
Identification of Potential Modulators of a Pathogenic G Protein-Gated Inwardly Rectifying K+ Channel 4 Mutant: In Silico Investigation in the Context of Drug Discovery for Hypertension

 , , ,
, , ,
Abstract
:
1. Introduction
2. Results and Discussion
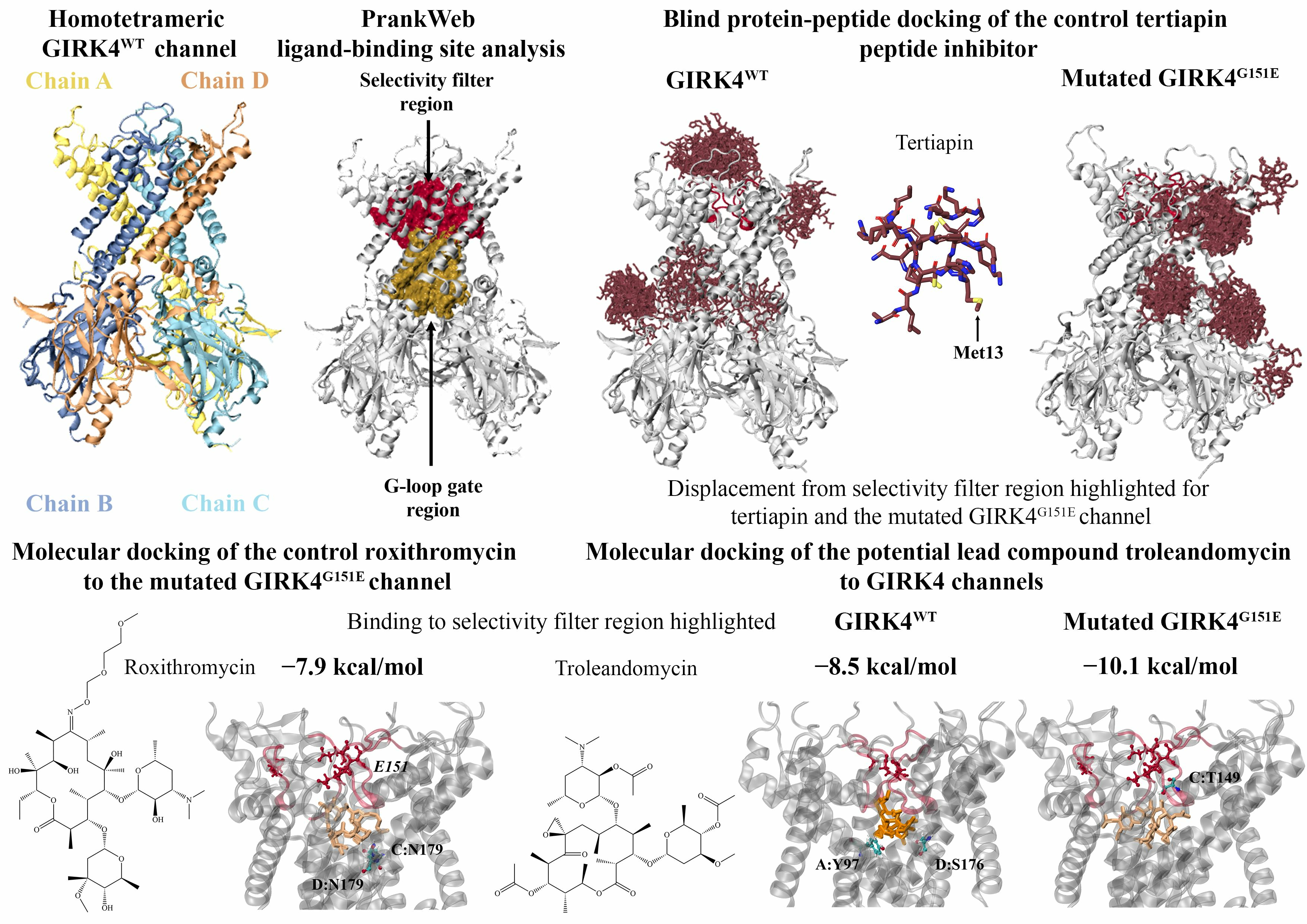
2.1. Protein–Peptide Docking of Tertiapin and Tertiapin-Q to Homotetrameric GIRK4
2.2. Identification of Potential Ligand-Binding Sites in the GIRK4WT and GIRK4G151E Channels
2.3. Molecular Docking to the Central Cavity Region of Homotetrameric GIRK4
2.4. Molecular Docking to the Region Encompassing the G-Loop Gate
3. Materials and Methods
3.1. Preparation of Protein Structures
3.2. Preparation of Ligands
3.3. Ligand-Binding Site Analysis and Molecular Docking
3.4. Protein–Peptide Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gurgenci, T.; Geraghty, S.; Wolley, M.; Yang, J. Screening for primary aldosteronism: How to adjust existing antihypertensive medications to avoid diagnostic errors. Aust. J. Gen. Pract. 2020, 49, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Mulatero, P.; Monticone, S.; Deinum, J.; Amar, L.; Prejbisz, A.; Zennaro, M.-C.; Beuschlein, F.; Rossi, G.P.; Nishikawa, T.; Morganti, A.; et al. Genetics, prevalence, screening and confirmation of primary aldosteronism: A position statement and consensus of the Working Group on Endocrine Hypertension of The European Society of Hypertension. J. Hypertens. 2020, 38, 1919–1928. [Google Scholar] [CrossRef] [PubMed]
- Funder, J.W.; Carey, R.M.; Mantero, F.; Murad, M.H.; Reincke, M.; Shibata, H.; Stowasser, M.; Young, W.F. The Management of Primary Aldosteronism: Case Detection, Diagnosis, and Treatment: An Endocrine Society Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 2016, 101, 1889–1916. [Google Scholar] [CrossRef] [PubMed]
- Scholl, U.I. Genetics of Primary Aldosteronism. Hypertension 2022, 79, 887–897. [Google Scholar] [CrossRef]
- Turcu, A.F.; Yang, J.; Vaidya, A. Primary aldosteronism—A multidimensional syndrome. Nat. Rev. Endocrinol. 2022, 18, 665–682. [Google Scholar] [CrossRef]
- Zennaro, M.-C.; Boulkroun, S.; Fernandes-Rosa, F. Genetic Causes of Functional Adrenocortical Adenomas. Endocr. Rev. 2017, 38, 516–537. [Google Scholar] [CrossRef]
- Scholl, U.I.; Nelson-Williams, C.; Yue, P.; Grekin, R.; Wyatt, R.J.; Dillon, M.J.; Couch, R.; Hammer, L.K.; Harley, F.L.; Farhi, A.; et al. Hypertension with or without adrenal hyperplasia due to different inherited mutations in the potassium channel KCNJ5. Proc. Natl. Acad. Sci. USA 2012, 109, 2533–2538. [Google Scholar] [CrossRef]
- Williams, T.A.; Monticone, S.; Mulatero, P. KCNJ5 Mutations Are the Most Frequent Genetic Alteration in Primary Aldosteronism. Hypertension 2015, 65, 507–509. [Google Scholar] [CrossRef]
