
Condensation of Benzyl Carbamate with Glyoxal in Polar Protic and Aprotic Solvents

Laboratory for Chemistry of Nitrogen Compounds, Institute for Problems of Chemical and Energetic Technologies, Siberian Branch of the Russian Academy of Sciences (IPCET SB RAS), Biysk 659322, Russia
Molecules 2023, 28(22), 7648; https://doi.org/10.3390/molecules28227648
Submission received: 3 November 2023
/
Revised: 15 November 2023
/
Accepted: 16 November 2023
/
Published: 17 November 2023
(This article belongs to the Special Issue Heterocyclic Chemistry in Europe)
Abstract
:The synthesis of substituted 2,4,6,8,10,12-hexaazaisowurtzitane via direct condensation is challenging. The selection of starting ammonia derivatives is very limited. The important step in developing alternative synthetic routes to these compounds is to investigate their formation process in detail. Here, we examined an acid-catalyzed condensation between benzyl carbamate and glyoxal in a ratio of 2:1 in a range of polar protic and aprotic solvents, and discovered a new process occurring during the cascade condensation of glyoxal with ammonia derivatives as well as discovered several processes hindering the formation of caged compounds. More specifically, a cyclic compound, N,N′-bis(carbobenzoxy)-3,6-diamino-1,4-dioxane-2,5-diol, was found to form at the early stage of condensation under low acidity conditions. The formation of this compound is governed by an easier condensation of alcohol groups compared to the amide ones. The condensation intermediates, N,N′-bis(carbobenzoxy)ethan-1,2-diol, N,N′,N″-tris(carbobenzoxy)ethanol, and N,N′,N″,N‴-tetrakis(carbobenzoxy)ethan, were obtained at a higher acidity. A range of solvents were identified: those that react with benzyl carbamate, those that promote the progress of side processes, and those that promote precipitation of condensation intermediates. A few byproducts were isolated and identified. It was found that DMSO exhibits a strong deactivating ability, while CH3CN exhibits a strong activating ability towards the acid-catalyzed condensation process of benzyl carbamate with glyoxal.
1. Introduction
The search for and development of synthetic methods for new, more powerful high-energy-density materials (HEDMs) is a relevant objective for chemists and materials scientists worldwide [1,2,3,4,5,6,7,8,9,10,11,12,13]. One of the most actively developing directions in the synthesis of these substances is the synthesis of caged nitramines of aza- and oxaazaisowurtzitanes.
The first works that focused on the synthesis of caged nitramines incorporating oxaazaisowurtzitane cages appeared in the early 1980s but did not gain publicity [14,15]. The publicly available studies on the synthesis of the compact strained compound, 2,4,6,8,10,12-hexabenzyl-2,4,6,8,10,12-hexaazatetracyclo[5.5.0.03,11.05,9]dodecane (2,4,6,8,10,12-hexabenzyl-2,4,6,8,10,12-hexaazaisowurtzitane, HBIW), and on its nitration to 2,4,6,8,10,12-hexanitro-2,4,6,8,10,12-hexaazatetracyclo[5.5.0.03,11.05,9]dodecane (2,4,6,8,10,12-hexanitro-2,4,6,8,10,12-hexaazaisowurtzitane, CL-20) (Figure 1 and Scheme 1) appeared in the mid-1990s only [16,17,18,19,20]. The energetic performance of CL-20 aroused an extreme interest of specialists engaged in the synthesis of HEDMs. CL-20 exhibits one of the highest detonation velocities (V0D = 9.36 (ε) km/s) among all explosives and the highest density among the known nitramines (ρ = 2.044 g/cm3) [18,19,21,22,23], as well as a positive enthalpy of formation (ΔHf = 454 kJ/mol), optimal oxygen balance (−11.0) and detonation pressure (420 kbar) [23,24,25,26,27,28]. CL-20 is superior to other high-energy explosive materials such as HMX, RDX, PETN, etc., CL-20 is touted as a potential component of solid propellants [29,30,31,32,33,34,35,36,37,38] and composite explosives [39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55].
At present, CL-20 is the most efficient HEDM among those being industrially produced. Active efforts are underway to find new synthetic methods and improve the known ones [15,23,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79]. In addition, the possibility is being explored to better the properties of CL-20 through upgrading the molecule (replacement of nitro groups by other explosophoric groups; crosslinking with other high-energy bulky molecules) [80,81,82,83,84,85,86,87].
CL-20 is commercially produced by the multistage transfunctionalization of HBIW, which in turn is prepared by the condensation of benzylamine with glyoxal in about 89−95% yield [15,23,88,89,90]. All the commercial synthetic processes of CL-20 involve the catalytic reductive debenzylation of HBIW to 4,10-dibenzyl-2,6,8,12-tetraacetyl-2,4,6,8,10,12-hexaazatetracyclo[5.5.0.03,11.05,9]dodecane (4,10-dibenzyl-2,6,8,12-tetraacetyl-2,4,6,8,10,12-hexaazaisowurtzitane, TADBIW) (Scheme 1) [15,23]. The reduction uses a Pd-containing catalyst in the presence of acetic anhydride and a bromine source [91]. Despite the high yields of TADBIW (80–90%) [92,93,94,95], this stage is the most considerable contributor to the cost of CL-20. The Pd catalyst is poisoned by the debenzylation products and requires frequent replacement [96,97].
The problem associated with the high cost of CL-20 substantially limits the application of this HEDM and can be settled in two ways: the first one is by upgrading the known synthetic methods and the second one is by developing conceptually new approaches to its synthesis. Despite researchers from all around the world mostly focusing their efforts on the first method for as many as several decades, they have failed to replace the debenzylation stage or appreciably reduce its cost. In this regard, the second method is growing more urgent.
