2.1. Theoretical Analysis of FLP Sites on Albite Surfaces
The basic structural units of albite (NaAlSi
3O
8) are [SiO
4] and [AlO
4] tetrahedrons, and each oxygen atom in the skeleton is shared with two adjacent tetrahedrons. Because of the charge imbalance caused by Al replacing Si, a Na
+ cation with balanced charge is introduced into the structure and is located in the pores of the tetrahedral skeletal structure. Albite minerals have two common exposed surfaces, (001) and (010), in a natural or grinding external force situation [
14,
15], and the structures are shown in
Figure 1.
Initially, we analyzed the active sites in the two surface structures of albite according to Lewis acid–base theory. The tetrahedral central atoms in albite structures Al and Si have four sp
3 hybrid orbitals, which are bonded to four O atoms on the (001) surface. [AlO
3] lacking one oxygen is exposed on the surface layer. Al is in the coordination unsaturated state and has an empty orbital, which can accept electron pairs; Al acts as a Lewis acid (LA). The oxygen atom exposed on the surface has an electron rich attribute and acts as a Lewis base (LB). [SiO
3] lacking oxygen is exposed on the (010) surface. Si is in the coordination unsaturated state, its valence orbit can accept electrons, and it acts as a LA; similarly, the oxygen atom exposed to the surface acts as a LB. Both LAs and LBs on the two surfaces are distributed on the skeleton of [AlO
4] and [SiO
4]. The formation of solid frustrated Lewis acid–base pairs requires not only independent LAs and LBs but also appropriate sterically encumbered matching pairs. Albite is a mineral with an ultra-microchannel structure, and the pore diameter parallel to the channel is mainly below 0.3 nm [
16,
17,
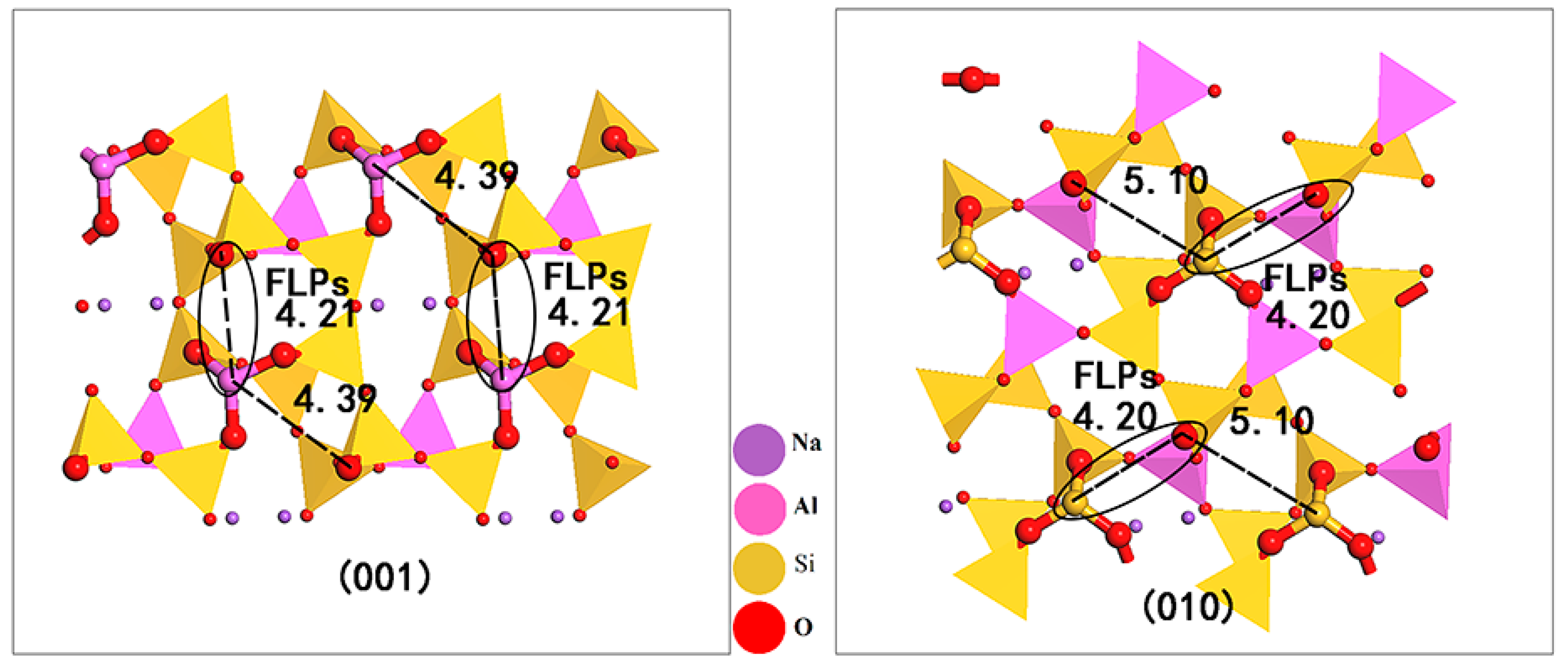
18]. Due to its undulating and uneven surface, many micropores are formed that are not parallel to the crystal surface; that is, the Lewis acid and base sites distributed on the surface skeleton of albite are not in the same horizontal plane. On the (001) surface, there were two sterically encumbered combinations: one group was located on the channel skeleton, with a distance of 4.21 Å, and the other group was located on the pore skeleton, with a distance of 4.39 Å. On the (010) surface, there were also two combinations: the distance on the channel skeleton was 5.10 Å, and the distance on the pore skeleton was 4.20 Å. Using the viewpoint that a distance between Lewis acid–base sites of approximately 4 Å is appropriate [
