26506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Chen, H.; Ma, L.; He, Z.; Wang, D.; Liu, Y.; Lin, Q.; Zhang, T.; Gray, N.; Kaniskan, H.Ü.; et al. Light-induced control of protein destruction by opto-PROTAC. Sci. Adv. 2020, 6, eaay5154. [Google Scholar] [CrossRef] [Green Version]
- Winter, G.E.; Mayer, A.; Buckley, D.L.; Erb, M.A.; Roderick, J.E.; Vittori, S.; Reyes, J.M.; di Iulio, J.; Souza, A.; Ott, C.J.; et al. BET Bromodomain Proteins Function as Master Transcription Elongation Factors Independent of CDK9 Recruitment. Mol. Cell 2017, 67, 5–18.e19. [Google Scholar] [CrossRef] [Green Version]
- Peter, B.; Eisenwort, G.; Sadovnik, I.; Bauer, K.; Willmann, M.; Rülicke, T.; Berger, D.; Stefanzl, G.; Greiner, G.; Hoermann, G.; et al. BRD4 degradation blocks expression of MYC and multiple forms of stem cell resistance in Ph + chronic myeloid leukemia. Am. J. Hematol. 2022, 97, 1215–1225. [Google Scholar] [CrossRef]
- Bauer, K.; Berghoff, A.S.; Preusser, M.; Heller, G.; Zielinski, C.C.; Valent, P.; Grunt, T.W. Degradation of BRD4—A promising treatment approach not only for hematologic but also for solid cancer. Am. J. Cancer Res. 2021, 11, 530–545. [Google Scholar]
- Xu, L.; Chen, Y.; Mayakonda, A.; Koh, L.; Chong, Y.K.; Buckley, D.L.; Sandanaraj, E.; Lim, S.W.; Lin, R.Y.-T.; Ke, X.-Y.; et al. Targetable BET proteins- and E2F1-dependent transcriptional program maintains the malignancy of glioblastoma. Proc. Natl. Acad. Sci. USA 2018, 115, E5086–E5095. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Peng, R.; Chen, S.; Shen, A.; Zhao, L.; Tang, W.; Wang, X.; Li, Z.; Zha, Z.; Yi, M.; et al. Versatile Nano-PROTAC-Induced Epigenetic Reader Degradation for Efficient Lung Cancer Therapy. Adv. Sci. 2022, 9, e2202239. [Google Scholar] [CrossRef]
- Hines, J.; Lartigue, S.; Dong, H.; Qian, Y.; Crews, C.M. MDM2-Recruiting PROTAC Offers Superior, Synergistic Antiproliferative Activity via Simultaneous Degradation of BRD4 and Stabilization of p53. Cancer Res 2019, 79, 251–262. [Google Scholar] [CrossRef] [Green Version]
- Qin, A.-C.; Jin, H.; Song, Y.; Gao, Y.; Chen, Y.-F.; Zhou, L.-N.; Wang, S.-S.; Lu, X.-S. The therapeutic effect of the BRD4-degrading PROTAC A1874 in human colon cancer cells. Cell Death Dis. 2020, 11, 805. [Google Scholar] [CrossRef]
- Qin, C.; Hu, Y.; Zhou, B.; Fernandez-Salas, E.; Yang, C.-Y.; Liu, L.; McEachern, D.; Przybranowski, S.; Wang, M.; Stuckey, J.; et al. Discovery of QCA570 as an Exceptionally Potent and Efficacious Proteolysis Targeting Chimera (PROTAC) Degrader of the Bromodomain and Extra-Terminal (BET) Proteins Capable of Inducing Complete and Durable Tumor Regression. J. Med. Chem. 2018, 61, 6685–6704. [Google Scholar] [CrossRef]
- Liu, C. The novel BET degrader, QCA570, is highly active against the growth of human NSCLC cells and synergizes with osimertinib in suppressing osimertinib-resistant EGFR-mutant NSCLC cells. Am. J. Cancer Res. 2022, 12, 779–792. [Google Scholar]
- Pillow, T.H.; Adhikari, P.; Blake, R.A.; Chen, J.; Del Rosario, G.; Deshmukh, G.; Figueroa, I.; Gascoigne, K.E.; Kamath, A.V.; Kaufman, S.; et al. Antibody Conjugation of a Chimeric BET Degrader Enables in vivo Activity. Chemmedchem 2019, 15, 17–25. [Google Scholar] [CrossRef] [Green Version]
- Sang, X.; Zhang, Y.; Fang, F.; Gao, L.; Tao, Y.; Li, X.; Zhang, Z.; Wang, J.; Tian, Y.; Li, Z.; et al. BRD4 Inhibitor GNE-987 Exerts Anticancer Effects by Targeting Super-Enhancer-Related Gene LYL1 in Acute Myeloid Leukemia. J. Immunol. Res. 2022, 2022, 7912484. [Google Scholar] [CrossRef]
- Vannam, R.; Sayilgan, J.; Ojeda, S.; Karakyriakou, B.; Hu, E.; Kreuzer, J.; Morris, R.; Lopez, X.I.H.; Rai, S.; Haas, W.; et al. Targeted degradation of the enhancer lysine acetyltransferases CBP and p300. Cell Chem. Biol. 2021, 28, 503–514.e12. [Google Scholar] [CrossRef]