The most promising approach to the synthesis of CL-20 is a two-stage synthesis involving a condensation stage that produces the 2,4,6,8,10,12-hexaazaisowurtzitane derivative and a stage of exhaustive nitration of the resultant cage to CL-20. The major challenge of this approach is that it is difficult to pick ammonia derivatives capable of furnishing the 2,4,6,8,10,12-hexaazaisowurtzitane derivative with easily nitratable N-substituents [14,15,23].
Hexaazaisowurtzitane derivatives can only be obtained in acceptable yields by the condensation of glyoxal with benzylamine (its derivatives), benzylamine-like compounds (a series of amines bearing an aromatic moiety linked via a methylene bridge to the amino group), allylamine (or similar compounds) and, as it turned out most recently, with a primary amine where the amino group is linked to the strained aliphatic ring such as cyclopropylamine and cyclobutylamine [15,23,98,99].
The key to solving the problems related to developing the two-stage synthetic method for CL-20 can only be a detailed study of the formation process of the 2,4,6,8,10,12-hexaazaisowurtzitane cage. Since HBIW is assumed to be formed as a result of trimerization of the corresponding diimine [17] and benzylamine is the best ammonia derivative for the synthesis of 2,4,6,8,10,12-hexaazaisowurtzitane, it is of interest to explore the condensation of glyoxal with structurally similar compounds that have the ability to form imines. The present study reports the results of an acid-catalyzed condensation between benzyl carbamate (1) and glyoxal in protic and aprotic solvents.
2. Results and Discussion
We previously investigated the condensation of benzamide [100] and some substituted sulfonamides [101,102,103,104] with glyoxal whereby a range of new oxaazaisowurtzitane derivatives was obtained and several process mechanisms and regularities were discovered. The present study proposes investigating the formation of aza- and oxaazaisowurtzitanes, as well as finding ammonia derivatives suitable for this process. Benzyl carbamate (1) was chosen herein as a new substrate for the study. This compound is similar in structure to benzylamine and is capable of yielding imines [105]. We failed to find information on the condensation between carbamic acid esters and glyoxal.
The acid-catalyzed condensation between carbamate 1 and glyoxal was carried out in a ratio of 2:1 in polar protic (H2O, HCOOH (FoOH) and AcOH) and aprotic solvents (AcOEt, Et2O, (CH3)2CO, CH3CN, CH2Cl2, THF and DMSO) at room temperature. The acid catalyst used was H2SO4. The reaction products were analyzed by HPLC. Preparative chromatography and recrystallization were employed to isolate pure compounds.
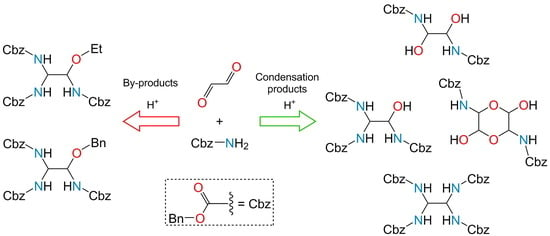
A few major reaction products were formed in the chosen solvents under moderate acidity conditions. All these products were isolated and identified: N,N′-bis(carbobenzoxy)-3,6-diamino-1,4-dioxane-2,5-diol (2); N,N′-bis(carbobenzoxy)ethan-1,2-diol (3); N,N′,N″-tris(carbobenzoxy)ethanol (4); and N,N′,N″,N‴-tetrakis(carbobenzoxy)ethan (5) (Scheme 2). The greatest difficulty was to isolate compounds 2–4 that are unstable in acidic and alkaline media. They cannot be heated and held in solvents for a long time.
Compound 2 was probably formed as a result of the condensation of two N-carbobenzoxyethan-1,2,2-triol (6) molecules (Scheme 3).
Three byproducts were isolated in addition to the condensation products of carbamate 1 with glyoxal. N,N′,N″-tris(carbobenzoxy)-2-ethoxyethan (7) was obtained in Et2O and AcOEt, which is a condensation product of compound 4 with EtOH (Scheme 4). EtOH required for the formation of this compound was formed by the hydrolysis of the solvents in the reaction mixture.
The hydrolysis of carbamate 1 was found to proceed in Et2O, THF, AcOEt and CH2Cl2 when the content of H2SO4 in the mixture was 2% and higher, as is corroborated by the formation of N,N′,N″-tris(carbobenzoxy)-2-benzoxyethan (8), a condensation product of compound 4 and benzyl alcohol (BnOH) (Scheme 5).
The condensation between carbamate 1 and glyoxal in FoOH did not almost take place. The major reaction product was a re-esterification product of benzyl formate (9) (Scheme 6). The process proceeded actively even without additional acidification with FoOH.
We investigated the condensation of carbamate 1 with glyoxal in aqueous H2SO4 first. Because carbamate 1 is poorly water-soluble, its dissolution was performed in aqueous H2SO4, afterwards glyoxal was added portionwise to the reaction mixture with stirring. The process was carried out for 3 h. In all cases, the portionwise addition of glyoxal produced an abundant precipitate. When the time was over, the reaction mixture was diluted with H2O and filtered off.
Table 1 summarizes the composition of the principal reaction products of the acid-catalyzed condensation between carbamate 1 and glyoxal over H2SO4 of different concentrations. The syntheses were affected for 3 h.
As is seen in Table 1, the condensation process in aqueous H2SO4 stopped at compound 5 which was formed in a small quantity and precipitated from the reaction mixture alongside compounds 2–4. Apart from the poor solubility of compounds 2–5 in aqueous H2SO4, the condensation was hindered by hydrolysis of the starting carbamate 1 through the ester group, as can be observed from the formation of compound 8. The hydrolysis process accelerated as the acidity was raised (Table 1, Entries 1–5).
Next, we examined the condensation in the chosen organic solvents. Table 2 summarizes the composition of the major reaction products of the acid-catalyzed condensation between carbamate 1 and glyoxal in a range of polar protic and aprotic organic solvents over H2SO4 of different concentrations. The syntheses were effected for 20 h.
