
Salazinic Acid and Norlobaridone from the Lichen Hypotrachyna cirrhata: Antioxidant Activity, α-Glucosidase Inhibitory and Molecular Docking Studies
 , , and
, , and
Abstract
:
1. Introduction
2. Results and Discussion
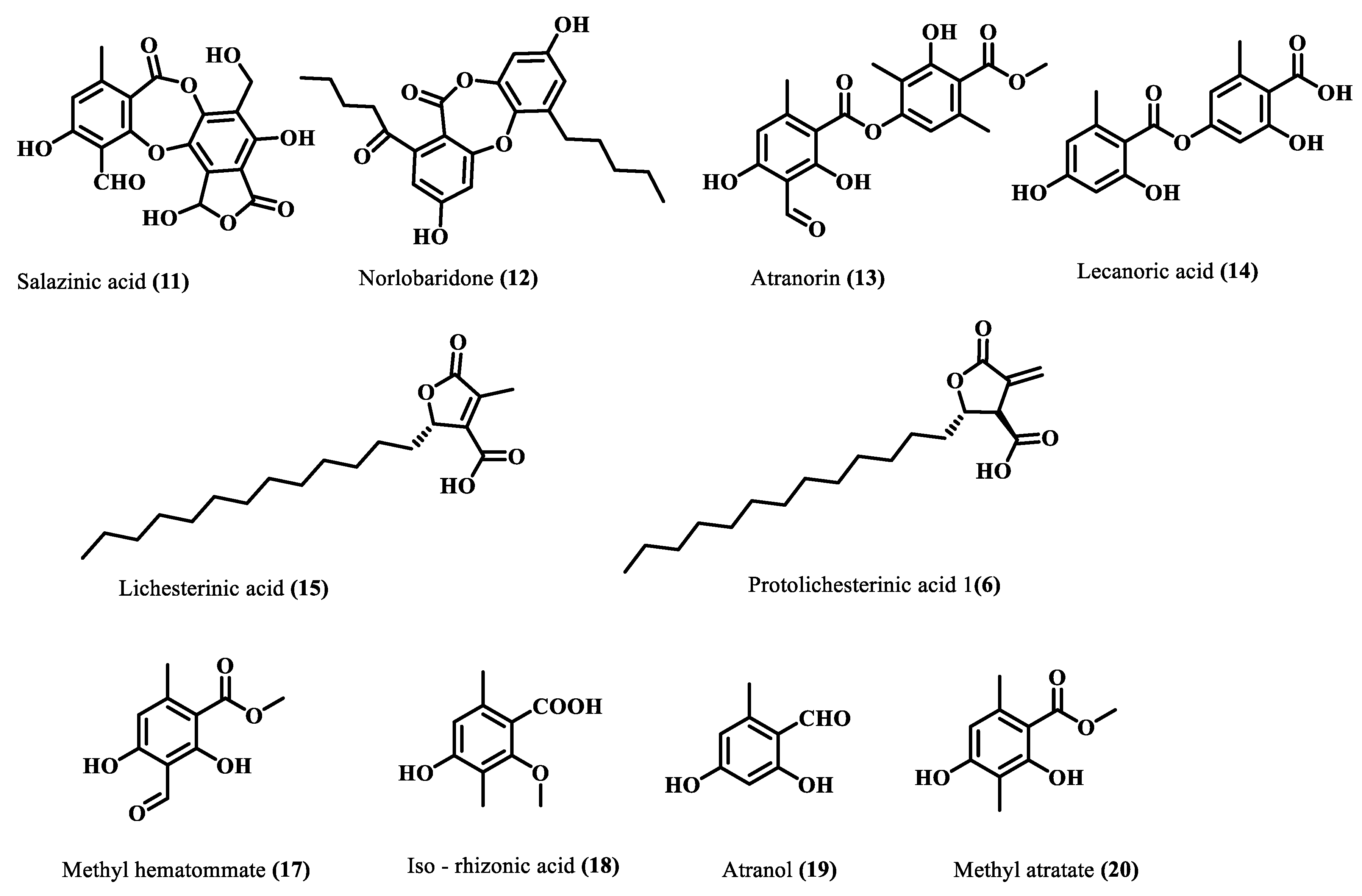
Chemical Profiling, Isolation, and Structure Elucidation
3. Biological Activity
3.1. ABTS·+ and DPPH Radicals Scavenging Activity Analysis
3.2. Protective Effect on Oxidative DNA Damage
3.3. Intestinal α-Glucosidase Activities
4. Molecular Modeling Studies
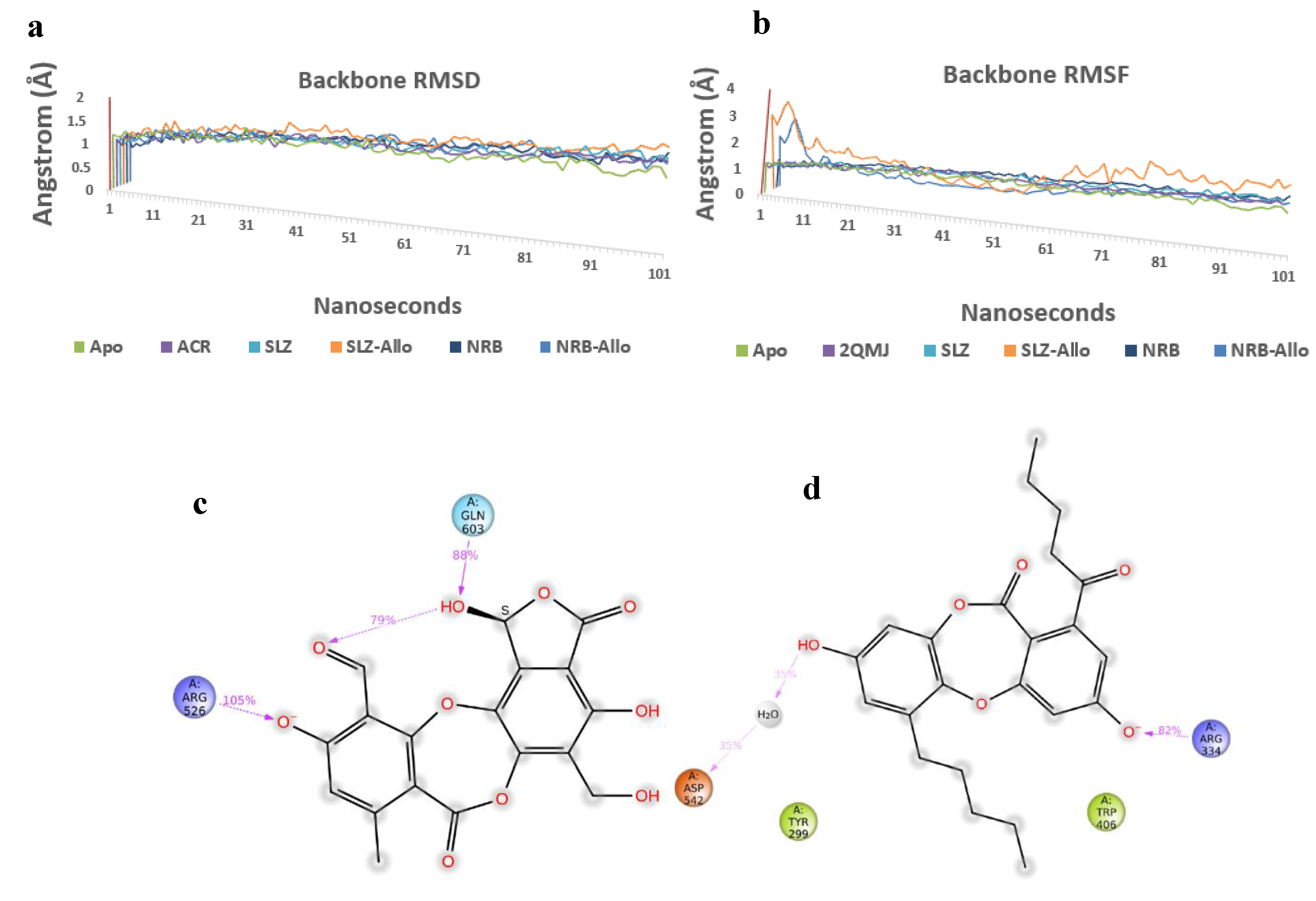
4.1. Molecular Interaction Analysis of Active Pocket
4.2. Allosteric Binding Studies
4.3. Molecular Dynamics Simulations
5. Materials and Methods
5.1. General
5.2. Instrumental UPLC Conditions
5.3. Lichen Collection and Identification
5.4. Extraction and Isolation
5.5. In Vitro Antioxidant and Antihyperglycemic Assay
5.5.1. DPPH Radical Scavenging Activity
5.5.2. ABTS Radical Scavenging Activity
5.5.3. Free Radical Induced DNA Damage
5.5.4. Intestinal α-Glucosidase Inhibition
5.6. Molecular Modeling Studies
5.7. Molecular Dynamics Protocol
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Honegger, R. Lichens and Their Allies Past and Present. In Plant Relationships. The Mycota 5, 3rd ed.; Scott, B., Mesarich, C., Eds.; Springer: Cham, Switzerland, 2023; pp. 133–183. [Google Scholar]
- Goga, M.; Elečko, J.; Marcinčinová, M.; Ručová, D.; Bačkorová, M.; Bačkor, M. Lichen metabolites: An overview of some secondary metabolites and their biological potential. In Co-Evolution of Secondary Metabolites. Reference Series in Phytochemistry; Merillon, J.M., Ramawat, K., Eds.; Springer: Cham, Switzerland, 2018. [Google Scholar] [CrossRef]
- Adenubi, O.T.; Famuyide, I.M.; McGaw, L.J.; Eloff, J.N. Lichens: An update on their ethnopharmacological uses and potential as sources of drug leads. J. Ethnopharmacol. 2022, 298, 115657. [Google Scholar] [CrossRef] [PubMed]
- Swathi, D.; Suchitha, Y.; TR, P.K.; Venugopal, T.M.; Vinayaka, K.S.; Mallikarjun, N.; Raghavendra, H.L. Antimicrobial, antihelmintic and insecticidal activity of a macrolichen Everniastrum cirrhatum (Fr.) Hale. Int. J. Drug Dev. Res. 2010, 2, 780–789. [Google Scholar]
- Kekuda, T.R.; Vinayaka, K.S.; Swathi, D.; Suchitha, Y.; Venugopal, T.M.; Mallikarjun, N. Mineral composition, total phenol content and antioxidant activity of a macrolichen Everniastrum cirrhatum (Fr.) Hale (Parmeliaceae). J. Chem. 2011, 8, 1886–1894. [Google Scholar]