12,
19,
20], the Lewis acid–base pairs on the channel skeleton of the (001) face and the pore skeleton of the (010) face are matched; therefore, there are FLPs on both surfaces of albite.
Second, to further verify the characteristics of FLPs on the surface of albite, we calculated the reaction barrier for activating H
2 (see
Figure 2). The results showed that FLPs on both surfaces could enable H
2 heterolysis. The dissociation activation energy of the (001) surface was 36.48 kJ/mol, and that of the (010) surface was 1.01 kJ/mol. Heterolysis fragments of H
− and H
+ were adsorbed on the LA and LB, respectively. Erker et al. studied the activation mechanism of small molecules H
2 by FLPs and thought that the activation of small molecules by FLPs was based on the polarization of the electrostatic field between the electron receptor and donor acid–base pair [
21,
22]. Using a Lewis acid–base electrostatic field, an activity region would be formed, and the energy barrier in the reaction process was generated in the preparatory entry stage of small molecules. Once small molecules entered the active region of FLPs, the reaction energy barrier disappeared, and a stable dissociated product was finally formed [
23]. We also found in the calculation that when H
2 is at a suitable distance from the Lewis acid–base pair on the two surfaces, direct dissociation adsorption occurs. When H
2 enters the active region formed by LA and LB, it is directly dissociated. The transition-state (TS) structure of H
2 dissociation (
Figure 2) shows that the H
2 molecule has not been dissociated; in fact, the activation energies of 36.48 kJ/mol and 1.01 kJ/mol are required for H
2 to enter the activity region from outside. The FLPs on the two surfaces have a strong activation effect on H
2, showing evident FLP characteristics. Thus, albite is a natural mineral with an ultra-microchannel structure, and its surface coordination unsaturated Si, Al, and O atoms are distributed on the skeleton formed by [AlO
4] and [SiO
4], which can form mutually independent LAs and LBs. Its ultra-microchannel structure causes the LAs and LBs to form matching subnanoscale sterically encumbered minerals, thus forming FLPs.
