
The Effect of Alkali Iodide Salts in the Inclusion Process of Phenolphthalein in β-Cyclodextrin: A Spectroscopic and Theoretical Study

Abstract
:1. Introduction
2. Materials and Methods
2.1. Solutions
2.2. Spectrophotometric Measurements
2.3. Theoretical Calculations
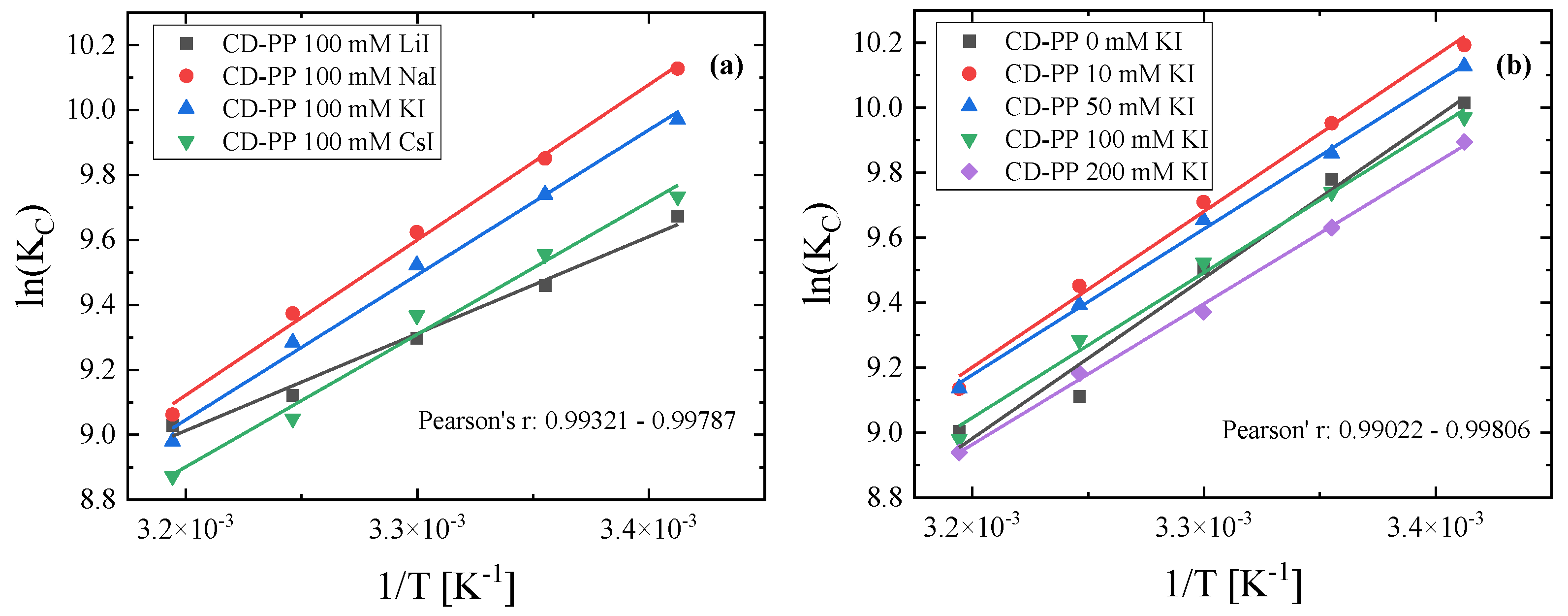
3. Results and Discussion
3.1. Calculation of the Complexation Coefficient
3.2. Thermodynamic Analysis
3.3. Spectroscopic Evidence of the Complex Formation
3.4. Molecular Docking and DFT Results
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Buvári, A.; Barcza, L. β-cyclodextrin Complexes of Different Type with Inorganic Compounds. Inorg. Chim Acta 1979, 33, L179–L180. [Google Scholar] [CrossRef]
- Yi, Z.-P.; Chen, H.-L.; Huang, Z.-Z.; Huang, Q.; Yu, J.-S. Contributions of weak interactions to the inclusion complexation of 3-hydroxynaphthalene-2-carboxylic acid and its analogues with cyclodextrins. J. Chem. Soc. Perkin Trans 2 2000, 2, 121–127. [Google Scholar] [CrossRef]
- Fifere, A.; Marangoci, N.; Maier, S.; Coroaba, A.; Maftei, D.; Pinteala, M. Theoretical study on β-cyclodextrin inclusion complexes with propiconazole and protonated propiconazole. Beilstein J. Org. Chem. 2012, 8, 2191–2201. [Google Scholar] [CrossRef] [Green Version]
- Gidwani, B.; Vyas, A. Hindawi Publishing Corporation. A Comprehensive Review on Cyclodextrin-Based Carriers for Delivery of Chemotherapeutic Cytotoxic Anticancer Drugs. BioMed. Res. Inter. 2015, 2015, 198268. [Google Scholar] [CrossRef] [Green Version]
- Baruch-Teblum, E.; Mastai, Y.; Landfester, K. Miniemulsion polymerization of cyclodextrin nanospheres for water purification from organic pollutants. Eur. Pol. J. 2010, 46, 1671–1678. [Google Scholar] [CrossRef]
- Astray, G.; Gonzalez-Barreiro, C.; Mejuto, J.; Rial-Otero, R.; Simal-Gandara, J. A review on the use of cyclodextrins in foods. Food Hydrocoll. 2009, 23, 163–1640. [Google Scholar] [CrossRef]
- Rasheed, A.; Kumar, A.; Sravanthi, C.K. Cyclodextrins as drug carrier molecule: A review. Sci. Pharm. 2008, 76, 567–598. [Google Scholar] [CrossRef]
- Su, J.; Chen, J.; Li, L.; Li, B.; Shi, L.; Chen, L.; Xu, Z. Formation of β-Cyclodextrin Inclusion Enhances the Stability and Aqueous Solubility of Natural Borneol. J. Food Sci. 2012, 77, C658–C664. [Google Scholar] [CrossRef]
- Crini, G. Review: A History of Cyclodextrins. Chem. Rev. 2014, 114, 10940–10975. [Google Scholar] [CrossRef]
- Lee, J.-U.; Lee, S.-S.; Lee, S.; Oh, H. Noncovalent Complexes of Cyclodextrin with Small Organic Molecules: Applications and Insights into Host–Guest Interactions in the Gas Phase and Condensed Phase. Molecules 2020, 25, 4048. [Google Scholar] [CrossRef]