The analysis of data outlined in Table 2 allows for some conclusions. For instance, an obvious relationship is observed between the condensation process activity and the acidity of the medium. In weak organic acids (Table 2, Entries 18 and 21) and in most of the organic solvents in question when the content of H2SO4 in the mixture was 0.3% (Table 2, Entries 1, 3, 6, 8, 13 and 15), compounds 2–4 were formed first. The formation of compound 2 required a considerably lower acidity than that of compound 5, indicating a higher condensation rate between the alcohol groups in the reaction medium compared to the condensation between the amide group of carbamate 1 and the alcohol groups. As the H2SO4 content in the mixture was raised to 2–7%, the condensation process was activated between compound 4 and carbamate 1 to give compound 5 in this case. This relationship is best visible in THF, DMSO and AcOH (Table 2, Entries 3–5, 15–17 and 21–23).
The main obstacles in the path of the acid-catalyzed condensation of carbamate 1 with glyoxal in the chosen solvents were side processes, low solubility of the condensation intermediates in the reaction mixture, and the deactivating effect of the solvent.
The side processes proceeded most actively in (CH3)2CO, AcOEt, FoOH, Et2O, CH2Cl2 and CH3CN.
In Et2O and AcOEt, the major byproduct was compound 7 (Table 2, Entries 1, 2, 6 and 7). The absence (a low yield) of the reaction products of EtOH with compound 3 can be suggestive of a low hydrolysis rate of the solvents under the reaction conditions. The formation of compound 3 was faster than the accumulation of EtOH in the reaction mixture. Compound 7 was formed in a considerable amount in unstable AcOEt at already 0.3% H2SO4, as opposed to Et2O (Table 2, Entry 6).
We noted previously that (CH3)2CO was readily engaged in a cascade of side condensation processes at an already small content of H2SO4 in the mixture [100]. However, the addition of 0.3–2% H2SO4 to the reaction mixture did not lead to an overly active formation of those compounds, and the condensation products of carbamate 1 and glyoxal were formed together with byproducts (Table 2, Entries 8–9). As the amount of H2SO4 was raised to 7%, the formation of byproducts accelerated so much that the reaction mass became resinified within 20 h, acquiring a dark-brown hue.
The condensation was slow in CH2Cl2 at 0.3% H2SO4 (Table 2, Entry 13). Only a small amount of compounds 2–5 was generated within 20 h. When the content of H2SO4 in the mixture was raised to 2%, the condensation process was activated and the hydrolysis process of carbamate 1 began concurrently to liberate BnOH (Table 2, Entry 14). Byproduct 8 was formed in 11% and compound 5 low soluble in CH2Cl2 precipitated in a great amount. The process performed at 7% H2SO4 in the reaction mixture resulted in a great quantity of byproducts, which were probably the condensation product of BnOH with carbamate 1 and the condensation products of BnOH with condensation intermediates of carbamate 1 and glyoxal.
Unlike (CH3)2CO, AcOEt, FoOH, Et2O and CH2Cl2, the condensation in CH3CN at 0.3–2% H2SO4 in the mixture took place actively without the formation of byproducts and hydrolysis products. However, when the acidity of the medium was raised to 7%, the amount of the resulting products 4 and 5 decreased abruptly, and products with a short retention time (HPLC), shorter than that of carbamate 1, were observed to form in the stock solution. Probably at least one of these products was a reaction product (a short HPLC retention time) of acetonitrile because it was not formed in the other solvents.
The condensation intermediates were observed to precipitate actively in Et2O, CH2Cl2, CH3CN and AcOH, shifting the reaction equilibrium.
When the process was carried out in Et2O at 2% H2SO4 in the reaction mixture, a great amount of compound 4 was formed that precipitated from the reaction mixture. That said, no formation of compound 5 was noticed, which is likely suggestive of an extremely low solubility of compound 4 in this solvent (Table 2, Entry 2).
The condensation carried out in CH2Cl2, CH3CN and AcOH was accompanied by the precipitation of compound 5. The greatest quantity of this compound within 20 h was formed in CH2Cl2 (Table 2, Entry 14) and CH3CN (Table 2, Entry 11) over 2% H2SO4 in the mixture, and in AcOH (Table 2, Entry 23) over 7% H2SO4.
When the process was performed in pure AcOH for 20 h, compound 2 precipitated in a small quantity (Table 2, Entry 21). AcOH appeared to possess sufficient acidity for the formation of compound 6 and its condensation to compound 2.
It can be hypothesized that the activating effect of the polar aprotic solvents on the acid-catalyzed condensation in question will depend on their dipole moments but this dependence was not observed. Having similar dipole moments, solvents like CH3CN (3.92 D) and DMSO (3.96 D) exerted an opposite effect on the condensation process.
The condensation process was the slowest in DMSO. Even when the content of H2SO4 in the mixture was 7%, no active formation of condensation products was recorded (Table 2, Entry 17). DMSO behaves itself in a similar manner when benzylamine is condensed with glyoxal [100]. The ability of this solvent to deactivate the acid-catalyzed condensation process was employed by us in the analysis of unstable condensation intermediates 2–4, which were dissolved in DMSO-D6 for 1H and 13C NMR analysis. It is quite probable that DMSO is likewise able to deactivate the condensation of other unstable compounds that structurally contain active hydroxyls.
The highest activating ability with respect to the condensation process was observed in CH3CN (Table 2, Entries 10–12). Compounds 4 and 5 were observed to form actively at already 0.3% H2SO4 in the mixture and began to precipitate from the reaction mixture when glyoxal was added portionwise.