- Bai, L.; Zhou, B.; Yang, C.-Y.; Ji, J.; McEachern, D.; Przybranowski, S.; Jiang, H.; Hu, J.; Xu, F.; Zhao, Y.; et al. Targeted Degradation of BET Proteins in Triple-Negative Breast Cancer. Cancer Res. 2017, 77, 2476–2487. [Google Scholar] [CrossRef] [Green Version]
- Saenz, D.T.; Fiskus, W.; Qian, Y.; Manshouri, T.; Rajapakshe, K.; Raina, K.; Coleman, K.G.; Crew, A.P.; Shen, A.; Mill, C.P.; et al. Novel BET protein proteolysis-targeting chimera exerts superior lethal activity than bromodomain inhibitor (BETi) against post-myeloproliferative neoplasm secondary (s) AML cells. Leukemia 2017, 31, 1951–1961. [Google Scholar] [CrossRef] [Green Version]
- Merika, M. Recruitment of CBP/p300 by the IFN beta enhanceosome is required for synergistic activation of transcription. Mol. Cell 1998, 1, 277–287. [Google Scholar] [CrossRef]
- Hull, E.E.; Montgomery, M.R.; Leyva, K.J. HDAC Inhibitors as Epigenetic Regulators of the Immune System: Impacts on Cancer Therapy and Inflammatory Diseases. BioMed Res. Int. 2016, 2016, 8797206. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Seto, E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb. Perspect. Med. 2016, 6, a026831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, K.; Wu, H.; Zhang, Z.; Leisten, E.D.; Nie, X.; Liu, B.; Wen, Z.; Zhang, J.; Cunningham, M.D.; Tang, W. Development of Selective Histone Deacetylase 6 (HDAC6) Degraders Recruiting Von Hippel–Lindau (VHL) E3 Ubiquitin Ligase. ACS Med. Chem. Lett. 2020, 11, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Zhao, Y.; Nie, X.; Wu, H.; Wang, B.; Almodovar-Rivera, C.M.; Xie, H.; Tang, W. A Cell-Based Target Engagement Assay for the Identification of Cereblon E3 Ubiquitin Ligase Ligands and Their Application in HDAC6 Degraders. Cell Chem. Biol. 2020, 27, 866–876.e8. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Lv, W.; He, M.; Deng, H.; Li, H.; Wu, W.; Rao, Y. Plasticity in designing PROTACs for selective and potent degradation of HDAC6. Chem. Commun. 2019, 55, 14848–14851. [Google Scholar] [CrossRef] [PubMed]
- An, Z.; Lv, W.; Su, S.; Wu, W.; Rao, Y. Developing potent PROTACs tools for selective degradation of HDAC6 protein. Protein Cell 2019, 10, 606–609. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Y.; Wang, J.; Zhao, L.Y.; Chen, X.; Zheng, G.; Zhang, X.; Liao, D. Discovery of histone deacetylase 3 (HDAC3)-specific PROTACs. Chem. Commun. 2020, 56, 9866–9869. [Google Scholar] [CrossRef]
- Cao, F.; de Weerd, S.; Chen, D.; Zwinderman, M.R.H.; van der Wouden, P.E.; Dekker, F.J. Induced protein degradation of histone deacetylases 3 (HDAC3) by proteolysis targeting chimera (PROTAC). Eur. J. Med. Chem. 2020, 208, 112800. [Google Scholar] [CrossRef]
- Schiedel, M.; Herp, D.; Hammelmann, S.; Swyter, S.; Lehotzky, A.; Robaa, D.; Oláh, J.; Ovádi, J.; Sippl, W.; Jung, M. Chemically Induced Degradation of Sirtuin 2 (Sirt2) by a Proteolysis Targeting Chimera (PROTAC) Based on Sirtuin Rearranging Ligands (SirReals). J. Med. Chem. 2017, 61, 482–491. [Google Scholar] [CrossRef]
- Schiedel, M. HaloTag-Targeted Sirtuin-Rearranging Ligand (SirReal) for the Development of Proteolysis-Targeting Chimeras (PROTACs) against the Lysine Deacetylase Sirtuin 2 (Sirt2)*. Chembiochem 2020, 21, 3371–3376. [Google Scholar] [CrossRef]
- Hong, J.Y.; Jing, H.; Price, I.R.; Cao, J.; Bai, J.J.; Lin, H. Simultaneous Inhibition of SIRT2 Deacetylase and Defatty-Acylase Activities via a PROTAC Strategy. ACS Med. Chem. Lett. 2020, 11, 2305–2311. [Google Scholar] [CrossRef]
- Hubbert, C.; Guardiola, A.; Shao, R.; Kawaguchi, Y.; Ito, A.; Nixon, A.; Yoshida, M.; Wang, X.-F.; Yao, T.-P. HDAC6 is a microtubule-associated deacetylase. Nature 2002, 417, 455–458. [Google Scholar] [CrossRef]
- Boyault, C.; Gilquin, B.; Zhang, Y.; Rybin, V.; Garman, E.; Meyer-Klaucke, W.; Matthias, P.; Müller, C.; Khochbin, S. HDAC6–p97/VCP controlled polyubiquitin chain turnover. EMBO J. 2006, 25, 3357–3366. [Google Scholar] [CrossRef] [Green Version]