- Ankita, H.; Tripathi, A.H.; Negi, N.; Gahtori, R.; Kumari, A.; Joshi, P.; Tewari, L.M.; Joshi, Y.; Bajpai, R.; Upreti, D.K.; et al. A Review of Anti-Cancer and Related Properties of Lichen-Extracts and Metabolites. Anti-Cancer Agents in Medicinal Chemistry 2022, 22, 115–142. [Google Scholar]
- Anil Kumar, H.S.K.; Prashith, T.R.; Vinayaka, K.S.; Venugopal, T.M. Anti-obesity (Pancreatic lipase inhibitory) activity of Everniastrum cirrhatum (Fr.) Hale (Parmeliaceae). Pharmacogn. J. 2011, 3, 65–68. [Google Scholar] [CrossRef]
- Sepulveda, B.; Benites, D.; Albornoz, L.; Simirgiotis, M.; Castro, O.; Garcia-Beltran, O.; Areche, C. Green ultrasound-assisted extraction of lichen substances from Hypotrachyna cirrhata. Ethyl lactate, a better extracting agent than methanol toxic organic solvent? Nat. Prod. Res. 2023, 37, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Pol, C.S.; Savale, S.A.; Khare, R.; Verma, N.; Behera, B.C. Antioxidative, cardioprotective, and Anticancer Potential of Two Lichenized Fungi, Everniastrum cirrhatum and Parmotrema reticulatum, from Western Ghats of India. Nat. Prod. Res. 2017, 23, 142–156. [Google Scholar]
- White, P.A.S.; Oliveira, R.C.M.; Oliveira, A.P.; Serafini, M.R.; Araújo, A.A.S.; Gelain, D.P.; Moreira, J.C.F.; Almeida, J.R.G.S.; Quintans, J.S.S.; Quintans-Junior, L.J. Antioxidant activity and mechanisms of action of natural compounds isolated from Lichens: A Systematic Review. Molecules 2014, 19, 14496–14527. [Google Scholar] [CrossRef]
- Elečko, J.; Vilková, M.; Frenák, R.; Routray, D.; Ručová, D.; Bačkor, M.; Goga, M. A Comparative study of isolated secondary metabolites from Lichens and their antioxidative Properties. Plants 2022, 11, 1077. [Google Scholar] [CrossRef]
- Hawrył, A.; Hawrył, M.; Hajnos-Stolarz, A.; Abramek, J.; Bogucka-Kocka, A.; Komsta, Ł. HPLC fingerprint analysis with the antioxidant and cytotoxic activities of selected Lichens combined with the chemometric calculations. Molecules 2020, 25, 4301. [Google Scholar] [CrossRef]
- Fernández-Moriano, C.; Gómez-Serranillos, M.P.; Crespo, A. Antioxidant potential of lichen species and their secondary metabolites. A systematic review. Pharm. Biol. 2016, 54, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Françoise, L.D.; Holger, T.; Marie-Laurence, A.; David, D.; Joël, B. Oxidative stress regulation in Lichens and its relevance for survival in coastal habitats. In Advances in Botanical Research; Jacquot, J.-P., Gadal, P., Bourgougnon, N., Eds.; Elsevier Eds. Sea Plants; Academic Press: Cambridge, MA, USA, 2014; Volume 71, pp. 467–504. [Google Scholar]