2.2. Effect on C-H Bond Activation of Methane
To deeply understand the adsorption and activation mechanism of CH
4 on the FLPs of the albite surface, we initially calculated the adsorption energies of CH
4, CH
3, H and CH
3/H on the two surfaces (see
Table 1). Using the table, the most stable CH
4 is adsorbed on the LA (Al,Si), and the most stable adsorption of the co-adsorption system of CH
4 dissociated species CH
3/H is located on the LA (Al,Si)/LB (O). Additionally, the adsorption energies of CH
4 and dissociated species CH
3, H on the two surfaces follow the trend of CH
4 < CH
3 < H. The adsorption of CH
3 and H on the (010) surface is stronger than that on the (001) surface, and their adsorption energies are 150.09 kJ/mol and 102.4 kJ/mol higher, respectively. The adsorption of CH
4 on the two surfaces had a similar phenomenon to that of H
2, showing different adsorption between the CH
4 and LA (Al,Si) distances and degrees of polarization, as shown in
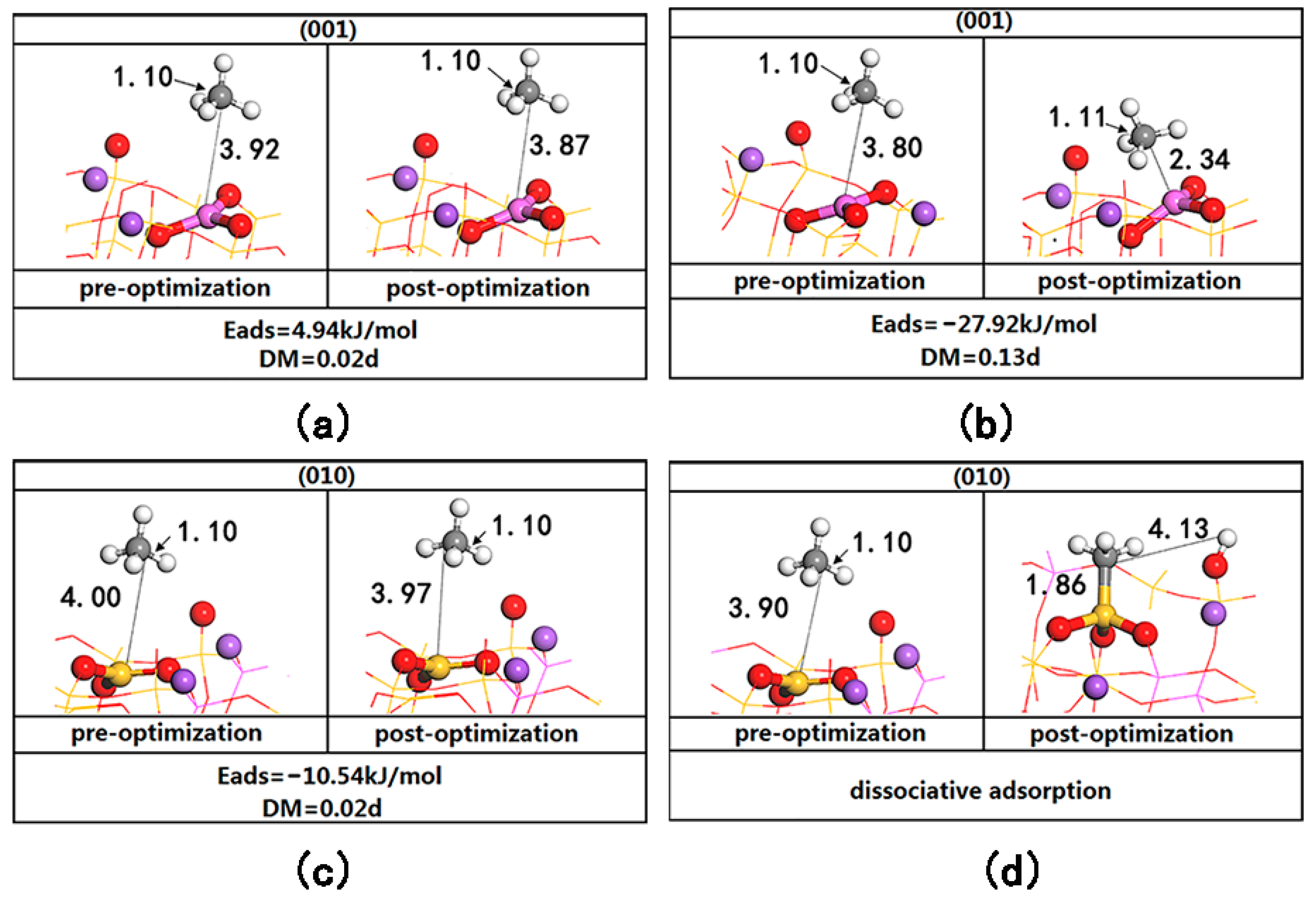
Figure 3. On the (001) surface, when CH
4 is far from the LA (Al) site (d
C-Al > 3.8 Å), the adsorption energy is positive, indicating instability (see
Figure 3a). When CH
4 is close to the LA (Al) site (d
C-Al < 3.8 Å), CH
4 is stably adsorbed at a position of 2.34 Å from C to Al (see
Figure 3b). The adsorption energy is −27.86 kJ/mol, generating a dipole moment of 0.13 D, indicating that CH
4 has been polarized; the C-H bond is weakened and stretched to 1.11 Å. On the (010) surface, when CH
4 is far from the LA (Si) site (d
C-Si > 3.9 Å), the adsorption of CH
4 on the surface is weak (see
Figure 3c). When CH
4 is close to the LA (Si) site (d
C-Si < 3.9 Å), heterolysis occurs directly, CH
3 is adsorbed on the LA (Si), and H is adsorbed on the LB (O), as shown in
Figure 3d. This is completely consistent with the phenomenon described by Grimme S. et al., in that “once the molecule enters the hole of FLPs, the reaction proceeds without a barrier” [
23]; that is, CH
4 can undergo direct dissociative adsorption in the active area of the Lewis acid–base pairs on the (010) surface.
To analyze the micro-mechanism of its dissociative adsorption process, we observed the dynamic change in the (010) surface adsorption configuration, as shown in
Figure 4, and the corresponding Hirshfeld charge distribution and CH
4 dipole moments were calculated (see
Table 2). Interestingly, there are three stages in the dissociative adsorption process of CH
4. In the first stage (a–d), as CH
4 gradually approaches the LA (Si), the LA (Si) is in a state of acquiring electrons, CH