- Choi, H.; Oh, Y.H.; Park, S.; Lee, S.S.; Oh, H.B.; Lee, S. Unveiling host–guest–solvent interactions in solution by identifying highly unstable host–guest configurations in thermal non equilibrium gas phase. Sci. Rep. 2022, 12, 8169. [Google Scholar] [CrossRef]
- Hu, Q.-D.; Tang, G.-P.; Chu, P.K. Cyclodextrin-based host–guest supramolecular nanoparticles for delivery: From design to applications. Acc. Chem. Res. 2014, 47, 2017–2025. [Google Scholar] [CrossRef]
- Zhan, W.; Wei, T.; Yu, Q.; Chen, H. Fabrication of supramolecular bioactive surfaces via β-cyclodextrin-based host–guest interactions. ACS Appl. Mater. Interfaces 2018, 10, 36585–36601. [Google Scholar] [CrossRef]
- Fde Sousa, B.; Oliveira, M.F.; Lula, I.S.; Sansiviero, M.T.C.; Corte, M.E.; Sinisterra, R.D. Study of inclusion compound in solution involving tetracycline and β-cyclodextrin by FT-IR-ATR. Vib. Spectrosc. 2008, 46, 57–62. [Google Scholar] [CrossRef]
- Mochida, K.; Kagita, A.; Matsui, Y.; Date, Y. Effects of inorganic salts on the Dissociation of a Complex of β-cyclodextrin with an Azo Dye in an Aqueous Soloution. BCSJ 1973, 46, 3703–3707. [Google Scholar] [CrossRef]
- Meyler, D.C. Side effects of drugs: The international encyclopedia of adverse drug reactions and interactions. Indian J. Pharmacol. 2016, 48, 224. [Google Scholar]
- Kuwabara, T.; Takamura, M.; Matsushita, A.; Ikeda, H.; Nakamura, A.; Ueno, A.; Toda, F. Phenolphthalein-Modified β-Cyclodextrin as a Molecule-Responsive Colorless-to-Color Change Indicator. J. Org. Chem. 1998, 63, 8729–8735. [Google Scholar] [CrossRef]
- Arsad, S.R.; Maarof, H.; Ibrahim, W.A.W.; Aboul-Enein, H.Y. Theoretical and Molecular Docking Study of Ketoconazole on Heptakis(2,3,6-tri-O-methyl)-β-cyclodextrin as Chiral Selector. Chirality 1998, 28, 209–214. [Google Scholar] [CrossRef]
- Jones, P.G. Crystal growing. Chem. Br. 1981, 17, 222–225. [Google Scholar]
- Kalampounias, A.G.; Papatheodorou, G.N. Ligand Field States and VibrationalModes of Solid and Molten Elpasolite: Cs2NaHoCl6. Z. Für Nat. A 2007, 62, 169–175. [Google Scholar] [CrossRef]
- Meretoudi, A.; Banti, C.N.; Siafarika, P.; Kalampounias, A.G.; Hadjikakou, S.K. Tetracycline Water Soluble Formulations with Enhanced Antimicrobial Activity. Antibiotics 2020, 9, 845. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, G.M.; Goodsell, D.S.; Pique, M.E.; Lindstrom, W.L.; Huey, R.; Forli, S.; Hart, W.E.; Halliday, S.; Belew, R.; Olson, A.J. AutoDock Version 4.2 Updated for version 4.2.6 Automated Docking of Flexible Ligands to Flexible Receptors. J. Chem. Inf. Model. 2007, 47, 1481–1492. [Google Scholar]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated Docking Using a Lamarckian Genetic Algorithm and an Empirical Binding Free Energy Function. J. Comput. Chem. 1998, 19, 163–1662. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Diaz, D.; Varga-Baca, I.; Gracia-Mora, J. β-cyclodextrin Inclusion Complexes with Iodine. J. Chem. Educ. 1994, 71, 708. [Google Scholar] [CrossRef]