- Kumar, K.; Siva, B.; Sarma, V.; Mohabe, S.; Reddy, A.M.; Boustie, J.; Tiwari, A.K.; Rao, N.R.; Babu, K.S. UPLC–MS/MS quantitative analysis and structural fragmentation study of five Parmotrema lichens from the Eastern Ghats. J. Pharm. Biomed. Anal. 2018, 156, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Kumar, T.K.; Siva, B.; Anand, A.; Anusha, K.; Mohabe, S.; Reddy, A.M.; Le Devehat, F.; Tiwari, A.K.; Boustie, J.; Babu, K.S. Comprehensive Lichenometabolomic exploration of Ramalina conduplicans Vain Using UPLC-Q-ToF-MS/MS: An identification of free radical scavenging and anti-hyperglycemic constituents. Molecules 2022, 27, 6720. [Google Scholar] [CrossRef]
- Nayaka, S. Methods and Techniques in Collection, Preservation and Identification of Lichens. In Plant Taxonomy and Biosystematics; Rana, T.S., Nair, K.N., Upreti, D.K., Eds.; New India Publishing Agency: New Delhi, India, 2014; pp. 101–128. [Google Scholar]
- Seger, C.; Sturm, S.; Stuppner, H. Mass spectrometry and NMR spectroscopy: Modern high-end detectors for high resolution separation techniques–state of the art in natural product HPLC-MS, HPLC-NMR, and CE-MS hyphenations. Nat. Prod. Rep. 2013, 30, 970–987. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.; Siva, B.; Rama Rao, N.; Suresh Babu, K. Rapid identification of limonoids from Cipadessa baccifera and Xylocarpus granatum using ESI-Q-ToF-MS/MS and their structure-fragmentation study. J. Pharm. Biomed. Anal. 2018, 152, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Demarque, D.P.; Crotti, A.E.M.; Vessecchi, R.; Lopes, J.L.C.; Lopes, N.P. Fragmentation reactions using electrospray ionization mass spectrometry: An important tool for the structural elucidation and characterization of synthetic and natural products. Nat. Prod. Rep. 2016, 33, 432–455. [Google Scholar] [CrossRef]
- Pavan Kumar, P.; Siva, B.; Anand, A.; Tiwari, A.K.; Vekata Rao, C.; Boustie, J.; Suresh Babu, K. Isolation, semi-synthesis, free-radicals scavenging, and advanced glycation end products formation inhibitory constituents from Parmotrema tinctorum. J. Asian Nat. Prod. Res. 2020, 22, 976–988. [Google Scholar] [CrossRef]