4 is in a state of donating electrons, and the LB (O) has no electron transfer; the CH
4 dipole moment gradually increases, and CH
4 is in a state of physical adsorption. In the second stage (e-f), when the distance between C and Si is 2.23 Å, CH
4 chemisorbs with the LA (Si). At this time, the LA (Si) still maintains a state of acquiring electrons, the LB (O) begins to enter a state of donating electrons, and the dipole moment of CH
4 rapidly increases from 0.24 D to 0.57 D. The C-H bond closest to the LB (O) is stretched to 1.23 Å. In the third stage (g-h), when the C-H bond is stretched to 1.34 Å, the C-H bond is broken, and the H-LB (O) bond forms at the same time. After reaching a stable state, CH
3 and H are adsorbed on the LA (Si) and the LB (O), respectively, with charges of −0.07 e and +0.18 e. Based on the above reaction details, when CH
4 is adsorbed on the LA (Si) on the albite (010) surface, LA (Si) can obtain electrons and has strong electrophilicity. Therefore, it initially attacks the electron-enriched C atom in CH
4, causing the electrons from the CH
4 bonding orbital σ to flow to the LA (Si) on the surface to form a Si-CH
4 intermediate; the C-H bond of CH
4 is weakened, which then promotes the nucleophilic attack of the LB (O) and provides electrons to the antibonding orbital σ* of the C-H bond of CH
4 until it ruptures. In the process of CH
4 activation, the LA, as an electrophilic reagent, plays an important role in the polarization of CH
4; a greater degree of polarization of CH
4 correlates to a stronger reaction activity. The LB, as a nucleophilic reagent, can obtain protons, and they synergistically complete the cleavage of CH
4. Since the electrophilicity of the LA on the (001) surface is weaker than that on the (010) surface, its polarization to CH
4 is relatively weak, and CH
4 cannot be directly cleaved.
Second, the reaction barriers of CH
4 activation by two surface FLPs were calculated (see
Figure 5); both surfaces show good activity. The reaction heat of CH
4 dissociation on the (001) surface is −71.72 kJ/mol, and an activation energy of 50.07 kJ/mol needs to be overcome. The reaction heat of CH
4 dissociation on the (010) surface is −292.61 kJ/mol, and the activation energy is 2.63 kJ/mol. From the two surface TS structures, CH
4 on the (001) surface has been dissociated, while CH
4 on the (010) surface has not been dissociated. CH
4 was polarized and contained the activation energy required for methane to enter the activity region from outside. Once CH
4 enters the activity region, heterolysis occurs according to the direct dissociation mechanism.