- Terekhova, I.V. Comparative thermodynamic study on complex formation of native and hydroxypropylated cyclodextrins with benzoic acid. Thermochim. Acta 2011, 44, 118–121. [Google Scholar] [CrossRef]
- Kano, K.; Tamiya, Y.; Hashimoto, A.S. Binding Forces in Complexation of p-Alkylphenols with β-cyclodextrin and Methylated β-cyclodextrins. J. Incl. Phenom. 1992, 13, 287–293. [Google Scholar] [CrossRef]
- Ross, P.D.; Rekharsky, M.V. Thermodynamics of Hydrogen Bond and Hydrophobic Interactions in Cyclodextrin Complexes. Biophys. J. 1996, 71, 2144–2154. [Google Scholar] [CrossRef] [Green Version]
- Connors, K.A. The Stability of Cyclodextrin Complexes in Solutions. Chem. Rev. 1997, 97, 1325–1358. [Google Scholar] [CrossRef]
- Prozeller, D.; Morsbach, S.; Landfester, K. Isothermal titration calorimetry as a complementary method for investigating nanoparticle-protein interactions. Nanoscale 2019, 11, 19265–19273. [Google Scholar] [CrossRef] [Green Version]
- Ameen, M.; Kunsági-Máté, H.; Bognár, S.; Szente, B.; Poór, L.; Lemli, M.B. Thermodynamic Characterization of the Interaction between the Antimicrobial Drug Sulfamethazine and Two Selected Cyclodextrins. Molecules 2019, 24, 4565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holm, R.; Schonbeck, C.; Somprasirt, P.; Westh, P.; Mu, H. A study of salt effects on the complexation between β-cyclodextrins and bile salts based on the Hofmeister series. J. Incl. Phenom. 2014, 80, 243–251. [Google Scholar] [CrossRef]
- Terekhova, I.V.; Romanova, A.O.; Kumeev, R.S.; Fedorov, M.V. Selective Na+/K+ Effects on the Formation of α-Cyclodextrin Complexes with Aromatic Carboxylic Acids: Competition for the Guest. J. Phys. Chem. B 2010, 114, 12607–12613. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Liu, L.; Guo, Q.X. A Theoretical Study on the Inclusion Complexation of Cyclodextrins with Inorganic Cations and Anions. J. Incl. Phenom. 2002, 43, 223–229. [Google Scholar] [CrossRef]
- Buvári, Á.; Barcza, L.; Kajtár, M. Complex formation of phenolphthalein and some related compounds with β-cyclodextrin. J. Chem. Soc. Perkin Trans 1988, 2, 1687–1690. [Google Scholar] [CrossRef]
- Zarzycki, P.; Lamparczyk, H. The equilibrium constant of β-cyclodextrin–phenolphtalein complex; influence of temperature and tetrahydrofuran. J. Pharm. Biomed. Anal. 1988, 18, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.T.; Nguyen, T.H.; Pham, T.N.H.; Huy, N.T.; Bay, M.V.; Pham, M.Q.; Nam, P.C.; Vu, V.V.; Ngo, S.T. Autodock Vina Adopts More Accurate Binding Poses but Autodock4 Forms Better Binding Affinity. J. Chem. Inf. Model 2020, 60, 204–211. [Google Scholar] [CrossRef]