- Chester, D.O.; Elix, J.A. Three New Aliphatic Acids from Lichens of Genus Parmelia (Subgenus Xanthoparmelia). Aust. J. Chem. 1979, 32, 2565–2569. [Google Scholar] [CrossRef]
- Goel, M.; Dureja, P.; Rani, A.; Uniyal, P.L.; Laatsch, H. Isolation, characterization and antifungal activity of major constituents of the Himalayan Lichen Parmelia reticulata Tayl. J. Agric. Food Chem. 2011, 59, 2299–2307. [Google Scholar] [CrossRef]
- Huneck, S.; Yoshimura, I. Identification of lichen substances. In Identification of Lichen Substances; Springer: Berlin/Heidelberg, Germany, 1996; pp. 11–123. [Google Scholar]
- Bouges, H.; Monchot, A.; Antoniotti, S. Enzyme-Catalysed conversion of atranol and derivatives into dimeric hydrosoluble materials: Application to the preparation of a low-atranol oakmoss absolute. Cosmetics 2018, 5, 69. [Google Scholar] [CrossRef]
- Prior, R.L.; Wu, X.; Schaich, K. Standardized methods for the determination of antioxidant capacity and phenolics in foods and dietary supplements. J. Agric. Food Chem. 2005, 53, 4290–4302. [Google Scholar] [CrossRef]
- Meir, S.; Kanner, J.; Akiri, B.; Philosoph-Hadas, S. Determination and involvement of aqueous reducing compounds in oxidative defense systems of various senescing leaves. J. Agric. Food Chem. 1995, 43, 1813–1819. [Google Scholar] [CrossRef]
- Siddeeg, A.; Al-Kehayez, N.M.; Abu-Hiamed, H.A.; Al-Sanea, E.A.; Al-Farga, A.M. Mode of action and determination of antioxidant activity in the dietary sources: An overview. Saudi J. Biol. Sci. 2021, 28, 1633–1644. [Google Scholar] [CrossRef]
- Lloyd, D.R.; Phillips, D.H. Oxidative DNA damage mediated by copper(II), iron(II) and nickel(II) Fenton reactions: Evidence for site-specific mechanisms in the formation of double-strand breaks, 8-hydroxydeoxyguanosine and putative intrastr and cross-links. Mutat. Res. Fundam. Mol. Mech. Mutagen. 1999, 424, 23–36. [Google Scholar] [CrossRef]
- Kumar, S.; Narwal, S.; Kumar, V.; Prakash, O. α-glucosidase inhibitors from plants: A natural approach to treat diabetes. Pharmacogn. Rev. 2011, 5, 19. [Google Scholar] [CrossRef]
- Prashith Kekuda, T.R.; Lavanya, D.; Pooja, R. Lichens as promising resources of enzyme inhibitors: A review. J. Drug Deliv. Ther. 2019, 9, 665–676. [Google Scholar]