From a thermodynamic point of view, the dissociation reaction heat of the (010) surface is far greater than that of the (001) surface, indicating that the bonding effect between dissociated fragments CH
3 and H with acid–base sites of the (010) surface is stronger than that of the (001) surface. From a kinetic point of view, the activation energy of CH
4 on the (010) surface is much lower than that on the (001) surface. Although the dissociation activation energy of CH
4 on the (010) surface is the lowest, the strong bonding effect between CH
3/H and the surface means that desorption requires a higher desorption energy, which is in accordance with the Brónsted Evans Polanyi rule [
24].
2.3. Effect on the Methyl C-C Coupling Mode
The albite catalyst is prepared by high-temperature calcination and grinding using an external force to physically mix and fully expose the two common exposed surfaces, (001) and (010). In fact, it is a double combination catalyst with (001) and (010) coexisting, and they participate in the catalytic reaction of methane at the same time.
Figure 6 shows the catalytic conversion path of methane on the albite catalyst; when methane enters the catalyst surface from the gas phase, the FLPs on both surfaces cause methane heterolysis according to the same activation mechanism, and the dissociated fragments CH
3 and H are located on LA and LB sites on both surfaces, respectively. Since the bonding effect with CH
3 and H adsorbed on the (001) surface is much weaker than that on the (010) surface, they preferentially desorb during temperature rise desorption, causing a spillover phenomenon; at the same time, because the Lewis acid site Al on the (001) surface has an empty orbital and CH
3− has a lone electron pair, after the two bond, that is, CH
3− adsorbed on (001) surface can neither give nor gain electrons, C-C coupling does not occur on (001) surface. Similarly, the spilled H
+ cannot gain electrons from Lewis acid site Al in (001) surface to form H
−. Therefore, the coupling reaction of CH
3 and H overflowers on (001) surface can only occur on (010) surface, that is, CH
3 and H preferentially desorbed become overflowers and diffuse to the (010) surface with an increase in the concentration gradient. In the overflow process, since the bonding effect of CH