- Abookleesh, F.; Mosa, F.E.S.; Barakat, K.; Ullah, A. Assessing Molecular Docking Tools to Guide the Design of Polymeric Materials Formulations: A Case Study of Canola and Soybean Protein. Polymers 2022, 14, 3690. [Google Scholar] [CrossRef]
- He, J.; Zheng, Z.-P.; Zhu, Q.; Guo, F.; Chen, J. Encapsulation Mechanism of Oxyresveratrol by β-Cyclodextrin and Hydroxypropyl-β-Cyclodextrin and Computational Analysis. Molecules 2017, 22, 1801. [Google Scholar] [CrossRef] [Green Version]
- Taraszewska, J.; Wójcik, J. Complexation of inorganic anions by β-cyclodextrin studied by polarography and 1H NMR. Supramol. Chem. 1993, 2, 337–343. [Google Scholar] [CrossRef]
- Buvári, Á.; Barcza, L. Complex formation of inorganic salts with β-cyclodextrin. J. Inc. Macrocycl. Chem 1989, 7, 379–389. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| T (K) | KC (×104 M−1) | ΔH (kcal/mol) | ΔG (kcal/mol) | ΔS (cal/molK) |
|---|---|---|---|---|
| 293 | 2.23 ± 0.08 | –9.82 ± 0.80 | −5.83 ± 0.02 | −13.56 ± 2.68 |
| 298 | 1.76 ± 0.07 | −5.79 ± 0.02 | ||
| 303 | 1.34 ± 0.06 | −5.72 ± 0.03 | ||
| 308 | 0.90 ± 0.06 | −5.57 ± 0.04 | ||
| 313 | 0.81 ± 0.05 | −5.60 ± 0.04 |
| KC (×104 M−1) | ΔG (kcal/mol) | ΔH (kcal/mol) | ΔS(kcal/molK) | |
|---|---|---|---|---|
| KI 10 mM | 2.59 ± 0.02 | −5.920 ± 0.005 | −9.53 ± 0.42 | −12.20 ± 1.40 |
| KI 50 mM | 2.27 ± 0.02 | −5.840 ± 0.005 | −9.01 ± 0.43 | −10.59 ± 1.41 |
| KI 100 mM | 1.99 ±0.02 | −5.760 ± 0.006 | −8.87 ± 0.43 | −10.41 ± 1.43 |
| KI 200 mM | 1.58 ± 0.02 | −5.630 ± 0.007 | −8.61 ± 0.43 | −9.75 ± 0.72 |
| LiI 100 mM | 2.27 ± 0.01 | −5.840 ± 0.003 | −5.94 ± 0.37 | −0.32 ± 1.21 |
| NaI 100mM | 2.28 ± 0.01 | −5.840 ± 0.003 | −9.50 ± 0.36 | −12.28 ± 1.21 |
| CsI 100 mM | 2.50 ± 0.02 | −5.900 ± 0.005 | −8.02 ± 0.93 | −7.95 ± 1.56 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kouderis, C.; Tsigoias, S.; Siafarika, P.; Kalampounias, A.G. The Effect of Alkali Iodide Salts in the Inclusion Process of Phenolphthalein in β-Cyclodextrin: A Spectroscopic and Theoretical Study. Molecules 2023, 28, 1147. https://doi.org/10.3390/molecules28031147
Kouderis C, Tsigoias S, Siafarika P, Kalampounias AG. The Effect of Alkali Iodide Salts in the Inclusion Process of Phenolphthalein in β-Cyclodextrin: A Spectroscopic and Theoretical Study. Molecules. 2023; 28(3):1147. https://doi.org/10.3390/molecules28031147
Chicago/Turabian StyleKouderis, Constantine, Stefanos Tsigoias, Panagiota Siafarika, and Angelos G. Kalampounias. 2023. "The Effect of Alkali Iodide Salts in the Inclusion Process of Phenolphthalein in β-Cyclodextrin: A Spectroscopic and Theoretical Study" Molecules 28, no. 3: 1147. https://doi.org/10.3390/molecules28031147