- Verma, N.; Behera, B.; Sharma, B.O. Glucosidase inhibitory and radical scavenging properties of lichen metabolites salazinic acid, sekikaic acid and usnic acid. Hacet. J. Biol. Chem. 2012, 40, 7–21. [Google Scholar]
- Nguyen, H.T.; Pham, D.D.; Nguyen, H.T.; Do, T.H.; Nguyen, N.H.; Duong, T.H. Design, modification, and bio-evaluation of salazinic acid derivatives. Arab. J. Chem. 2022, 15, 103535. [Google Scholar]
- Ding, H.; Hu, X.; Xu, X.; Zhang, G.; Gong, D. Inhibitory mechanism of two allosteric inhibitors, oleanolic acid and ursolic acid on α-glucosidase. Int. J. Biol. Macromol. 2018, 107, 1844–1855. [Google Scholar] [CrossRef]
- Kozakov, D.; Grove, L.E.; Hall, D.R.; Bohnuud, T.; Mottarella, S.E.; Luo, L.; Xia, B.; Beglov, D.; Vajda, S. The FT Map family of web servers for determining and characterizing ligand-binding hot spots of proteins. Nat. Protoc. 2015, 10, 733–755. [Google Scholar] [CrossRef] [PubMed]
- Brenke, R.; Kozakov, D.; Chuang, G.Y.; Beglov, D.; Hall, D.; Landon, M.R.; Mattos, C.; Vajda, S. Fragment-based identification of druggable ‘hot spots’ of proteins using Fourier domain correlation techniques. Bioinformatics 2009, 25, 621–627. [Google Scholar] [CrossRef]
- Awasthi, D.D. A Compendium of the Macrolichens from India, Nepal and Sri Lanka; Bishen Singh Mahendra Pal Singh: Dehradun, India, 2007. [Google Scholar]
- Tiwari, A.K.; Manasa, K.; Kumar, D.A.; Zehra, A. Raw horse gram seeds possess more in vitro antihyperglycaemic activities and antioxidant properties than their sprouts. Nutrafoods 2013, 12, 47–54. [Google Scholar] [CrossRef]
- Deepthi, S.; Anusha, K.; Anand, A.; Manasa, A.; Babu, K.S.; Mudiam, M.K.R.; Tiwari, A.K. Micronutrients and phytochemicals content in various rice (Oryza sativa Linn.) samples control carbohydrate digestion variedly and present differential antioxidant activities: An in vitro appraisal. Indian J. Tradit. Knowl. 2020, 19, 821–831. [Google Scholar]
- Wojdyło, A.; Nowicka, P. Profile of phenolic compounds of Prunus armeniaca L. leaf extract determined by LC-ESI-QTOF-MS/MS and their antioxidant, anti-diabetic, anti-cholinesterase, and anti-inflammatory potency. Antioxidants 2021, 10, 1869. [Google Scholar] [CrossRef]
- Sim, L.; Quezada-Calvillo, R.; Sterchi, E.E.; Nichols, B.L.; Rose, D.R. Human intestinal maltase–glucoamylase: Crystal structure of the N-terminal catalytic subunit and basis of inhibition and substrate specificity. J. Mol. Biol. 2008, 375, 782–792. [Google Scholar] [CrossRef]