- Haakenson, J.; Zhang, X. HDAC6 and Ovarian Cancer. Int. J. Mol. Sci. 2013, 14, 9514–9535. [Google Scholar] [CrossRef]
- Aldana-Masangkay, G.I.; Sakamoto, K.M. The Role of HDAC6 in Cancer. J. Biomed. Biotechnol. 2011, 2011, 875824. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Cao, L.; Xu, C.; Liu, F.; Xiang, G.; Liu, X.; Jiao, J.; Niu, Y. The immunohistochemical expression and potential prognostic value of HDAC6 and AR in invasive breast cancer. Hum. Pathol. 2018, 75, 16–25. [Google Scholar] [CrossRef]
- Iizuka, T. hTERT promoter polymorphism, -1327C>T, is associated with the risk of epithelial cancer. Springerplus 2013, 2, 249. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Yang, K.; Zhang, Z.; Leisten, E.D.; Li, Z.; Xie, H.; Liu, J.; Smith, K.A.; Novakova, Z.; Barinka, C.; et al. Development of Multifunctional Histone Deacetylase 6 Degraders with Potent Antimyeloma Activity. J. Med. Chem. 2019, 62, 7042–7057. [Google Scholar] [CrossRef]
- Jiang, Y.; Hsieh, J. HDAC3 controls gap 2/mitosis progression in adult neural stem/progenitor cells by regulating CDK1 levels. Proc. Natl. Acad. Sci. USA 2014, 111, 13541–13546. [Google Scholar] [CrossRef] [Green Version]
- Bhaskara, S.; Knutson, S.K.; Jiang, G.; Chandrasekharan, M.B.; Wilson, A.J.; Zheng, S.; Yenamandra, A.; Locke, K.; Yuan, J.-L.; Bonine-Summers, A.R.; et al. Hdac3 Is Essential for the Maintenance of Chromatin Structure and Genome Stability. Cancer Cell 2010, 18, 436–447. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Wu, C.; Yang, J.; Zhao, Y.; Jia, H.; Xue, M.; Da Xu, D.; Yang, F.; Fu, D.; Wang, C.; et al. Insulin-like growth factor 1-induced enolase 2 deacetylation by HDAC3 promotes metastasis of pancreatic cancer. Signal Transduct. Target. Ther. 2020, 5, 53. [Google Scholar] [CrossRef]
- Muller, B.M. Differential expression of histone deacetylases HDAC1, 2 and 3 in human breast cancer--overexpression of HDAC2 and HDAC3 is associated with clinicopathological indicators of disease progression. BMC Cancer 2013, 13, 215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno, D.A.; Scrideli, C.A.; Cortez, M.A.A.; De Paula Queiroz, R.; Valera, E.T.; Da Silva Silveira, V.; Yunes, J.A.; Brandalise, S.R.; Tone, L.G. Research paper: Differential expression of HDAC3, HDAC7 and HDAC9 is associated with prognosis and survival in childhood acute lymphoblastic leukaemia. Br. J. Haematol. 2010, 150, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Li, J. HDAC3 deteriorates colorectal cancer progression via microRNA-296-3p/TGIF1/TGFbeta axis. J. Exp. Clin. Cancer Res. 2020, 39, 248. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T. SIRT2, a tubulin deacetylase, acts to block the entry to chromosome condensation in response to mitotic stress. Oncogene 2007, 26, 945–957. [Google Scholar] [CrossRef] [Green Version]
- Serrano, L.; Martínez-Redondo, P.; Marazuela-Duque, A.; Vazquez, B.N.; Dooley, S.J.; Voigt, P.; Beck, D.B.; Kane-Goldsmith, N.; Tong, Q.; Rabanal, R.M.; et al. The tumor suppressor SirT2 regulates cell cycle progression and genome stability by modulating the mitotic deposition of H4K20 methylation. Genes Dev. 2013, 27, 639–653. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Ye, Y.; Yang, X.; Liu, B.; Wang, Z.; Chen, S.; Jiang, K.; Zhang, W.; Jiang, H.; Mustonen, H.K.; et al. SIRT 2-dependent IDH 1 deacetylation inhibits colorectal cancer and liver metastases. EMBO Rep. 2020, 21, e48183. [Google Scholar] [CrossRef]
- Park, J.W.; Han, J.-W. Targeting epigenetics for cancer therapy. Arch. Pharmacal Res. 2019, 42, 159–170. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Ren, B.; Yang, J.; Wang, H.; Yang, G.; Xu, R.; You, L.; Zhao, Y. The role of histone methylation in the development of digestive cancers: A potential direction for cancer management. Signal Transduct. Target. Ther. 2020, 5, 143. [Google Scholar] [CrossRef]