The analysis of the HPLC data for the condensation products of carbamate 1 and glyoxal in the chosen solvents allows for the conclusion that the condensation process in some solvents does not stop at the precipitation of compounds 4 and 5 but proceeds “deeper” to produce a set of more complex condensation products having a longer retention time (HPLC). The compounds (about 20 in number) were identical for all the solvents and did not precipitate from the reaction mixture. The formation of these compounds occurred in Et2O and THF at 2–7% H2SO4 in the mixture, in CH2Cl2 and AcOH at 2% H2SO4, in CH3CN at 0.3–2% H2SO4, and negligibly in FoOH. These products were formed most actively at the lowest acidity in CH3CN.
3. Materials and Methods
The reagents were purchased from commercial suppliers and used as received, unless mentioned otherwise. Commercially available compounds were used without further purification, unless stated otherwise. Melting points were determined on a Stuart SMP30 melting point apparatus (Bibby Scientific Ltd., Staffordshire, UK). Infrared (IR) spectra were recorded on a Simex FT-801 Fourier transform infrared spectrometer (Simex, Novosibirsk, Russia) in KBr pellets or in a liquid film. 1H and 13C NMR spectra were recorded on a Bruker AV-400 instrument (Bruker Corporation, Billerica, MA, USA) at 400 MHz and 100 MHz. Chemical shifts are expressed in ppm (δ). Elemental analysis was performed on a ThermoFisher FlashEA 1112 elemental analyzer (ThermoFisher, Waltham, MA, USA). For preparative chromatography, silica gel Kieselgel 60 (0.063–0.2 mm, Macherey-Nagel GmbH & Co. KG, Dueren, Germany) was used. HPLC analysis was performed on an Agilent 1200 chromatograph (Agilent Technologies, Waldbronn, Germany) with a diode array detector. The separation was carried out on a Hypersil ODS (100 × 2.1 mm, a 5 µm mesh) column. Mixed solvents A (0.2% phosphoric acid) and B (acetonitrile) were used as the mobile phase. The mobile phase composition was varied in the gradient mode: the concentration of solvent B was linearly raised from 20 to 50% for 5 min, then from 50 to 100% for 5 to 70 min, and maintained at this level for another 10 min. The flow rate of the eluent was 0.25 mL/min, the column temperature was 25 °C, the detection was run at a 210-nm wavelength, and the sample volume was 3 µL. The column conditioning time between successive injections was 15 min.
4. Experimental
4.1. N,N′-Bis(carbobenzoxy)-3,6-diamino-1,4-dioxane-2,5-diol (2)
Carbamate 1 (3 g, 0.020 mol) was added to AcOH (84 mL) with vigorous stirring. Next, after the carbamate was dissolved, glyoxal (1.44 g, 40%, 0.010 mol) was added to the reaction mixture, and the whole was allowed to be stirred at room temperature for 60 h and then filtered. The filter cake was washed with (CH3)2CO (4 × 5 mL), transferred to a beaker and muddled in (CH3)2CO (30 mL) at reflux for 20 min, and then filtered. The filter cake was washed with (CH3)2CO (2 × 6 mL) and dried to constant weight at room temperature (0.44 g; containing 77% of compound 2). The dry sediment was further dissolved in a minimum quantity of DMSO at 60–70 °C, diluted twofold with acetonitrile and allowed to crystallize at room temperature for 24 h. Upon time completion, the mixture was filtered and the filter cake was washed with (CH3)2CO (3 × 5 mL) and dried to constant weight at room temperature to furnish compound 2 as a white powder.
Yield: 0.104 g (94.8% assay HPLC), 0.236 mmol (4.7% calculated on a glyoxal basis). Mp = 189–192 °C. IR (KBr): ν = 3369, 3311, 3060, 3029, 2969, 2895, 1699, 1525, 1452, 1360, 1343, 1243, 1145, 1086, 1044, 986, 964, 911, 881, 856, 784, 745, 697, 662 cm−1. 1H NMR (DMSO-D6): δ = 4.84 (s, 2H), 5.05 (q, J1 = 12.9, J2 = 17.1, Hz, 4H), 5.41 (d, J = 9.7 Hz, 2H), 6.99 (d, J = 2.5 Hz, 2H), 7.25 (d, J = 9.6 Hz, 2H), 7.35–7.32 (m, 10H) ppm. 13C{1H} NMR (DMSO-D6): δ = 66.0, 72.2, 90.0, 128.1, 128.3, 128.8, 137.3, 155.9 ppm. Elemental analysis, calcd (%) for C20H22N2O8 (418.40): C, 57.41; H, 5.30; N, 6.70; O, 30.59; found: C, 55.84; H, 5.22; N, 6.58; O, 29.63 (see Supplementary Materials).
4.2. N,N′-Bis(carbobenzoxy)ethan-1,2-diol (3)
H2SO4 (0.202 mL, 0.37 g, 94%) was added to Et2O (80 mL) with vigorous stirring and cooling with ice water. The reaction mixture was then heated to 15–20 °C; afterwards, carbamate 1 (4 g, 0.026 mol) was added thereto with stirring. After the carbamate was dissolved, glyoxal (1.92 g, 40%, 0.013 mol) was added portionwise to the mixture for 10 min at 22–25 °C. Once the portionwise addition was completed, the mixture was left to stir at room temperature for 45 min and then filtered. The filter cake was washed with Et2O (3 × 5 mL) and H2O (4 × 7 mL), transferred to a beaker and muddled in H2O (40 mL) for 1 h at room temperature, and then filtered. The filter cake was washed with H2O (3 × 7 mL) and dried to constant weight at room temperature (0.823 g; containing 53% of compound 3). The resultant sediment was then muddled in (CH3)2CO (150 mL) for 1.5 h and then filtered. The filtrate was evaporated in a rotary evaporator at a bath temperature that was not above 25 °C. The residue was recrystallized from (CH3)2CO (without heating), and the filter cake was washed with Et2O (2 × 5 mL) and dried at room temperature to constant weight to give compound 3 as a white crystalline powder.