- Cui, M.; Cantwell, L.; Zorn, A.; Logothetis, D.E. Kir Channel Molecular Physiology, Pharmacology, and Therapeutic Implications. Handb. Exp. Pharmacol. 2021, 267, 277–356. [Google Scholar] [CrossRef]
- Jeremic, D.; Sanchez-Rodriguez, I.; Jimenez-Diaz, L.; Navarro-Lopez, J.D. Therapeutic potential of targeting G protein-gated inwardly rectifying potassium (GIRK) channels in the central nervous system. Pharmacol. Ther. 2021, 223, 107808. [Google Scholar] [CrossRef]
- Walsh, K. Targeting GIRK Channels for the Development of New Therapeutic Agents. Front. Pharmacol. 2011, 2, 64. [Google Scholar] [CrossRef] [PubMed]
- Campos-Ríos, A.; Rueda-Ruzafa, L.; Lamas, J.A. The Relevance of GIRK Channels in Heart Function. Membranes 2022, 12, 1119. [Google Scholar] [CrossRef] [PubMed]
- Kano, H.; Toyama, Y.; Imai, S.; Iwahashi, Y.; Mase, Y.; Yokogawa, M.; Osawa, M.; Shimada, I. Structural mechanism underlying G protein family-specific regulation of G protein-gated inwardly rectifying potassium channel. Nat. Commun. 2019, 10, 2008. [Google Scholar] [CrossRef] [PubMed]
- Cui, M.; Xu, K.; Gada, K.D.; Shalomov, B.; Ban, M.; Eptaminitaki, G.C.; Kawano, T.; Plant, L.D.; Dascal, N.; Logothetis, D.E. A novel small-molecule selective activator of homomeric GIRK4 channels. J. Biol. Chem. 2022, 298, 102009. [Google Scholar] [CrossRef]
- Tauber, P.; Penton, D.; Stindl, J.; Humberg, E.; Tegtmeier, I.; Sterner, C.; Beuschlein, F.; Reincke, M.; Barhanin, J.; Bandulik, S.; et al. Pharmacology and Pathophysiology of Mutated KCNJ5 Found in Adrenal Aldosterone-Producing Adenomas. Endocrinology 2014, 155, 1353–1362. [Google Scholar] [CrossRef]
- Mussa, A.; Camilla, R.; Monticone, S.; Porta, F.; Tessaris, D.; Verna, F.; Mulatero, P.; Einaudi, S. Polyuric-polydipsic syndrome in a pediatric case of non-glucocorticoid remediable familial hyperaldosteronism. Endocr. J. 2012, 59, 497–502. [Google Scholar] [CrossRef]
- Kobayashi, T.; Ikeda, K.; Kojima, H.; Niki, H.; Yano, R.; Yoshioka, T.; Kumanishi, T. Ethanol opens G-protein-activated inwardly rectifying K+ channels. Nat. Neurosci. 1999, 2, 1091–1097. [Google Scholar] [CrossRef]
- Yow, T.T.; Pera, E.; Absalom, N.; Heblinski, M.; Johnston, G.A.; Hanrahan, J.R.; Chebib, M. Naringin directly activates inwardly rectifying potassium channels at an overlapping binding site to tertiapin-Q. Br. J. Pharmacol. 2011, 163, 1017–1033. [Google Scholar] [CrossRef]
- Kaufmann, K.; Romaine, I.; Days, E.; Pascual, C.; Malik, A.; Yang, L.; Zou, B.; Du, Y.; Sliwoski, G.; Morrison, R.D.; et al. ML297 (VU0456810), the first potent and selective activator of the GIRK potassium channel, displays antiepileptic properties in mice. ACS Chem. Neurosci. 2013, 4, 1278–1286. [Google Scholar] [CrossRef]