3 on the (001) surface is much smaller than that of H, CH
3 desorbs first.
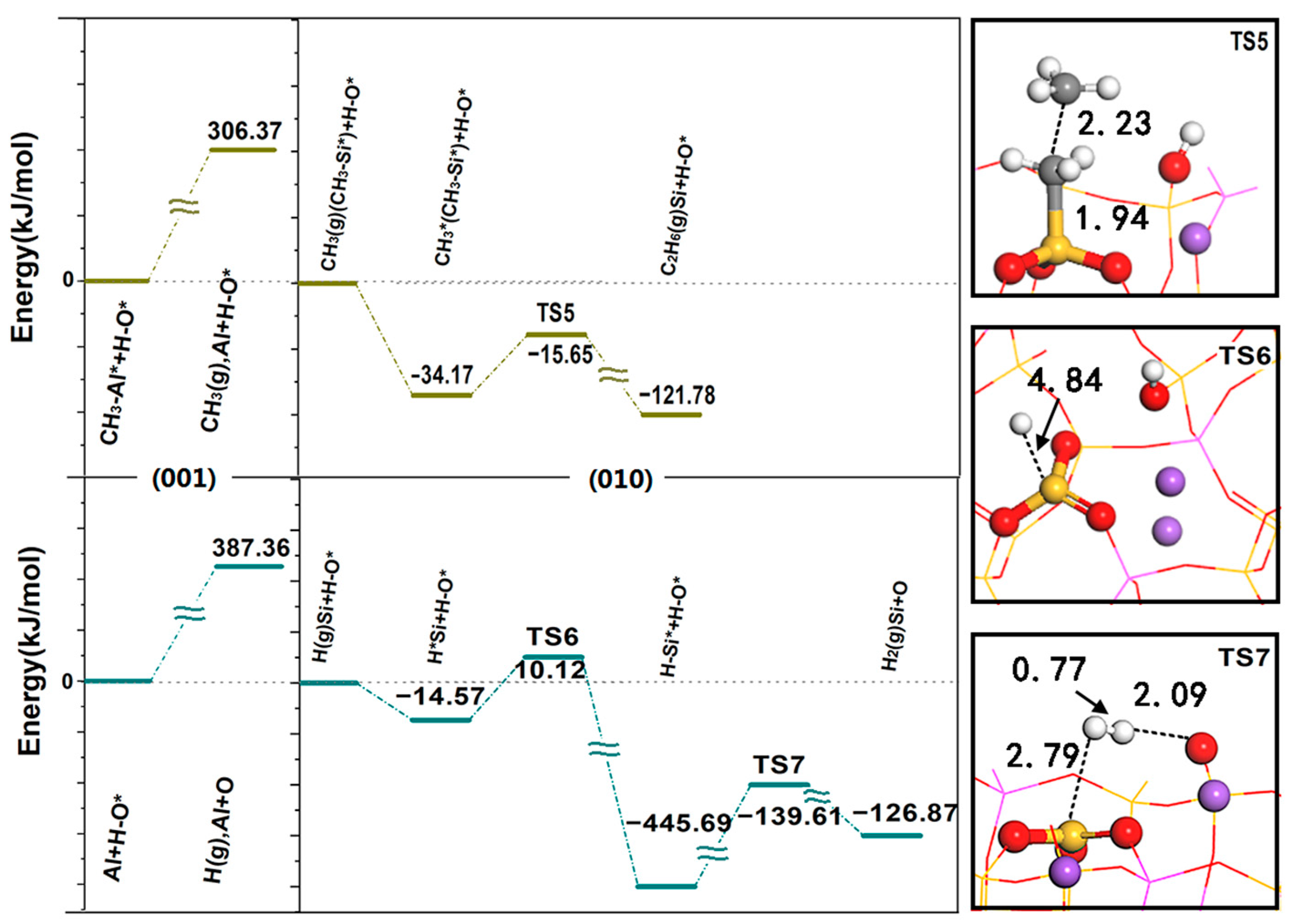
Figure 7 shows the desorption energy of CH
3 spillover and H spillover on the (001) surface and the reaction energy barrier on the (010) surface. The desorption of CH
3 requires an activation energy of 306.37 kJ/mol, and the diffusion to the (010) surface is coupled with the adsorbed methyl group through the E-R mechanism. The formation of C
2H
6 is an exothermic process and only requires an activation energy of 18.51 kJ/mol, indicating that C-C coupling has a strong bonding effect and shows good activity. CH
3 is E-R coupled on the surface, which is advantageous both in thermodynamics and kinetics, and part of the C
2H
6 generated by coupling continues to dehydrogenate and then generates C
2H
4. The above results are consistent with the experimental results. At 1073 K, the analysis of the relationship between CH
4 conversion, C
2 hydrocarbon selectivity and space velocity (GHSV) shows that CH
4 conversion decreases with increasing GHSV; CH
4 activation is fully achieved on the surface FLPs because a higher GHSV corresponds to a shorter contact time and lower CH
4 conversion rate. The phenomenon of increasing C
2H
6 and decreasing C
2H
4 with increasing GHSV further confirms that the coupling of CH
3 to produce C
2H
6 occurs on the surface rather than in the gas phase. C
2H
4 is the product of dehydrogenation of C
2H
6 in the gas phase [
13]. When H overflows and diffuses to the (010) surface, and since the electronegativity of the H atom (2.1) is greater than that of the Si atom (1.8), H spillover can easily acquire unpaired electrons from the LA (Si) to form H
−. This reaction process produces a strong exothermic reaction with a reaction heat of −431.12 kJ/mol and an activation energy of 24.69 kJ/mol. After that, H adsorbed on the LA (Si) reacts with proton H adsorbed on the LB (O) to generate H
2 through the L-H mechanism; the required activation energy is 306.08 kJ/mol, and the closed cycle of methane conversion is completed. Since the desorption energy required for H spillover on the (001) surface (387.36 kJ/mol) is the highest during the whole reaction process, this is the rate-determining step in the whole cycle process.