- Jeong, G.-Y.; Park, M.K.; Choi, H.-J.; An, H.W.; Park, Y.-U.; Choi, H.-J.; Park, J.; Kim, H.-Y.; Son, T.; Lee, H.; et al. NSD3-Induced Methylation of H3K36 Activates NOTCH Signaling to Drive Breast Tumor Initiation and Metastatic Progression. Cancer Res 2021, 81, 77–90. [Google Scholar] [CrossRef]
- Tellez, C.S.; Picchi, M.A.; Juri, D.; Do, K.; Desai, D.H.; Amin, S.G.; Hutt, J.A.; Filipczak, P.T.; Belinsky, S.A. Chromatin remodeling by the histone methyltransferase EZH2 drives lung pre-malignancy and is a target for cancer prevention. Clin. Epigenetics 2021, 13, 44. [Google Scholar] [CrossRef]
- Sun, Y.; Zhang, Y.; Chen, X.; Yu, A.; Du, W.; Huang, Y.; Wu, F.; Yu, L.; Li, J.; Wen, C.; et al. Discovery of a potent and selective proteolysis targeting chimera (PROTAC) degrader of NSD3 histone methyltransferase. Eur. J. Med. Chem. 2022, 239, 114528. [Google Scholar] [CrossRef]
- Xu, C.; Meng, F.; Park, K.-S.; Storey, A.J.; Gong, W.; Tsai, Y.-H.; Gibson, E.; Byrum, S.D.; Li, D.; Edmondson, R.D.; et al. A NSD3-targeted PROTAC suppresses NSD3 and cMyc oncogenic nodes in cancer cells. Cell Chem. Biol. 2021, 29, 386–397.e9. [Google Scholar] [CrossRef]
- Hsu, J.H.-R.; Rasmusson, T.; Robinson, J.; Pachl, F.; Read, J.; Kawatkar, S.; O’Donovan, D.H.; Bagal, S.; Code, E.; Rawlins, P.; et al. EED-Targeted PROTACs Degrade EED, EZH2, and SUZ12 in the PRC2 Complex. Cell Chem. Biol. 2020, 27, 41–46.e17. [Google Scholar] [CrossRef]
- Potjewyd, F.; Turner, A.-M.W.; Beri, J.; Rectenwald, J.M.; Norris-Drouin, J.L.; Cholensky, S.H.; Margolis, D.M.; Pearce, K.H.; Herring, L.E.; James, L.I. Degradation of Polycomb Repressive Complex 2 with an EED-Targeted Bivalent Chemical Degrader. Cell Chem. Biol. 2019, 27, 47–56.e15. [Google Scholar] [CrossRef]
- Wang, J.; Yu, X.; Gong, W.; Liu, X.; Park, K.-S.; Ma, A.; Tsai, Y.-H.; Shen, Y.; Onikubo, T.; Pi, W.-C.; et al. EZH2 noncanonically binds cMyc and p300 through a cryptic transactivation domain to mediate gene activation and promote oncogenesis. Nature 2022, 24, 384–399. [Google Scholar] [CrossRef]
- Lu, C.; Han, H.D.; Mangala, L.S.; Ali-Fehmi, R.; Newton, C.S.; Ozbun, L.; Armaiz-Pena, G.N.; Hu, W.; Stone, R.L.; Munkarah, A.; et al. Regulation of Tumor Angiogenesis by EZH2. Cancer Cell 2010, 18, 185–197. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Chen, X.; Liu, X.; Lu, D.; Li, S.; Qu, L.; Yin, F.; Luo, H.; Zhang, Y.; Luo, Z.; et al. Discovery of precision targeting EZH2 degraders for triple-negative breast cancer. Eur. J. Med. Chem. 2022, 238, 114462. [Google Scholar] [CrossRef]
- Tu, Y.; Sun, Y.; Qiao, S.; Luo, Y.; Liu, P.; Jiang, Z.-X.; Hu, Y.; Wang, Z.; Huang, P.; Wen, S. Design, Synthesis, and Evaluation of VHL-Based EZH2 Degraders to Enhance Therapeutic Activity against Lymphoma. J. Med. Chem. 2021, 64, 10167–10184. [Google Scholar] [CrossRef]
- Shen, Y.; Gao, G.; Yu, X.; Kim, H.; Wang, L.; Xie, L.; Schwarz, M.; Chen, X.; Guccione, E.; Liu, J.; et al. Discovery of First-in-Class Protein Arginine Methyltransferase 5 (PRMT5) Degraders. J. Med. Chem. 2020, 63, 9977–9989. [Google Scholar] [CrossRef]
- Dolle, A. Design, Synthesis, and Evaluation of WD-Repeat-Containing Protein 5 (WDR5) Degraders. J. Med. Chem. 2021, 64, 10682–10710. [Google Scholar] [CrossRef]
- Yu, X.; Li, D.; Kottur, J.; Shen, Y.; Kim, H.S.; Park, K.-S.; Tsai, Y.-H.; Gong, W.; Wang, J.; Suzuki, K.; et al. A selective WDR5 degrader inhibits acute myeloid leukemia in patient-derived mouse models. Sci. Transl. Med. 2021, 13, eabj1578. [Google Scholar] [CrossRef] [PubMed]
- Vougiouklakis, T.; Hamamoto, R.; Nakamura, Y.; Saloura, V. The NSD family of protein methyltransferases in human cancer. Epigenomics 2015, 7, 863–874. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Piao, L.; Zhuang, Q.; Yuan, X.; Liu, Z.; He, X. The role of histone lysine methyltransferase NSD3 in cancer. OncoTargets Ther. 2018, 11, 3847–3852. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Ivanov, A.A.; Su, R.; Gonzalez-Pecchi, V.; Qi, Q.; Liu, S.; Webber, P.; McMillan, E.; Rusnak, L.; Pham, C.; et al. The OncoPPi network of cancer-focused protein–protein interactions to inform biological insights and therapeutic strategies. Nat. Commun. 2017, 8, 14356. [Google Scholar] [CrossRef] [PubMed]