- Wen, W.; Wu, W.; Romaine, I.M.; Kaufmann, K.; Du, Y.; Sulikowski, G.A.; Weaver, C.D.; Lindsley, C.W. Discovery of ‘molecular switches’ within a GIRK activator scaffold that afford selective GIRK inhibitors. Bioorg Med. Chem. Lett. 2013, 23, 4562–4566. [Google Scholar] [CrossRef]
- Xu, Y.; Cantwell, L.; Molosh, A.I.; Plant, L.D.; Gazgalis, D.; Fitz, S.D.; Dustrude, E.T.; Yang, Y.; Kawano, T.; Garai, S.; et al. The small molecule GAT1508 activates brain-specific GIRK1/2 channel heteromers and facilitates conditioned fear extinction in rodents. J. Biol. Chem. 2020, 295, 3614–3634. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Washiyama, K.; Ikeda, K. Inhibition of G Protein-Activated Inwardly Rectifying K+ Channels by Various Antidepressant Drugs. Neuropsychopharmacology 2004, 29, 1841–1851. [Google Scholar] [CrossRef] [PubMed]
- Kuzhikandathil, E.V.; Oxford, G.S. Classic D1 dopamine receptor antagonist R-(+)-7-chloro-8-hydroxy-3-methyl-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine hydrochloride (SCH23390) directly inhibits G protein-coupled inwardly rectifying potassium channels. Mol. Pharmacol. 2002, 62, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Ung, P.M.-U.; Zahoránszky-Kőhalmi, G.; Zakharov, A.V.; Martinez, N.J.; Simeonov, A.; Glaaser, I.W.; Rai, G.; Schlessinger, A.; Marugan, J.J.; et al. Identification of a G-Protein-Independent Activator of GIRK Channels. Cell Rep. 2020, 31, 107770. [Google Scholar] [CrossRef]
- Niu, Y.; Tao, X.; Touhara, K.K.; MacKinnon, R. Cryo-EM analysis of PIP(2) regulation in mammalian GIRK channels. Elife 2020, 9, e60552. [Google Scholar] [CrossRef]
- Tao, X.; Avalos, J.L.; Chen, J.; MacKinnon, R. Crystal structure of the eukaryotic strong inward-rectifier K+ channel Kir2.2 at 3.1 A resolution. Science 2009, 326, 1668–1674. [Google Scholar] [CrossRef]
- Morais-Cabral, J.H.; Zhou, Y.; MacKinnon, R. Energetic optimization of ion conduction rate by the K+ selectivity filter. Nature 2001, 414, 37–42. [Google Scholar] [CrossRef]
- Corradi, V.; Bukiya, A.N.; Miranda, W.E.; Cui, M.; Plant, L.D.; Logothetis, D.E.; Tieleman, D.P.; Noskov, S.Y.; Rosenhouse-Dantsker, A. A molecular switch controls the impact of cholesterol on a Kir channel. Proc. Natl. Acad. Sci. USA 2022, 119, e2109431119. [Google Scholar] [CrossRef]
- Jin, W.; Lu, Z. Synthesis of a stable form of tertiapin: A high-affinity inhibitor for inward-rectifier K+ channels. Biochemistry 1999, 38, 14286–14293. [Google Scholar] [CrossRef]
- Jin, W.; Klem, A.M.; Lewis, J.H.; Lu, Z. Mechanisms of inward-rectifier K+ channel inhibition by tertiapin-Q. Biochemistry 1999, 38, 14294–14301. [Google Scholar] [CrossRef]
