
Monitoring Exposure to Five Chemical Warfare Agents Using the Dried Urine Spot Technique and Liquid Chromatography-Mass Spectrometry/Mass Spectrometry—In Vivo Determination of Sarin Metabolite in Mice

Abstract
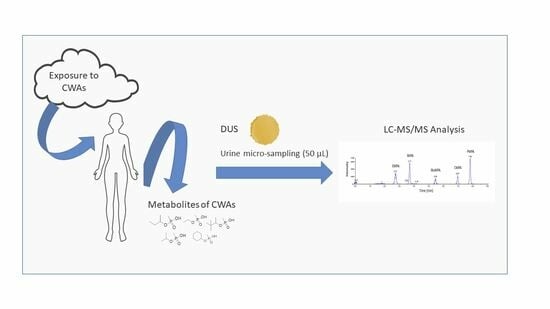
:
1. Introduction
2. Results
2.1. Development and Optimization of the Extraction Procedure (Urine Volume, “In-Tip” Extraction Method, Solvent and Volume)
2.2. Validation of the Method
2.2.1. Calibration Model
2.2.2. Selectivity (Interferences)
2.2.3. LOD and LOQ in Urine Samples
2.2.4. Recoveries
2.2.5. Accuracy and Precision
2.2.6. Stability
2.2.7. Matrix Effects in LC-MS/MS
2.2.8. DUS vs. “Dilute and Shoot”
2.3. Exposure of Mice to GB—In Vivo Experiment
2.4. Non-Targeted “Screening”
3. Summary and Conclusions
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Preparation of Standard Solutions
4.3. Instrument Conditions
4.4. DUS Sample Preparation
4.5. Animals
4.6. In Vivo Experiment
4.7. Method Validation
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Chauhan, S.; D’Cruz, R.; Faruqi, S.; Singh, K.K.; Varma, S.; Singh, M.; Karthik, V. Chemical warfare agents. Environ. Toxicol. Pharmacol. 2008, 26, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Bigley, A.N.; Raushel, F.M. The evolution of phosphotriesterase for decontamination and detoxification of organophosphorus chemical warfare agents. Chem. Biol. Interact. 2019, 308, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Noort, D.; Hulst, A.G.; Platenburg, D.H.J.M.; Polhuijs, M.; Benschop, H.P. Quantitative analysis of O-isopropyl methylphosphonic acid in serum samples of Japanese citizens allegedly exposed to sarin: Estimation of internal dosage. Arch. Toxicol. 1998, 72, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Black, R.M. History and perspectives of bioanalytical methods for chemical warfare agent detection. J. Chromatogr. B. 2010, 878, 1207–1215. [Google Scholar] [CrossRef]
- Harald, J.; van der Schans, M.J.; Koller, M.; Spruit, E.T.; Worek, F.; Thiermann, H.; Noort, D. Fatal sarin poisoning in Syria 2013: Forensic verification within an international laboratory network. Forensic Toxicol. 2018, 36, 61–71. [Google Scholar] [CrossRef]
- Nakagawa, T.; Tu, A.T. Murders with VX: Aum Shinrikyo in Japan and the assassination of Kim Jong-Nam in Malaysia. Forensic Toxicol. 2018, 36, 542–544. [Google Scholar] [CrossRef]
- Harald, J.; Thiermann, H. Poisoning by organophosphorus nerve agents and pesticides: An overview of the principal strategies and current progress of mass spectrometry-based procedures for verification. J. Mass Spectrom. Adv. Clin. Lab. 2021, 19, 20–31. [Google Scholar] [CrossRef]
- Marder, D.; Dagan, S.; Yishai-Aviram, L.; Loewenthal, D.; Chapman, S.; Adani, R.; Lazar, S.; Weissberg, A.; Gura, S. Instantaneous monitoring of free sarin in whole blood by dry blood spot–thermal desorption–GC–FPD/MS analysis. J. Chromatogr. B 2020, 1136, 121911. [Google Scholar] [CrossRef]
- Brown, H.M.; McDaniel, T.J.; Doppalapudi, K.R.; Mulligan, C.C.; Fedick, P.W. Rapid, in situ detection of chemical warfare agent simulants and hydrolysis products in bulk soils by low-cost 3D-printed cone spray ionization mass spectrometry. Analyst 2021, 146, 3127–3136. [Google Scholar] [CrossRef]
- Aszyk, J.; Byliński, H.; Namieśnik, J.; Kot-Wasik, A. Main strategies, analytical trends and challenges in LC-MS and ambient mass spectrometry–based metabolomics. Trends Anal. Chem. 2018, 108, 278–295. [Google Scholar] [CrossRef]
- Gaugler, S.; Luginbühl, M.; Stoeth, F.; Martin, M.; Weinmann, W.; König, S. High resolution, high accuracy non-targeted LC-HR-MS/MS dried urine spot screening for drug of abuse testing. Toxicol. Anal. 2022, 34, S115. [Google Scholar] [CrossRef]