- Schrödinger Release 2022-3: Desmond Molecular Dynamics System, D. E. Shaw Research, New York, NY, 2021; Maestro-Desmond interoperability Tools; Schrödinger: New York, NY, USA, 2021.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S.No | Compound Name | RT (min) | Formula | Mass (m/z) Calculated | Mass (m/z) Observed | Error (Δ ppm) | Fragmentation Pattern |
|---|---|---|---|---|---|---|---|
| 01 | Orsellinic acid # (8) | 5.75 | C8H7O4 | 167.0344 | 167.0341 | −1.8 | 123.0440 (C7H7O2) |
| 02 | Consalazinic acid (11) # | 5.92 | C18H13O10 | 389.0509 | 389.0506 | −0.8 | 371.0398(C18H11O9), 345.0593(C17H13O8) 327.0503(C17H11O7), 309.0400(C17H9O6) 297.0398(C16H9O6), 281.0449(C16H9O5) 265.0501(C16H9O4), 253.0500(C15H9O4) 227.0344(C15H9O4), 225.0554(C14H9O3) 209.0603(C14H9O2), 197.0594 (C13H9O2) 171.0409 (C11H7O2). |
| 03 | ND | 6.63 | C10H9O7 | 241.0348 | 241.0342 | −2.5 | 223.0234(C10H7O6), 209.0082 (C9H5O6) 190.9971 (C9H3O5), 165.0185 (C8H5O4). |
| 04 | Thamnolic acid # | 6.94 | C19H15O11 | 419.0614 | 419.0607 | −1.7 | 401.0466 (C19H13O10), 389.0490 (C18H13O10) 375.0553 (C18H15O9), 327.0514 (C17H11O7) 313.0349 (C16H9O7), 297.0410 (C16H9O6) 269.0433 (C15H9O5), 253.0497 (C15H9O4) 241.0588 (C14H9O4), 225.0558 (C14H9O3) 209.0472 (C10H9O5), 195.0453 (C13H7O2), 167.0440 (C12H7O1). |
| 05 | Connorstictic acid # | 7.25 | C18H13O9 | 373.0560 | 373.0553 | −1.9 | 343.0449 (C17H11O8) 329.0657 (C17H13O7), 255.0655 (C15H11O4), 227.0655 (C14H11O3), 212.0486 (C13H8O3), 203.0729 (C12H11O3), 199.0709 (C13H11O2), 161.0638(C10H9O2), 137.0609 (C8H9O2). |
| 06 | Atranol * (19) | 8.00 | C8H7O3 | 151.0395 | 151.0392 | −2.0 | 123.0441 (C7H7O2), 105.0339 (C7H5O). |
| 07 | Unidentified | 8.47 | C20H17O11 | 433.0771 | 433.0771 | 0.0 | 401.0508 (C19H13O10), 357.0617 (C18H13O8), 339.0524 (C18H11O7), 297.0406 (C16H9O6), 281.0456 (C16H9O5), 253.0512 (C15H9O4), 209.0424 (C10H9O5), 179.0328 (C9H7O4), 151.0050 (C7H3O4). |
| 08 | Salazinic acid * (11) | 8.64 | C18H11O10 | 387.0352 | 387.0352 | 0.0 | 343.0459 (C17H11O8), 325.0355 (C13H9O10) 313.0353 (C16H9O7), 243.0295 (C13H7O5) 227.0344 (C13H7O4), 225.0552 (C14H9O3) 213.0551 (C13H9O3), 151.0394 (C8H7O3) 121.0287 (C7H5O2). |
| 09 | Haematommic acid # (9) | 9.06 | C9H7O5 | 195.0293 | 195.0290 | −1.5 | 151.0391 (C8H7O3), 123.0442 (C7H7O2). |