- Saloura, V.; Vougiouklakis, T.; Zewde, M.; Deng, X.; Kiyotani, K.; Park, J.-H.; Matsuo, Y.; Lingen, M.; Suzuki, T.; Dohmae, N.; et al. WHSC1L1-mediated EGFR mono-methylation enhances the cytoplasmic and nuclear oncogenic activity of EGFR in head and neck cancer. Sci. Rep. 2017, 7, 40664. [Google Scholar] [CrossRef] [Green Version]
- Cao, R.; Wang, L.; Wang, H.; Xia, L.; Erdjument-Bromage, H.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of Histone H3 Lysine 27 Methylation in Polycomb-Group Silencing. Science 2002, 298, 1039–1043. [Google Scholar] [CrossRef] [Green Version]
- Kuzmichev, A.; Nishioka, K.; Erdjument-Bromage, H.; Tempst, P.; Reinberg, D. Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev. 2002, 16, 2893–2905. [Google Scholar] [CrossRef] [Green Version]
- Jiang, T.; Wang, Y.; Zhou, F.; Gao, G.; Ren, S.; Zhou, C. Prognostic value of high EZH2 expression in patients with different types of cancer: A systematic review with meta-analysis. Oncotarget 2015, 7, 4584–4597. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.H.; Roberts, C.W. Targeting EZH2 in cancer. Nat. Med. 2016, 22, 128–134. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Hu, X.; Wang, Q.; Wu, X.; Zhang, Q.; Wei, W.; Su, X.; He, H.; Zhou, S.; Hu, R.; et al. Design and Synthesis of EZH2-Based PROTACs to Degrade the PRC2 Complex for Targeting the Noncatalytic Activity of EZH2. J. Med. Chem. 2021, 64, 2829–2848. [Google Scholar] [CrossRef]
- Yang, Y.; Bedford, M.T. Protein arginine methyltransferases and cancer. Nat. Rev. Cancer 2013, 13, 37–50. [Google Scholar] [CrossRef]
- Stopa, N.; Krebs, J.E.; Shechter, D. The PRMT5 arginine methyltransferase: Many roles in development, cancer and beyond. Cell. Mol. Life Sci. 2015, 72, 2041–2059. [Google Scholar] [CrossRef]
- Wu, Q.; Schapira, M.; Arrowsmith, C.H.; Barsyte-Lovejoy, D. Protein arginine methylation: From enigmatic functions to therapeutic targeting. Nat. Rev. Drug Discov. 2021, 20, 509–530. [Google Scholar] [CrossRef]
- Shailesh, H.; Zakaria, Z.Z.; Baiocchi, R.; Sif, S. Protein arginine methyltransferase 5 (PRMT5) dysregulation in cancer. Oncotarget 2018, 9, 36705–36718. [Google Scholar] [CrossRef] [Green Version]
- Qin, Y. PRMT5 enhances tumorigenicity and glycolysis in pancreatic cancer via the FBW7/cMyc axis. Cell Commun. Signal. 2019, 17, 30. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Yao, B.; Gui, T.; Guo, C.; Wu, X.; Li, J.; Ma, L.; Deng, Y.; Xu, P.; Wang, Y.; et al. PRMT5-dependent transcriptional repression of c-Myc target genes promotes gastric cancer progression. Theranostics 2020, 10, 4437–4452. [Google Scholar] [CrossRef]
- Beketova, E.; Owens, J.L.; Asberry, A.M.; Hu, C.-D. PRMT5: A putative oncogene and therapeutic target in prostate cancer. Cancer Gene Ther. 2021, 29, 264–276. [Google Scholar] [CrossRef]
- Barczak, W.; Jin, L.; Carr, S.M.; Munro, S.; Ward, S.; Kanapin, A.; Samsonova, A.; La Thangue, N.B. PRMT5 promotes cancer cell migration and invasion through the E2F pathway. Cell Death Dis. 2020, 11, 572. [Google Scholar] [CrossRef]
- Huang, W.-J.; Ruan, S.; Wen, F.; Lu, X.-N.; Gu, S.-P.; Chen, X.-X.; Liu, M.; Shu, P. Multidrug Resistance of Gastric Cancer: The Mechanisms and Chinese Medicine Reversal Agents. Cancer Manag. Res. 2020, 12, 12385–12394. [Google Scholar] [CrossRef]
- Bryan, A.F.; Wang, J.; Howard, G.C.; Guarnaccia, A.D.; Woodley, C.M.; Aho, E.R.; Rellinger, E.J.; Matlock, B.K.; Flaherty, D.K.; Lorey, S.L.; et al. WDR5 is a conserved regulator of protein synthesis gene expression. Nucleic Acids Res. 2020, 48, 2924–2941. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Liu, Z.; Klement, J.D.; Yang, D.; Merting, A.D.; Poschel, D.; Albers, T.; Waller, J.L.; Shi, H.; Liu, K. WDR5-H3K4me3 epigenetic axis regulates OPN expression to compensate PD-L1 function to promote pancreatic cancer immune escape. J. Immunother. Cancer 2021, 9, e002624. [Google Scholar] [CrossRef] [PubMed]