- Michely, J.A.; Meyer, M.R.; Maurer, H. Power of Orbitrap-based LC-high resolution-MS/MS for comprehensive drug testing in urine with or without conjugate cleavage or using dried urine spots after on-spot cleavage in comparison to established LC–MS or GC–MS procedures. Drug Test Anal. 2018, 10, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Yuan, S.; Yu, Z.; Zhao, Y.; Zhang, S.; Wu, H.; Yan, H.; Xiang, P. Development of an LC-MS/MS method for determining 5-MeO-DIPT in dried urine spots and application to forensic cases. J. Forensic Leg. Med. 2020, 72, 101963. [Google Scholar] [CrossRef] [PubMed]
- Pizzolato, T.M.; Lopez de Alda, M.J.; Barceló, D. LC-based analysis of drugs of abuse and their metabolites in urine. Trends Anal. Chem. 2007, 26, 609–624. [Google Scholar] [CrossRef]
- Pablo, A.; Breaud, A.R.; Clarke, W. Automated analysis of dried urine spot (DUS) samples for toxicology screening. Clin. Biochem. 2020, 75, 70–77. [Google Scholar] [CrossRef]
- Michely, J.A.; Meyer, M.R.; Maurer, H. Dried urine spots—A novel sampling technique for comprehensive LC-MSn drug screenin. Anal. Chim. Acta 2017, 982, 112–121. [Google Scholar] [CrossRef]
- Moretti, M.; Freni, F.; Carelli, C.; Previderé, C.; Grignani, P.; Vignali, C.; Cobo-Golpe, M.; Morini, L. Analysis of Cannabinoids and Metabolites in Dried Urine Spots (DUS). Molecules 2021, 26, 5334. [Google Scholar] [CrossRef]
- Yishai Aviram, L.; Magen, M.; Chapman, S.; Neufeld Cohen, A.; Lazar, S.; Dagan, S. Dry Blood Spot sample collection for post-exposure monitoring of chemical warfare agents—In vivo determination of phosphonic acids using LC-MS/MS. J. Chromatogr. B 2018, 1093–1094, 60–65. [Google Scholar] [CrossRef]
- Yishai Aviram, L.; Loewenthal, D.; Weissberg, A.; Marder, D.; Gura, S.; Chapman, S.; Gez, R.; Lazar, S.; Dagan, S. Determination of free G-type nerve agents in blood: In situ derivatization on a dried blood spot (DBS) paper followed by LC–MS/MS analysis. Forensic Toxicol. 2020, 38, 327–339. [Google Scholar] [CrossRef]
- Golime, R.; Chandra, B.; Palit, M.; Dubey, D.K. Adductomics: A promising tool for the verification of chemical warfare agents’ exposures in biological samples. Arch. Toxicol. 2019, 93, 1473–1484. [Google Scholar] [CrossRef]
- Bruin-Hoegée, M.; Fidder, A.; van Groningen, T.; van der Schans, M.J.; Noort, D.; van Asten, A. On-site detection and laboratory verification of the presence of nerve agent biomarkers using dried blood spots. Forensic Chem. 2023, 35, 100526. [Google Scholar] [CrossRef]
- Aramendia, M.; Vanhaecke, F.; Resano, M. Direct trace-elemental analysis of urine samples by laser ablation-inductively coupled plasma mass spectrometry after sample deposition on clinical filter paper. Anal. Chem. 2012, 84, 8682–8690. [Google Scholar] [CrossRef] [PubMed]
- Roen, B.T.; Sellevag, S.R.; Lundanes, E. On-line solid phase extraction-liquid chromatography-mass spectrometry for trace determination of nerve agent degradation products in water samples. Anal. Chim. Acta 2013, 761, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Deventer, K.; Pozo, O.J.; Verstraete, A.G.; Van Eenoo, P. Dilute-and-shoot-liquid chromatography-mass spectrometry for urine analysis in doping control and analytical toxicology. Trends Anal. Chem. 2014, 55, 1–13. [Google Scholar] [CrossRef]
- Roen, B.T.; Sellevag, S.R.; Lundanes, E. Quantifiacation of nerve agent biomarkers in human serum and urine. Anal. Chem. 2014, 86, 11833–11840. [Google Scholar] [CrossRef]
- Blanca, M.; Shifrovitch, A.; Dachir, S.; Lazar, S.; Elgarisi, M.; Prihed, H.; Baranes, S.; Egoz, I.; Avraham, M.; Dekel Jaoui, H.; et al. Extended retrospective detection of regenerated sarin (GB) in rabbit blood and the IMPA metabolite in urine: A pharmacokinetics study. Arch. Toxicol 2021, 95, 2403–2412. [Google Scholar] [CrossRef]