| 10 | Constictic acid # (2) | 10.49 | C19H13O10 | 401.0509 | 401.0524 | 3.7 | 357.0619 (C18H13O8), 313.0722 (C17H13O6), 253.0509 (C15H9O4), 243.0304 (C13H7O5), 227.0350 (C13H7O4), 225.0556 (C14H9O3). |
| 11 | Methyl atratate * (20) | 10.57 | C10H11O4 | 195.0657 | 195.0658 | 0.5 | 181.0500 (C9H9O4), 163.0394 (C9H7O3), 137.0602 (C8H9O2), 119.0496 (C8H7O). |
| 12 | Lecanoric acid * (14) | 10.76 | C16H13O7 | 317.0661 | 317.0663 | 0.6 | 167.0338 (C8H7O4), 149.0233 (C8H5O3) 123.0440 (C7H7O2), 105.0334 (C7H5O). |
| 13 | Gyrophoric acid # (4) | 13.45 | C24H19O10 | 467.0978 | 467.0978 | 0.0 | 317.0663 (C16H13O7), 167.0340 (C8H7O4) 149.0237 (C8H5O3), 123.0443 (C7H7O2). |
| 14 | Norlobaridone * (12) | 14.40 | C23H25O6 | 397.1651 | 397.1655 | 1.0 | 353.1755 (C22H25O4), 329.1755 (C20H25O4) 313.1076 (C18H17O5), 296.1050 (C18H16O4) 272.1048 (C16H16O4), 201.0546 (C12H9O3) 188.0468 (C11H8O3), 159.0441 (C10H7O2). |
| 15 | Dehydroalectoronic acid # (3) | 14.71 | C28H29O9 | 509.1812 | 509.1811 | −0.2 | 491.1705 (C28H27O8), 465.1921 (C27H29O7) 447.1801 (C27H27O6), 423.1574 (C28H23O4) 405.1404 (C24H21O6), 381.1403 (C22H21O6) 367.1207 (C21H19O6), 247.0965 (C14H15O4), 218.0951 (C13H14O3). |
| 16 | Usnic acid # (10) | 15.73 | C18H15O7 | 343.0818 | 343.0806 | −3.5 | 328.0593 (C17H12O7), 313.0359 (C16H9O7) 259.0595 (C14H11O5), 231.0652 (C13H11O4) 215.0350 (C12H7O4), 177.0180 (C9H5O4). |
| 17 | Atranorin * (13) | 15.80 | C19H17O8 | 373.0923 | 373.0935 | 3.2 | 195.0651 (C10H11O4), 177.0182 (C9H5O4), 163.0390 (C9H7O3), 133.0285 (C8H5O2). |
| 18 | Chloroatranorin # (5) | 16.21 | C19H16O8Cl | 407.0556 | 407.0540 | −3.9 | 223.0970 (C12H15O4), 210.9796 (C9H4O4Cl), 205.0863 (C12H13O3), 195.0652 (C10H11O4) 163.0392 (C9H7O3), 138.9950 (C7H4O1Cl). |
| 19 | Stenosporic acid # (6) | 16.23 | C23H27O7 | 415.1757 | 415.1758 | 0.2 | 223.0969 (C12H15O4), 205.0864 (C12H13O3) 179.1072 (C11H15O2), 163.0393 (C9H7O3). |
| 20 | Roccellaric acid # (7) | 16.65 | C19H33O4 | 325.2379 | 325.2382 | 0.9 | 281.2481 (C18H33O2). |
| 21 | Protolichesterinic acid * (16) | 16.75 | C19H31O4 | 323.2222 | 323.2224 | −1.5 | 279.2323 (C18H31O2). |
| 22 | Lichesterinic acid * (15) | 16.90 | C19H31O4 | 323.2222 | 323.2217 | 0.6 | 279.2322(C18H31O2). |
| Compound Name (2 mg/mL) | ABTS % Inhibition (IC50) µg/mL | DPPH % Inhibition (IC50) µg/mL | Intestinal α-Glucosidase % Inhibition (IC50) µg/mL | DNA Damage Assay (% Protection) |
|---|---|---|---|---|
| HC extract | 95.79 ± 2.13 | 98.23 ± 2.02 | 61.23 ± 0.46 | 89.63 ± 0.89 |