- Refik, K.; Elizabeth, J.C.; David, J.A.; Mark, C.B. Tertiapin-Q blocks recombinant and native large conductance K+ channels in a use-dependent manner. J. Pharmacol. Exp. Ther. 2005, 314, 1353. [Google Scholar] [CrossRef]
- Kitamura, H.; Yokoyama, M.; Akita, H.; Matsushita, K.; Kurachi, Y.; Yamada, M. Tertiapin potently and selectively blocks muscarinic K(+) channels in rabbit cardiac myocytes. J. Pharmacol. Exp. Ther. 2000, 293, 196–205. [Google Scholar]
- Hilder, T.A.; Chung, S.-H. Conductance properties of the inwardly rectifying channel, Kir3.2: Molecular and Brownian dynamics study. Biochim. Biophys. Acta (BBA) Biomembr. 2013, 1828, 471–478. [Google Scholar] [CrossRef]
- Patel, D.; Kuyucak, S.; Doupnik, C.A. Structural Determinants Mediating Tertiapin Block of Neuronal Kir3.2 Channels. Biochemistry 2020, 59, 836–850. [Google Scholar] [CrossRef]
- Li, D.; Chen, R.; Chung, S.H. Molecular dynamics of the honey bee toxin tertiapin binding to Kir3.2. Biophys. Chem. 2016, 219, 43–48. [Google Scholar] [CrossRef]
- Doupnik, C.A.; Parra, K.C.; Guida, W.C. A computational design approach for virtual screening of peptide interactions across K+ channel families. Comput. Struct. Biotechnol. J. 2015, 13, 85–94. [Google Scholar] [CrossRef]
- Ramu, Y.; Klem, A.M.; Lu, Z. Short Variable Sequence Acquired in Evolution Enables Selective Inhibition of Various Inward-Rectifier K+ Channels. Biochemistry 2004, 43, 10701–10709. [Google Scholar] [CrossRef]
- Choi, M.; Scholl, U.I.; Yue, P.; Björklund, P.; Zhao, B.; Nelson-Williams, C.; Ji, W.; Cho, Y.; Patel, A.; Men, C.J.; et al. K+ Channel Mutations in Adrenal Aldosterone-Producing Adenomas and Hereditary Hypertension. Science 2011, 331, 768–772. [Google Scholar] [CrossRef]
- Xu, X.; Nelson, J.W. Solution structure of tertiapin determined using nuclear magnetic resonance and distance geometry. Proteins 1993, 17, 124–137. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Xiao, S.; Xie, X.; Zhou, H.; Pang, C.; Li, S.; Zhang, H.; Logothetis, D.E.; Zhan, Y.; An, H. Three pairs of weak interactions precisely regulate the G-loop gate of Kir2.1 channel. Proteins Struct. Funct. Bioinform. 2016, 84, 1929–1937. [Google Scholar] [CrossRef] [PubMed]
- Scholl, U.I.; Abriola, L.; Zhang, C.; Reimer, E.N.; Plummer, M.; Kazmierczak, B.I.; Zhang, J.; Hoyer, D.; Merkel, J.S.; Wang, W.; et al. Macrolides selectively inhibit mutant KCNJ5 potassium channels that cause aldosterone-producing adenoma. J. Clin. Investig. 2017, 127, 2739–2750. [Google Scholar] [CrossRef] [PubMed]