Yield: 0.298 g (95.4% assay HPLC), 0.827 mmol (6.0% calculated on a compound 1 basis). Mp = 147–149 °C. IR (KBr): ν = 3307, 3273, 3061, 3035, 2942, 1688, 1544, 1497, 1451, 1409, 1380, 1312, 1282, 1236, 1156, 1045, 1010, 968, 910, 779, 731, 694 cm−1. 1H NMR (DMSO-D6): δ = 4.88 (t, J = 5.1 Hz, 2H), 5.03 (q, J1 = 12.6, J2 = 19.1, 4H), 5.73 (br. s, 2H), 7.30–7.37 (m, 10H), 7.46 (d, J = 5.2 Hz, 2H) ppm. 13C{1H} NMR (DMSO-D6): δ = 65.7, 77.0, 128.2, 128.8, 137.5, 156.0 ppm. Elemental analysis, calcd (%) for C18H20N2O6 (360.36): C, 59.99; H, 5.59; N, 7.77; O, 26.64; found: C, 59.96; H, 5.62; N, 7.72; O, 26.56 (see Supplementary Materials).
4.3. N,N′,N″-Tris(carbobenzoxy)ethanol (4)
H2SO4 (0.150 mL, 0.275 g, 94%) was added to Et2O (20 mL) with vigorous stirring and cooling with ice water. The reaction mixture was then heated to 15–20 °C; afterwards, carbamate 1 (0.717 g, 4.743 mmol) was added to the mixture with stirring. After the carbamate was dissolved, glyoxal (0.344 g, 40%, 2.370 mmol) was added portionwise to the mixture for 10 min at 22–25 °C. Once the portionwise addition was completed, the mixture was left to stir at room temperature for 20 h and then filtered. The filter cake was washed with Et2O (3 × 5 mL) and H2O (4 × 7 mL), transferred to a beaker and muddled in H2O (40 mL) for 1 h at room temperature, and then filtered. The filter cake was washed with H2O (3 × 7 mL) and dried to constant weight at room temperature (0.152 g; containing 78% of compound 4). The resultant sediment was then muddled in (CH3)2CO (15 mL) at room temperature for 1.5 h and then filtered. The filtrate was evaporated in a rotary evaporator at a bath temperature that was not above 25 °C. The resultant sediment was dissolved in a minimum quantity of (CH3)2CO (without heating); afterwards, compound 4 was crystallized out with hexane. For this, the solution had hexane added drop by drop until active crystallization started and was left to crystallize at room temperature for 24 h. Upon time completion, the suspension was filtered, and the filter cake was washed with Et2O (2 × 8 mL) and dried to constant weight at room temperature to afford compound 4 as a white crystalline powder.
Yield: 0.067 g (94.0% assay HPLC), 0.128 mmol (8.1% calculated on a compound 1 basis). Mp = 175–177 °C. IR (KBr): ν = 3436, 3307, 3061, 3032, 2951, 2891, 1693, 1662, 1536, 1513, 1454, 1381, 1337, 1295, 1225, 1156, 1086, 1016, 967, 913, 844, 782, 737, 695 cm−1. 1H NMR (DMSO-D6): δ = 4.99–5.08 (m, 6H), 5.14 (br. s, 2H), 6.10 (s, 1H), 7.28–7.44 (m, 17H), 7.61 (d, J = 6.6 Hz, 1H) ppm. 13C{1H} NMR (DMSO-D6): δ = 62.8, 65.9, 66.0, 75.6, 128.2, 128.8, 137.4, 155.6, 155.9, 156.1 ppm. Elemental analysis, calcd (%) for C26H27N3O7 (493.508): C, 63.28; H, 5.51; N, 8.51; O, 22.69; found: C, 62.85; H, 5.54; N, 8.44; O, 22.39 (see Supplementary Materials).
4.4. N,N′,N″,N‴-Tetrakis(carbobenzoxy)ethan (5)
H2SO4 (0.819 mL, 1.5 g, 94%) was added to AcOH (20 mL) with vigorous stirring and cooling with ice water. The reaction mixture was then heated to 15–20 °C; afterwards, carbamate 1 (0.717 g, 4.743 mmol) was added to the mixture with stirring. After the carbamate was dissolved, glyoxal (0.344 g, 40%, 2.370 mmol) was added portionwise to the mixture for 10 min at 22–25 °C. Once the portionwise addition was completed, the mixture was left to stir at room temperature for 20 h and then filtered. The filter cake was washed with AcOH (2 × 4 mL) and Et2O (3 × 5 mL) and dried to constant weight at room temperature (0.211 g; containing 82% of compound 5). The sediment was transferred to a beaker and muddled in DMF (20 mL) at room temperature for 1 h, and then filtered. The filter cake was washed with Et2O (2 × 5 mL) and dried to constant weight at room temperature. The resultant sediment was then dissolved in a minimum quantity of hot DMSO at 50–60 °C, afterwards compound 5 was crystallized out with acetonitrile. For this, the solution was cooled down to room temperature, diluted eightfold with CH3CN and then allowed to undergo crystallization at room temperature for 24 h. Upon time completion, the suspension was filtered, and the filter cake was washed with Et2O (3 × 5 mL) and dried to constant weight at room temperature to furnish compound 5 as a white crystalline powder.
Yield: 0.125 g (96.0% assay HPLC), 0.191 mmol (16.1% calculated on a compound 1 basis). Mp = 278–280 °C. IR (KBr): ν = 3305, 3091, 3034, 2951, 2897, 1698, 1547, 1518, 1454, 1339, 1298, 1238, 1139, 1012, 981, 914, 844, 785, 757, 740, 697, 673 cm−1. 1H NMR (DMSO-D6): δ = 5.02 (s, 8H), 5.32 (br. s, 2H), 7.34 (s, 20H), 5.32 (br. s, 4H) ppm. 13C{1H} NMR (DMSO-D6): δ = 61.6, 66.0, 128.2, 128.8, 137.2, 155.7 ppm. Elemental analysis, calcd (%) for C34H34N4O8 (626.65): C, 65.17; H, 5.47; N, 8.94; O, 20.43; found: C, 64.43; H, 5.43; N, 8.85; O, 20.37 (see Supplementary Materials).