| Salazinic acid (11) | 79.28 ± 2.07 (2.11 ± 0.04) | 54.20 ± 3.03 | 82.10 ± 1.76 (1.62 ± 0.07) | 95.44 ± 0.31 |
| Norlobaridone (12) | 99.95 ± 0.39 (0.59 ± 0.02) | 21.57 ± 1.32 | 82.80 ± 1.96 (1.41 ± 0.01) | 82.80 ± 9.93 |
| Atranorin (13) | 99.99 ± 0.51 (0.068 ± 0.02) | 55.91 ± 0.46 | 76.69 ± 3.6 (2.07 ± 0.01) | 96.87 ± 4.30 |
| Lecanoric acid (14) | 98.55 ± 0.06 (0.32 ± 0.02) | 16.45 ± 1.71 | 55.61 ± 1.27 | 99.49 ± 0.56 |
| Lichesterinic acid (15) | 16.37 ± 1.35 | 47.31 ± 2.49 | 48.40 ± 1.56 | 90.55 ± 2.47 |
| Protolichesterinic acid (16) | 22.88 ± 0.90 | 29.45 ± 1.87 | 44.79 ± 0.78 | 90.45 ± 0.02 |
| Methyl hematommate (17) | 94.70 ± 0.48 (0.53 ± 0.03 | 54.03 ± 2.96 | 35.64 ± 0.78 | 98.39 ± 0.11 |
| Iso- rhizonic acid (18) | 97.96 ± 1.41 (16.37 ± 0.06) | 45.05 ± 1.79 | 75.31 ± 3.92 (1.99 ± 0.03) | 91.48 ± 0.30 |
| Atranol (19) | 99.23 ± 0.51 (0.11 ± 0.07) | 78.46 ± 1.12 (1.73 ± 0.02) | 28.71 ± 0.54 | 94.16 ± 2.45 |
| Methylatratate (20) | 96.87 ± 1.02 (1.44 ± 0.05) | 63.02 ± 3.66 (3.02 ± 0.07) | 33.14 ± 1.17 | 92.15 ± 0.35 |
| Ascorbic acid (2 mg/mL) | 99.05 ± 0.62 (0.079 ± 0.01) | 83.68 ± 2.72 (1.59 ± 0.05) | - | - |
| Acarbose (2 mg/mL) | - | - | 67.40 ± 3.08 (2.58 ± 0.85) | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, T.K.; Siva, B.; Kiranmai, B.; Alli, V.J.; Jadav, S.S.; Reddy, A.M.; Boustie, J.; Le Devehat, F.; Tiwari, A.K.; Suresh Babu, K. Salazinic Acid and Norlobaridone from the Lichen Hypotrachyna cirrhata: Antioxidant Activity, α-Glucosidase Inhibitory and Molecular Docking Studies. Molecules 2023, 28, 7840. https://doi.org/10.3390/molecules28237840
Kumar TK, Siva B, Kiranmai B, Alli VJ, Jadav SS, Reddy AM, Boustie J, Le Devehat F, Tiwari AK, Suresh Babu K. Salazinic Acid and Norlobaridone from the Lichen Hypotrachyna cirrhata: Antioxidant Activity, α-Glucosidase Inhibitory and Molecular Docking Studies. Molecules. 2023; 28(23):7840. https://doi.org/10.3390/molecules28237840
Chicago/Turabian StyleKumar, Tatapudi Kiran, Bandi Siva, Basani Kiranmai, Vidya Jyothi Alli, Surender Singh Jadav, Araveeti Madhusudana Reddy, Joël Boustie, Françoise Le Devehat, Ashok Kumar Tiwari, and Katragadda Suresh Babu. 2023. "Salazinic Acid and Norlobaridone from the Lichen Hypotrachyna cirrhata: Antioxidant Activity, α-Glucosidase Inhibitory and Molecular Docking Studies" Molecules 28, no. 23: 7840. https://doi.org/10.3390/molecules28237840