- Cui, M.; Alhamshari, Y.; Cantwell, L.; Ei-Haou, S.; Eptaminitaki, G.C.; Chang, M.; Abou-Assali, O.; Tan, H.; Xu, K.; Masotti, M.; et al. A benzopyran with antiarrhythmic activity is an inhibitor of Kir3.1-containing potassium channels. J. Biol. Chem. 2021, 296, 100535. [Google Scholar] [CrossRef] [PubMed]
- Fusi, F.; Trezza, A.; Tramaglino, M.; Sgaragli, G.; Saponara, S.; Spiga, O. The beneficial health effects of flavonoids on the cardiovascular system: Focus on K+ channels. Pharmacol. Res. 2020, 152, 104625. [Google Scholar] [CrossRef] [PubMed]
- Bonvino, N.P.; Liang, J.; McCord, E.D.; Zafiris, E.; Benetti, N.; Ray, N.B.; Hung, A.; Boskou, D.; Karagiannis, T.C. OliveNet™: A comprehensive library of compounds from Olea europaea. Database 2018, 2018, bay016. [Google Scholar] [CrossRef]
- Pitsillou, E.; Beh, R.C.; Liang, J.J.; Tang, T.S.; Zhou, X.; Siow, Y.Y.; Ma, Y.; Hu, Z.; Wu, Z.; Hung, A.; et al. EpiMed Coronabank Chemical Collection: Compound selection, ADMET analysis, and utilisation in the context of potential SARS-CoV-2 antivirals. J. Mol. Graph. Model. 2023, 125, 108602. [Google Scholar] [CrossRef] [PubMed]
- Trezza, A.; Cicaloni, V.; Porciatti, P.; Langella, A.; Fusi, F.; Saponara, S.; Spiga, O. From in silico to in vitro: A trip to reveal flavonoid binding on the Rattus norvegicus Kir6.1 ATP-sensitive inward rectifier potassium channel. PeerJ 2018, 6, e4680. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; De Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- The UniProt Consortium; Bateman, A.; Martin, M.-J.; Orchard, S.; Magrane, M.; Ahmad, S.; Alpi, E.; Bowler-Barnett, E.H.; Britto, R.; Bye-A-Jee, H.; et al. UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2023, 51, D523–D531. [Google Scholar] [CrossRef]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Kim, S.; Lee, J.; Jo, S.; Brooks, C.L., III; Lee, H.S.; Im, W. CHARMM-GUI ligand reader and modeler for CHARMM force field generation of small molecules. J. Comput. Chem. 2017, 38, 1879–1886. [Google Scholar] [CrossRef]
- Liang, J.J.; Pitsillou, E.; Ververis, K.; Guallar, V.; Hung, A.; Karagiannis, T.C. Investigation of small molecule inhibitors of the SARS-CoV-2 papain-like protease by all-atom microsecond modelling, PELE Monte Carlo simulations, and in vitro activity inhibition. Chem. Phys. Lett. 2022, 788, 139294. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.M.; Mittal, J.; Feig, M.; MacKerell, A.D. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ(1) and χ(2) dihedral angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; A Shoemaker, B.; A Thiessen, P.; Yu, B.; et al. PubChem 2023 update. Nucleic Acids Res. 2023, 51, D1373–D1380. [Google Scholar] [CrossRef]