- Neilsen, B.K.; Chakraborty, B.; McCall, J.L.; Frodyma, D.E.; Sleightholm, R.L.; Fisher, K.W.; Lewis, R.E. WDR5 supports colon cancer cells by promoting methylation of H3K4 and suppressing DNA damage. BMC Cancer 2018, 18, 673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, Z.; Song, E.J.; Kawasawa, Y.I.; Li, J.; Dovat, S.; Song, C. WDR5 high expression and its effect on tumorigenesis in leukemia. Oncotarget 2016, 7, 37740–37754. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Dong, W.; Xie, R.; Wu, J.; Su, Q.; Li, W.; Yao, K.; Chen, Y.; Zhou, Q.; Zhang, Q.; et al. HSF1 facilitates the multistep process of lymphatic metastasis in bladder cancer via a novel PRMT5-WDR5-dependent transcriptional program. Cancer Commun. 2022, 42, 447–470. [Google Scholar] [CrossRef]
- Punzi, S.; Balestrieri, C.; D’Alesio, C.; Bossi, D.; Dellino, G.I.; Gatti, E.; Pruneri, G.; Criscitiello, C.; Lovati, G.; Meliksetyan, M.; et al. WDR5 inhibition halts metastasis dissemination by repressing the mesenchymal phenotype of breast cancer cells. Breast Cancer Res. 2019, 21, 123. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Li, K.; Liu, Z.; Liu, J.; Wang, Y.; Sun, R.; Li, Z.; Qiu, B.; Zhang, X.; Ren, G.; et al. WDR5 facilitates EMT and metastasis of CCA by increasing HIF-1α accumulation in Myc-dependent and independent pathways. Mol. Ther. 2021, 29, 2134–2150. [Google Scholar] [CrossRef]
- Peñalosa-Ruiz, G.; Bousgouni, V.; Gerlach, J.P.; Waarlo, S.; van de Ven, J.V.; Veenstra, T.E.; Silva, J.C.; van Heeringen, S.J.; Bakal, C.; Mulder, K.W.; et al. WDR5, BRCA1, and BARD1 Co-regulate the DNA Damage Response and Modulate the Mesenchymal-to-Epithelial Transition during Early Reprogramming. Stem Cell Rep. 2019, 12, 743–756. [Google Scholar] [CrossRef] [Green Version]
- Cai, W.L.; Chen, J.F.-Y.; Chen, H.; Wingrove, E.; Kurley, S.J.; Chan, L.H.; Zhang, M.; Arnal-Estape, A.; Zhao, M.; Balabaki, A.; et al. Human WDR5 promotes breast cancer growth and metastasis via KMT2-independent translation regulation. eLife 2022, 11, e78163. [Google Scholar] [CrossRef]
- Lu, K.; Tao, H.; Si, X.; Chen, Q. The Histone H3 Lysine 4 Presenter WDR5 as an Oncogenic Protein and Novel Epigenetic Target in Cancer. Front. Oncol. 2018, 8, 502. [Google Scholar] [CrossRef]
- Malek, R.; Gajula, R.P.; Williams, R.D.; Nghiem, B.; Simons, B.W.; Nugent, K.; Wang, H.; Taparra, K.; Lemtiri-Chlieh, G.; Yoon, A.R.; et al. TWIST1-WDR5-Hottip Regulates Hoxa9 Chromatin to Facilitate Prostate Cancer Metastasis. Cancer Res 2017, 77, 3181–3193. [Google Scholar] [CrossRef] [Green Version]
- Aalfs, J.D.; Kingston, R.E. What does ‘chromatin remodeling’ mean? Trends Biochem. Sci. 2000, 25, 548–555. [Google Scholar] [CrossRef]
- Clapier, C.R.; Cairns, B.R. The Biology of Chromatin Remodeling Complexes. Annu. Rev. Biochem. 2009, 78, 273–304. [Google Scholar] [CrossRef]
- Xiao, L.; Parolia, A.; Qiao, Y.; Bawa, P.; Eyunni, S.; Mannan, R.; Carson, S.E.; Chang, Y.; Wang, X.; Zhang, Y.; et al. Targeting SWI/SNF ATPases in enhancer-addicted prostate cancer. Nature 2021, 601, 434–439. [Google Scholar] [CrossRef]
- Kargbo, R.B. SMARCA2/4 PROTAC for Targeted Protein Degradation and Cancer Therapy. ACS Med. Chem. Lett. 2020, 11, 1797–1798. [Google Scholar] [CrossRef]
- Zoppi, V.; Hughes, S.J.; Maniaci, C.; Testa, A.; Gmaschitz, T.; Wieshofer, C.; Koegl, M.; Riching, K.M.; Daniels, D.L.; Spallarossa, A.; et al. Iterative Design and Optimization of Initially Inactive Proteolysis Targeting Chimeras (PROTACs) Identify VZ185 as a Potent, Fast, and Selective von Hippel–Lindau (VHL) Based Dual Degrader Probe of BRD9 and BRD7. J. Med. Chem. 2018, 62, 699–726. [Google Scholar] [CrossRef]