This spillover phenomenon not only enables highly efficient coupling of methyl groups but also plays two important roles: (1) to remove CH3 and H, which are firmly adsorbed on the surface of (010), to regenerate the acid–base sites and (2) to avoid deep dehydrogenation, which is conducive to the selectivity of C2 hydrocarbons. Because the heterolytic activation of CH4 by FLPs on the surface of albite is a synergistic catalytic mechanism, the dehydrogenation activation of methane can only be achieved using the joint action of the LAs and LBs. After the Lewis acid–base sites are regenerated, the methane conversion cycle reaction can continue; the desorption order of the dissociated fragments adsorbed on the Lewis acid–base sites directly affects the reaction direction of conversion. When the LB site preferentially regenerates, the carbon species adsorbed on the LA site undergo dehydrogenation, whereas when the carbon species adsorbed on the LA site preferentially desorb, the occurrence of deep dehydrogenation is avoided. Since the bonding effect between the (001) face of CH3 and the surface is much smaller than that of H, the preferential desorption of CH3 avoids further dehydrogenation. At the same time, CH3 preferentially overflowed is coupled on the (010) surface to form ethane, which also avoids further dehydrogenation of CH3 adsorbed on the (010) surface. This reasonably explains the experimental results that the selectivity of C2 hydrocarbons is maintained across up to 99% and there are zero carbon deposits in the temperature range of 873~1173 K.
2.4. Effect of Doping Modification on CH4 Conversion
From the above analysis, the albite catalyst shows good catalytic ability for methane C-H bond activation and methyl C-C coupling, has high selectivity for C
2 hydrocarbons and does not easily undergo carbon deposition. However, we also find that the conversion of CH
4 is relatively low, and it increases with increasing temperature. This phenomenon shows that the conversion of CH
4 is closely related to temperature. Since the FLP catalytic reaction is generally a synergistic mechanism, the regeneration of acid–base sites is crucial to the cyclic reaction. For albite catalysts, the desorption of dissociated fragment H on the (001) surface is the rate-determining step of the cyclic catalytic reaction. Under nonoxygen conditions, temperature is the main factor affecting the desorption of H, and higher desorption energy can lead to lower conversion. Therefore, on the basis of a lower desorption energy of CH
3 than that of H, a reduction in the desorption energy of H helps to improve methane conversion. In the albite structure, the Na
+ in the pores mainly balances the residual negative charges on the skeleton and affects the electrostatic field formed by the LA and LB. According to the principle of field strength superposition, doping high valence metal ions instead of Na
+ plays a role in regulating the electrostatic field of FLPs. In this experiment, we mixed the prepared albite catalyst with anhydrous lead chloride in different proportions, calcined it at 773 K, and formed a Pb
2+-doped Pb/Ab catalyst through ion exchange. To investigate the influence of Pb doping on the CH
4 conversion rate, we calculated the desorption energy of H and CH
3 before and after (001) surface doping, and the calculated results were analyzed and compared with the experimental values of CH
4 conversion and C
2 hydrocarbon selectivity before and after doping at 1073 K (see
Figure 8). The desorption energy of CH
3 after doping is still less than that of H, and the activation barrier of CH
4 is slightly increased, increasing from 50.07 kJ/mol to 59.21 kJ/mol; this result means that doping has less effect on the catalytic activity of CH
4, and the desorption energy of H is reduced from 387.36 kJ/mol to 337.46 kJ/mol, showing a decrease of 49.9 kJ/mol. Corresponding to the experimental results at 1073 K, the conversion of methane after doping was promoted by 2.45 times from 3.32% to 8.12%, and the selectivity remained above 99%. After doping, reducing the bonding effect of the (001) surface H has an evident effect on improving the conversion of CH
4. Therefore, we believe that a nonreduction (or very low reduction) in the activity and a smaller desorption energy of the CH
3 spillover than that of the H spillover using modification maximumly reduces the bonding effect between H and the surface to greatly improve the conversion rate. The above are our preliminary research results, and the application of albite catalysts requires further in-depth research.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}