- Dallakyan, S.; Olson, A.J. Small-Molecule Library Screening by Docking with PyRx. In Chemical Biology: Methods and Protocols; Hempel, J.E., Williams, C.H., Hong, C.C., Eds.; Springer: New York, NY, USA, 2015; pp. 243–250. [Google Scholar]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminformatics 2011, 3, 33. [Google Scholar] [CrossRef]
- Jakubec, D.; Skoda, P.; Krivak, R.; Novotny, M.; Hoksza, D. PrankWeb 3: Accelerated ligand-binding site predictions for experimental and modelled protein structures. Nucleic Acids Res. 2022, 50, W593–W597. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger LLC. Schrödinger Release 2022-2, Maestro; Schrödinger LLC: New York, NY, USA, 2022. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Schrödinger LLC. The PyMOL Molecular Graphics System, Version 1.8; Schrödinger LLC: New York, NY, USA, 2015. [Google Scholar]
- Zhou, P.; Jin, B.; Li, H.; Huang, S.Y. HPEPDOCK: A web server for blind peptide-protein docking based on a hierarchical algorithm. Nucleic Acids Res. 2018, 46, W443–W450. [Google Scholar] [CrossRef]
- Krissinel, E.; Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GIRK4WT | GIRK4G151E | |||
|---|---|---|---|---|
| Compound | Binding Affinity (kcal/mol) | Interactions | Binding Affinity (kcal/mol) | Interactions |
| EIPA | −6.7 | A: Y97 (H-bond), S176 (H-bond) | −6.3 | B: Y97 (H-bond), E147 (H-bond) |
| KB-R7943 | −7.6 | C: S143 (H-bond) | −7.4 | C: W108 (H-bond), E147 (H-bond), N179 (H-bond) |
| Nifedipine | −6.1 | - | −6.1 | B: S176 (H-bond) |
| Verapamil | −7.2 | - | −6.4 | - |
| Roxithromycin | −8.4 | A: Y97 (H-bond), N179 (H-bond) | −7.9 | C: N179 (H-bond), D: N179 (H-bond) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pitsillou, E.; Logothetis, A.N.O.; Liang, J.J.; El-Osta, A.; Hung, A.; AbuMaziad, A.S.; Karagiannis, T.C. Identification of Potential Modulators of a Pathogenic G Protein-Gated Inwardly Rectifying K+ Channel 4 Mutant: In Silico Investigation in the Context of Drug Discovery for Hypertension. Molecules 2023, 28, 7946. https://doi.org/10.3390/molecules28247946
Pitsillou E, Logothetis ANO, Liang JJ, El-Osta A, Hung A, AbuMaziad AS, Karagiannis TC. Identification of Potential Modulators of a Pathogenic G Protein-Gated Inwardly Rectifying K+ Channel 4 Mutant: In Silico Investigation in the Context of Drug Discovery for Hypertension. Molecules. 2023; 28(24):7946. https://doi.org/10.3390/molecules28247946
Chicago/Turabian StylePitsillou, Eleni, Alexander N. O. Logothetis, Julia J. Liang, Assam El-Osta, Andrew Hung, Asmaa S. AbuMaziad, and Tom C. Karagiannis. 2023. "Identification of Potential Modulators of a Pathogenic G Protein-Gated Inwardly Rectifying K+ Channel 4 Mutant: In Silico Investigation in the Context of Drug Discovery for Hypertension" Molecules 28, no. 24: 7946. https://doi.org/10.3390/molecules28247946