4.5. N,N′,N″-Tris(carbobenzoxy)-2-ethoxyethan (7)
H2SO4 (0.218 mL, 0.4 g, 94%) was added to AcOH (20 mL) with vigorous stirring and cooling with ice water. The reaction mixture was then heated to 15–20 °C; afterwards, carbamate 1 (0.717 g, 4.743 mmol) was added to the mixture with stirring. After the carbamate was dissolved, glyoxal (0.344 g, 40%, 2.370 mmol) was added portionwise to the mixture for 10 min at 22–25 °C. Once the portionwise addition was completed, the mixture was left to stir at room temperature for 20 h and then filtered. The filter cake was washed with AcOH (2 × 5 mL) and Et2O (4 × 5 mL) and dried to constant weight at room temperature (0.255 g; containing 61% of compound 7). The resultant sediment was then muddled in (CH3)2CO (15 mL) at room temperature for 1.5 h and then filtered. The filtrate was evaporated in a rotary evaporator at a bath temperature that was not above 30 °C. The residue was recrystallized from (CH3)2CO and dried to constant weight at room temperature to give compound 7 as a white crystalline powder.
Yield: 0.166 g (97.1% assay HPLC), 0.309 mmol (19.5% calculated on a compound 1 basis. Mp = 194–196 °C. IR (KBr): ν = 3308, 3062, 3032, 2970, 2927, 2897, 1692, 1535, 1514, 1454, 1380, 1337, 1297, 1274, 1162, 1099, 1052, 1017, 910, 843, 780, 737, 694 cm−1. 1H NMR (DMSO-D6): δ = 1.06 (t, J = 6.4 Hz, 3H), 3.36–3.55 (m, 2H), 4.97–5.10 (m, 7H), 5.22 (br. s, 1H), 7.35 (s, 15H), 7.53 (d, J = 6.7 Hz, 1H), 7.64 (d, J = 5.6 Hz, 1H), 7.76 (d, J = 9.0 Hz, 1H) ppm. 13C{1H} NMR (DMSO-D6): δ = 15.4, 61.6, 63.1, 65.84, 65.98, 66.05, 81.66, 128.14, 128.27, 128.33, 128.79, 128.82, 137.2, 137.4, 155.6, 155.8, 156.7 ppm. Elemental analysis, calcd (%) for C28H31N3O7 (521.56): C, 64.48; H, 5.99; N, 8.06; O, 21.47; found: C, 64.47; H, 6.02; N, 8.00; O, 21.33 (see Supplementary Materials).
4.6. N,N′,N″-Tris(carbobenzoxy)-2-benzoxyethan (8)
H2SO4 (1.8 mL, 3.3 g, 94%) was added to H2O (2 mL) with vigorous stirring and cooling with ice water. The resultant mixture was then heated to room temperature and carbamate 1 (0.358 g, 2.368 mmol) was added with stirring. After the carbamate was dissolved, glyoxal (0.172 g, 40%, 1.185 mmol) was added portionwise to the mixture for 10 min at 22–25 °C. Once the portionwise addition was completed, the mixture was allowed to stir at room temperature for 3 h. Upon time completion, the reaction mixture was diluted with ice water (30 mL), stirred for 10 min and filtered. The filter cake was washed with H2O (5 × 7 mL) and dried to constant weight at room temperature (0.322 g; containing 25.1% of compound 8). The resultant sediment was separated by preparative chromatography. Mixed CHCl3:CH3CN:AcOH in a ratio of 10:1:0.15 v/v was used as the eluent. Some of the sediment (compound 7) was insoluble in the eluent and should be filtered off prior to preparative chromatography. A fraction comprising at least 85% of compound 8 (Rf 0.69; HPLC control) was collected. Chloroform and acetonitrile were evaporated from the collected fraction in a rotary evaporator at a bath temperature not above 35 °C. Most of the AcOH was evaporated from the recovery flask with rubber bellow at room temperature. The resultant sediment with acetic acid traces was recrystallized from the same eluent as used in preparative chromatography, and the filter cake was washed with Et2O (2 × 5 mL) and dried to constant weight at room temperature to furnish compound 8 as a white powder.
Yield: 0.044 g (98.5% assay HPLC), 0.074 mmol (12.5% calculated on a compound 1 basis with allowance for hydrolysis of compound 1 to benzyl alcohol). Mp = 201–203 °C. IR (KBr): ν = 3320, 3089, 3062, 3034, 2959, 2882, 1710, 1693, 1556, 1530, 1514, 1453, 1379, 1341, 1297, 1270, 1238, 1150, 1101, 1050, 1023, 997, 973, 910, 825, 775, 754, 731, 695, 676 cm−1. 1H NMR (DMSO-D6): δ = 4.51 (q, J1 = 11.8, J2 = 36.6, 4H), 5.04–5.13 (m, 7H), 5.32 (br. s, 1H), 7.29–7.34 (m, 20H), 7.66 (dd, J1 = 6.7, J2 = 27.3, 2H) 7.95 (d, J = 9.0 Hz, 1H) ppm. 13C{1H} NMR (DMSO-D6): δ = 61.6, 65.90, 65.99, 66,13, 69.3, 81.9, 127.82, 127.96, 128.16, 128.24, 128.33, 128.6, 128.80, 128.82, 137.24, 137.29, 137.33, 138.5, 155.6, 155.8, 156.8 ppm. Elemental analysis, calcd (%) for C33H33N3O7 (583.63): C, 67.91; H, 5.70; N, 7.20; O, 19.19; found: C, 67.85; H, 5.76; N, 7.15; O, 19.10 (see Supplementary Materials).