- Gechijian, L.N.; Buckley, D.L.; Lawlor, M.; Reyes, J.; Paulk, J.; Ott, C.J.; Winter, G.E.; Erb, M.A.; Scott, T.; Xu, M.; et al. Functional TRIM24 degrader via conjugation of ineffectual bromodomain and VHL ligands. Nat. Chem. Biol. 2018, 14, 405–412. [Google Scholar] [CrossRef]
- Kadoch, C.; Hargreaves, D.C.; Hodges, H.C.; Elias, L.; Ho, L.; Ranish, J.; Crabtree, G.R. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat. Genet. 2013, 45, 592–601. [Google Scholar] [CrossRef] [Green Version]
- Farnaby, W.; Koegl, M.; Roy, M.J.; Whitworth, C.; Diers, E.; Trainor, N.; Zollman, D.; Steurer, S.; Karolyi-Oezguer, J.; Riedmueller, C.; et al. Publisher Correction: BAF complex vulnerabilities in cancer demonstrated via structure-based PROTAC design. Nat. Chem. Biol. 2019, 15, 846. [Google Scholar] [CrossRef]
- Weisberg, E.; Chowdhury, B.; Meng, C.; Case, A.E.; Ni, W.; Garg, S.; Sattler, M.; Azab, A.K.; Sun, J.; Muz, B.; et al. BRD9 degraders as chemosensitizers in acute leukemia and multiple myeloma. Blood Cancer J. 2022, 12, 110. [Google Scholar] [CrossRef]
- Le Douarin, B.; Zechel, C.; Garnier, J.M.; Lutz, Y.; Tora, L.; Pierrat, P.; Heery, D.; Gronemeyer, H.; Chambon, P.; Losson, R. The N-terminal part of TIF1, a putative mediator of the ligand-dependent activation function (AF-2) of nuclear receptors, is fused to B-raf in the oncogenic protein T18. EMBO J. 1995, 14, 2020–2033. [Google Scholar] [CrossRef]
- Tsai, W.-W.; Wang, Z.; Yiu, T.T.; Akdemir, K.C.; Xia, W.; Winter, S.; Tsai, C.-Y.; Shi, X.; Schwarzer, D.; Plunkett, W.; et al. TRIM24 links a non-canonical histone signature to breast cancer. Nature 2010, 468, 927–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Chen, H.; Ding, B.; Jiang, W. High expression of TRIM24 predicts worse prognosis and promotes proliferation and metastasis of epithelial ovarian cancer. J. Ovarian Res. 2022, 15, 19. [Google Scholar] [CrossRef] [PubMed]
- Groner, A.C.; Cato, L.; de Tribolet-Hardy, J.; Bernasocchi, T.; Janouskova, H.; Melchers, D.; Houtman, R.; Cato, A.C.; Tschopp, P.; Gu, L.; et al. TRIM24 Is an Oncogenic Transcriptional Activator in Prostate Cancer. Cancer Cell 2016, 29, 846–858. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Huang, Y.; Yang, D.; Li, X.; Liang, J.; Lin, L.; Zhang, M.; Zhong, K.; Liang, B.; Li, J. Overexpression of TRIM24 Is Associated with the Onset and Progress of Human Hepatocellular Carcinoma. PLoS ONE 2014, 9, e85462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, M.; Liu, M.; Kurosaki, T.; Suzuki, M.; Arai, T.; Sawabe, M.; Kasuya, Y.; Kato, M.; Fujimura, T.; Fukuhara, H.; et al. Association of rs6983561 polymorphism at 8q24 with prostate cancer mortality in a Japanese population. Clin. Genitourin. Cancer 2011, 9, 46–52. [Google Scholar] [CrossRef]
- Mullen, J.; Kato, S.; Sicklick, J.K.; Kurzrock, R. Targeting ARID1A mutations in cancer. Cancer Treat. Rev. 2021, 100, 102287. [Google Scholar] [CrossRef]
- Mathur, R. ARID1A loss in cancer: Towards a mechanistic understanding. Pharmacol. Ther. 2018, 190, 15–23. [Google Scholar] [CrossRef]
- Allo, G.; Bernardini, M.Q.; Wu, R.-C.; Shih, I.-M.; Kalloger, S.; Pollett, A.; Gilks, C.B.; Clarke, B.A. ARID1A loss correlates with mismatch repair deficiency and intact p53 expression in high-grade endometrial carcinomas. Mod. Pathol. 2013, 27, 255–261. [Google Scholar] [CrossRef] [Green Version]
- Jiang, T.; Chen, X.; Su, C.; Ren, S.; Zhou, C. Pan-cancer analysis of ARID1A Alterations as Biomarkers for Immunotherapy Outcomes. J. Cancer 2020, 11, 776–780. [Google Scholar] [CrossRef] [Green Version]
- Mathur, R.; Alver, B.H.; Roman, A.K.S.; Wilson, B.G.; Wang, X.; Agoston, A.T.; Park, P.J.; Shivdasani, R.A.; Roberts, C.W.M. ARID1A loss impairs enhancer-mediated gene regulation and drives colon cancer in mice. Nat. Genet. 2016, 49, 296–302. [Google Scholar] [CrossRef] [Green Version]