- Dagan, S.; Marder, D.; Tzanani, N.; Drug, E.; Prihed, H.; Yishai-Aviram, L. Evaluation of Matrix Complexity in Nontargeted Analysis of Small-Molecule Toxicants by Liquid Chromatography–High-Resolution Mass Spectrometry. Anal. Chem. 2023, 95, 7924–7932. [Google Scholar] [CrossRef]
- Peters, F.T.; Drummer, O.H.; Musshoff, F. Validation of new methods. Forensic Sci. Int. 2007, 165, 216–224. [Google Scholar] [CrossRef]
- Matuszewski, B.K.; Constanzer, M.L.; Chavez-Eng, C.M. Strategies for the assessment of matrix effect in quantitative bioanalytical methods based on HPLC–MS/MS. Anal. Chem. 2003, 75, 3019–3030. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Dominant MRM Transition (Negative ESI) | tR (min) | LOD (ng/mL) | LOQ (ng/mL) |
|---|---|---|---|---|
| EMPA (VX acid) | 123 > 95 | 2.7 | 10 | 30 |
| IMPA (GC-acid) | 137 > 79 | 3.7 | 3 | 9 |
| iBuMPA (RVX-acid) | 151 > 77 | 5.5 | 1 | 3 |
| CMPA (GF-acid) | 177 > 95 | 6.6 | 0.5 | 3 |
| PMPA (GD-acid) | 179 > 95 | 7.8 | 0.5 | 3 |
| Compound | 10 ng/mL in Urine | 100 ng/mL in Urine | ||||
|---|---|---|---|---|---|---|
| Recovery (%) | Accuracy (%) | Precision (%) | Recovery (%) | Accuracy (%) | Precision (%) | |
| EMPA | 41.7 | 23.1 | 9.2 | 40 | 21.2 | 12.5 |
| IMPA | 53.3 | 20.6 | 8.5 | 44 | 19.7 | 12.5 |
| iBuMPA | 55.5 | 14.6 | 8.9 | 80 | 12.6 | 11.7 |
| CMPA | 49.1 | 24.4 | 8.1 | 56 | 19.1 | 13.3 |
| PMPA | 54.1 | 16.3 | 8.5 | 54 | 1.9 | 2.9 |
| Characteristics | DBS [19] | DUS (This Study) |
|---|---|---|
| Sample volume, µL | 20 | 50 |
| Sample preparation | Extract: 400 µL MeOH Evaporation to dryness, add 400 µL water | Extract (in a polypropylene tip): 300 µL MeOH:H2O Filter the extract |
| LOD (ng/mL): | ||
| EMPA (VX acid) | 1 | 10 |
| IMPA (GB—acid) | 0.5 | 3 |
| iBuMPA (RVX-acid) | 1 | 1 |
| CMPA (GF-acid) | 1 | 0.5 |
| PMPA (GD-acid) | 0.3 | 0.5 |
| Long-term sample stability on the paper | ˃~1 month | ˃5 months |
| Concentration in an in vivo experiment (ng/mL): | 1LD50, 5 rats, IM.: | 1LD50, 5 mice, IN.: |
| 2 h after the exposure (Avg) | 28 | 8800 |
| 20 h after the exposure (Avg) | 3700 | |
| 24 h after the exposure (Avg) | 3.3 |
| Metabolite Structure | Negative Ion Formula | MRM (ESI−) | Transition Intensity Ratio |
|---|---|---|---|
EMPA (VX Acid) | C3H9PO3− | 123.0 > 95.0 | 3 |
| 123.0 > 79.0 | 1 | ||
IMPA (GB Acid) | C4H11PO3− | 137.0 > 95.0 | 6 |
| 137.0 > 79.0 | 1 | ||
iBuMPA (RVX Acid) | C5H13PO3− | 151.1 > 77.0 | 2 |
| 151.1 > 79.0 | 1 | ||
CMPA (GF Acid) | C7H15PO3− | 177.1 > 95.0 | 2 |
| 177.1 > 79.0 | 1 | ||
PMPA (GD Acid) | C7H17PO3− | 179.1 > 95.0 | 2 |
| 179.1 > 79.0 | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yishai Aviram, L.; Dagan, S.; Hindi, A.; Chapman, S.; Gez, R.; Drug, E. Monitoring Exposure to Five Chemical Warfare Agents Using the Dried Urine Spot Technique and Liquid Chromatography-Mass Spectrometry/Mass Spectrometry—In Vivo Determination of Sarin Metabolite in Mice. Molecules 2023, 28, 7687. https://doi.org/10.3390/molecules28237687
Yishai Aviram L, Dagan S, Hindi A, Chapman S, Gez R, Drug E. Monitoring Exposure to Five Chemical Warfare Agents Using the Dried Urine Spot Technique and Liquid Chromatography-Mass Spectrometry/Mass Spectrometry—In Vivo Determination of Sarin Metabolite in Mice. Molecules. 2023; 28(23):7687. https://doi.org/10.3390/molecules28237687
Chicago/Turabian StyleYishai Aviram, Lilach, Shai Dagan, Ariel Hindi, Shira Chapman, Rellie Gez, and Eyal Drug. 2023. "Monitoring Exposure to Five Chemical Warfare Agents Using the Dried Urine Spot Technique and Liquid Chromatography-Mass Spectrometry/Mass Spectrometry—In Vivo Determination of Sarin Metabolite in Mice" Molecules 28, no. 23: 7687. https://doi.org/10.3390/molecules28237687