5. Conclusions
The acid-catalyzed condensation between benzyl carbamate and glyoxal in a molar ratio of 2:1 was investigated in detail in a range of polar protic and aprotic solvents. Benzyl carbamate proved to be an active amide sufficiently stable towards acid hydrolysis and had the ability to engage in a condensation reaction with glyoxal under relatively low acidity conditions. For instance, in a relatively weak AcOH, the condensation between two benzyl carbamate molecules and two glyoxal molecules proceeded to yield a cyclic compound, N,N′-bis(carbobenzoxy)-3,6-diamino-1,4-dioxane-2,5-diol (2). Increasing the acidity of the solvents by adding H2SO4 to the reaction mixture allowed the activation of the condensation process and the preparation of products containing a greater quantity of secondary amido groups: N,N′-bis(carbobenzoxy)ethan-1,2-diol (3), N,N′,N″-tris(carbobenzoxy)ethanol (4) and N,N′,N″,N‴-tetrakis(carbobenzoxy)ethan (5).
The main obstacles in the path of the acid-catalyzed condensation of benzyl carbamate with glyoxal were discovered herein, which are side processes, the low solubility of condensation intermediates in the reaction mixture, and the deactivating effect of the solvent.
Side processes were noted to take place in most solvents under study and their rate depended on the acidity of the medium. N,N′,N″-tris(carbobenzoxy)-2-ethoxyethan (7) was formed in AcOEt and Et2O, re-esterification of benzyl carbamate took place actively in FoOH to give benzyl formate (9), while a range of byproducts was formed in (CH3)2CO by the transformation of (CH3)2CO. A great many byproducts with a short retention time (HPLC) were observed to form in solvents such as CH2Cl2 and CH3CN when the content of H2SO4 in the mixture was 7%. Hydrolysis of benzyl carbamate took place in H2O and CH2Cl2 to furnish a byproduct, N,N′,N″-tris(carbobenzoxy)-2-benzoxyethan (8).
The condensation intermediates were observed to precipitate actively in Et2O, CH2Cl2, CH3CN and AcOH, shifting the reaction equilibrium.
It has been established that there is no direct correlation between the dipole moments of the polar aprotic solvents and the activating effect with respect to the acid-catalyzed condensation process of benzyl carbamate with glyoxal.
DMSO had the strongest deactivating effect on the acid-catalyzed condensation process of benzyl carbamate with glyoxal. This feature of DMSO can probably be utilized for the stabilization of other compounds bearing active hydroxyls.
It can thus be concluded that H2O, Et2O, CH2Cl2, AcOEt, (CH3)2CO and DMSO are the worst solvents for the cascade condensation of benzyl carbamate with glyoxal. The use of these solvents for an acid-catalyzed condensation of other ammonia derivatives having a similar or lower basicity will probably be difficult as well.
Among the solvents chosen, CH3CN had the strongest activating effect on the acid-catalyzed condensation process between benzyl carbamate and glyoxal. The addition of as low as 0.3% H2SO4 to the reaction mixture is enough for an active formation of N,N′,N″,N‴-tetrakis(carbobenzoxy)ethan (5) and a range of more complex condensation products having a longer retention time than compound 5 (HPLC). The high activating ability of CH3CN with respect to the acid-catalyzed condensation reaction is likely a major factor that makes this solvent the best for the synthesis of 2,4,6,8,10,12-hexaazaisowurtzitane, which is particularly corroborated by the highest yields of HBIW and other hexaazaisowurtzitanes in this solvent.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules28227648/s1, Figures S1–S13: 1H and 13C NMR of compounds 2, 3, 4, 5, 7 and 8.
Funding
This research was funded by the Ministry of Science and Higher Education of the Russian Federation (grant No. 075-15-2020-803 with the Zelinsky Institute of Organic Chemistry of the RAS).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article and Supplementary Materials.
Acknowledgments
The work was conducted using instruments provided by the Biysk Regional Center for Shared Use of Scientific Equipment of the SB RAS (IPCET SB RAS, Biysk).
Conflicts of Interest
The author declares no conflict of interest.