- Xu, G.; Chhangawala, S.; Cocco, E.; Razavi, P.; Cai, Y.; Otto, J.E.; Ferrando, L.; Selenica, P.; Ladewig, E.; Chan, C.; et al. ARID1A determines luminal identity and therapeutic response in estrogen-receptor-positive breast cancer. Nat. Genet. 2020, 52, 198–207. [Google Scholar] [CrossRef]
- Shen, J.; Ju, Z.; Zhao, W.; Wang, L.; Peng, Y.; Ge, Z.; Nagel, Z.D.; Zou, J.; Wang, C.; Kapoor, P.; et al. ARID1A deficiency promotes mutability and potentiates therapeutic antitumor immunity unleashed by immune checkpoint blockade. Nat. Med. 2018, 24, 556–562. [Google Scholar] [CrossRef]



| Target | PROTAC | PROTAC Structure | E3 Ligase | Cancer | Ref. |
|---|---|---|---|---|---|
| BRD4 | ARV-825 |  | CRBN | Lymphoma Acute myeloid leukemia Neuroblastoma Cholangiocarcinom Thyroid cancer Gastric cancer Ovarian cancer Multiple myeloma Triple-negative breast cancer | [32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49] |
| BRD4 | MZ1 |  | VHL | Triple-negative breast cancer Ovarian cancer Diffuse large B-cell lymphoma Neuroblastoma, Colon cancer HER2-positive breast cancer | [50,51,52,53,54,55] |
| BRD4 | ARV-771 |  | VHL | Prostate cancer Liver cancer Non-small cell lung cancer Esophageal Squamous Cell Carcinoma | [56,57,58,59,60,61,62] |
| BRD4 | dBET1 |  | CRBN | Colon cancer, Breast cancer Ovarian cancer Acute Myeloid Leukemia Melanoma | [63,64,65,66] |
| BRD4 | dBET6 |  | CRBN | Chronic myeloid leukemia Lung cancer Glioblastoma | [67,68,69,70,71] |
| BRD4 | A1874 |  | MMD2 | Colon cancer Non-small cell lung cancer | [72,73] |
| BRD4 | QCA570 |  | CRBN | Non-small cell lung cancer Acute leukemia | [74,75] |
| BRD4 | GNE-987 |  | VHL | Acute Myeloid Leukemia | [74,75] |
| CREBBP | dCBP-1 |  | CRBN | Multiple myeloma | [76] |
| Target | PROTAC | PROTAC Structure | E3 Ligase | Cancer | Ref. |
|---|---|---|---|---|---|
| HDAC6 | Wh185 |  | VHL | Multiple myeloma | [82] |
| HDAC6 | YZ166 |  | CRBN | Multiple myeloma | [83] |
| HDAC6 | dHDAC6 |  | CRBN | Multiple myeloma | [84] |
| HDAC6 | NP8 |  | CRBN | Multiple myeloma | [85] |
| HDAC3 | XZ9002 |  | VHL | Breast cancer | [86] |
| HDAC3 | HD-TAC7 |  | CRBN | Leukaemia | [87] |
| SIRT2 | Sirt1133−747 |  | CRBN | Cervical cancer | [88] |
| SIRT2, | HT7-parkin |  | CRBN | Cervical cancer | [89] |
| SIRT2 | TM-P2-Thal |  | CRBN | Breast cancer | [90] |
| SIRT2 | TM-P4-Thal |  | CRBN | Breast cancer | [90] |
| Target | PROTAC | PROTAC Structure | E3 Ligase | Cancer | Ref. |
|---|---|---|---|---|---|
| NSD3 | MS9715 |  | VHL | Blood cancer Lung cancer | [111] |
| NSD3 | SYL2158 |  | VHL | Blood cancer Lung cancer | [112] |
| PRC2 | PROTAC-2 |  | VHL | Diffuse large B-cell lymphoma Rhabdoid cancer | [113] |
| PRC2 | UNC6852 |  | VHL | Cervical cancer Diffuse large B-cell lymphoma | [114] |
| EZH2 | MS177 |  | CRBN | MLL-r leukaemias | [115] |
| EZH2 | E7 |  | CRBN | Diffuse large B-cell lymphoma Prostate cancer Ovarian cancer | [116] |
| EZH2 | U3i |  | CRBN | Triple-negative breast cancer | [117] |
| EZH2 | YM281 |  | VHL | Lymphoma | [118] |
| EZH2 | YM181 |  | VHL | Lymphoma | [118] |
| PRMT5 | Ms4322 |  | VHL | Breast cancer Cervical cancer Lung cancer Leukemia ER (+) breast cancer | [119] |
| WRD5 | MS33 |  | VHL | Acute Myeloid Leukemia | [120] |
| WRD5 | MS67 |  | VHL | [121] |




{kind=link}
{kind=link}
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, X.; Wang, A.; Shi, Y.; Dai, M.; Liu, M.; Cai, H.-B. PROTACs in Epigenetic Cancer Therapy: Current Status and Future Opportunities. Molecules 2023, 28, 1217. https://doi.org/10.3390/molecules28031217
Liu X, Wang A, Shi Y, Dai M, Liu M, Cai H-B. PROTACs in Epigenetic Cancer Therapy: Current Status and Future Opportunities. Molecules. 2023; 28(3):1217. https://doi.org/10.3390/molecules28031217
Chicago/Turabian StyleLiu, Xuelian, Anjin Wang, Yuying Shi, Mengyuan Dai, Miao Liu, and Hong-Bing Cai. 2023. "PROTACs in Epigenetic Cancer Therapy: Current Status and Future Opportunities" Molecules 28, no. 3: 1217. https://doi.org/10.3390/molecules28031217