References
- Xue, Q.; Bi, F.; Zhang, J.; Zhang, J.; Wang, B.; Wu, M. Synthesis and characterization of two 1,2,4-oxadiazole-furazan-based nitrate ester compounds as potential energetic plasticizers. FirePhysChem 2023, 3, 16–22. [Google Scholar] [CrossRef]
- Parakhin, V.V.; Smirnov, G.A. Research progress on design, synthesis and performance of energetic polynitro hexaazaisowurtzitane derivatives: Towards improved CL-20 analogues. FirePhysChem 2023, in press. [Google Scholar] [CrossRef]
- Yadav, A.K.; Jujam, M.; Ghule, V.D.; Dharavath, S. High-performing, insensitive and thermally stable energetic materials from zwitterionic gem-dinitromethyl substituted C-C bonded 1,2,4-triazole and 1,3,4-oxadiazole. Chem. Commun. 2023, 59, 4324–4327. [Google Scholar] [CrossRef]
- Zhang, W.; Yang, Y.; Wang, Y.; Fei, T.; Wang, Y.; Sun, C.; Pang, S. Challenging the limits of the oxygen balance of a pyrazole ring. Chem. Eng. J. 2023, 451, 138609. [Google Scholar] [CrossRef]
- Wang, T.; Lu, Z.; Bu, S.; Kuang, B.; Zhang, L.; Yi, Z.; Wang, K.; Zhu, S.; Zhang, J. Combination of nitrogen-rich skeleton and coordination group: Synthesis of a high-energy primary explosive based on 1H-tetrazole-5-carbohydrazide. Def. Technol. 2023, in press. [Google Scholar] [CrossRef]
- Wang, K.; Li, X.; Yang, K.; Huo, H.; Xue, Q.; Wang, B.; Bi, F. A novel synthetic method of 1,1,4,4-tetramethyl-2-tetrazene (TMTZ) via photocatalytic reaction. FirePhysChem 2022, 2, 267–271. [Google Scholar] [CrossRef]
- Xiong, J.; Cai, J.; Lai, Q.; Yin, P.; Pang, S. Asymmetric assembly of pyrazole and 1,2,3-triazole with a methylene bridge: Regioisomerism and energetic properties. Chem. Commun. 2022, 58, 10647–10650. [Google Scholar] [CrossRef]
- Ma, W.; Zhang, Z.Q.; Ma, Q.; Tang, J.; Yang, W.; Yang, H.; Cheng, G.; Fan, G.J. Bicyclic high-energy and low-sensitivity regioisomeric energetic compounds based on polynitrobenzene and pyrazoles. Cryst. Growth Des. 2023, 23, 1127–1132. [Google Scholar] [CrossRef]
- Konnov, A.A.; Klenov, M.S.; Churakov, A.M.; Dalinger, I.L.; Strelenko, Y.A.; Fedyanin, I.V.; Lempert, D.B.; Pivkina, A.N.; Kon’kova, T.S.; Matyushin, Y.N.; et al. Novel energetic furazans containing isomeric N-(azoxy)-dinitropyrazole moieties: Synthesis, characterization and comparison of properties. Energetic Mater. Front. 2023, 4, 1–9. [Google Scholar] [CrossRef]
- Pandey, K.; Bhatia, P.; Dolui, P.; Ghule, V.D.; Kumar, D. Connecting energetic nitropyrazole and nitrobenzene moieties with C-C bonds using suzuki cross-coupling reaction: A novel route to thermally stable energetic materials. Asian J. Org. Chem. 2022, 11, e202200543. [Google Scholar] [CrossRef]
- Leonov, N.E.; Emel’yanov, A.E.; Klenov, M.S.; Churakov, A.M.; Strelenko, Y.A.; Pivkina, A.N.; Fedyanin, I.V.; Lempert, D.B.; Kon’kova, T.S.; Matyushin, Y.N.; et al. Novel (1H-tetrazol-5-yl-NNO-azoxy)furazans and their energetic salts: Synthesis, characterization and energetic properties. Mendeleev Commun. 2022, 32, 714–716. [Google Scholar] [CrossRef]
- Sheremetev, A.B.; Mel’nikova, S.F.; Kokareva, E.S.; Nekrutenko, R.E.; Strizhenko, K.V.; Suponitsky, K.Y.; Pham, T.D.; Pivkina, A.N.; Sinditskii, V.P. Nitroxy- and azidomethyl azofurazans as advanced energetic materials. Def. Technol. 2022, 18, 1369–1381. [Google Scholar] [CrossRef]
- Larin, A.A.; Ananyev, I.V.; Dubasova, E.V.; Teslenko, F.E.; Monogarov, K.A.; Khakimov, D.V.; He, C.; Pang, S.; Gazieva, G.A.; Fershtat, L.L. Simple and energetic: Novel combination of furoxan and 1,2,4-triazole rings in the synthesis of energetic materials. Energ. Mater. Front. 2022, 3, 146–153. [Google Scholar] [CrossRef]
- Paromov, A.E.; Sysolyatin, S.V. Oxaazatetracyclo[5.5.0.03,11.05,9]dodecanes—A promising foundation for the design of ther-mally stable, high-density energetic compounds. Chem. Heterocycl. Compd. 2017, 53, 630–637. [Google Scholar] [CrossRef]
- Gong, X.; Sun, C.; Pang, S.; Zhang, J.; Li, Y.; Zhao, X. Research Progress in Study of Isowurtzitane Derivatives. Chin. J. Org. Chem. 2012, 3, 486–496. [Google Scholar] [CrossRef]
- Kodama, T.; Tojo, M.; Ikeda, M. Hexaazaisowurtzitane Derivatives and Process for Producing the Same. WO Patent 9623792 A1, 17 May 2000. [Google Scholar]
- Nielsen, A.T.; Nissan, R.A.; Vanderah, D.J. Polyazapolycyclics by condensation of aldehydes with amines. 2. Formation of 2,4,6,8,10,12-hexabenzyl-2,4,6,8,10,12-hexaazatetracyclo[5.5.0.05.9.03,11]dodecanes from glyoxal and benzylamines. J. Org. Chem. 1990, 55, 1459–1466. [Google Scholar] [CrossRef]
- Nielsen, A.T. Caged Polynitramine Compound. U.S. Patent 5693794, 2 December 1997. [Google Scholar]
- Nielsen, A.T.; Chafin, A.P.; Christian, S.L.; Moore, D.W.; Nadler, M.P.; Nissan, R.A.; Vanderah, D.J.; Flippen-Anderson, J.L. Synthesis of polyazapolycyclic caged polynitramines. Tetrahedron 1998, 54, 11793–11812. [Google Scholar] [CrossRef]
- Sakovich, G.V.; Sysolyatin, S.V.; Kozyrev, N.V.; Makarovets, N.A. Explosive Composition. RU Patent 2252925, 27 May 2005. [Google Scholar]
- Viswanath, D.S.; Ghosh, T.K.; Boddu, V.M. Hexanitrohexaazaisowurtzitane (HNIW, CL-20). In Emerging Energetic Materials: Synthesis, Physicochemical, and Detonation Properties; Viswanath, D.S., Ghosh, T.K., Boddu, V.M., Eds.; Springer: Dordrecht, The Netherlands, 2018; pp. 59–100. [Google Scholar] [CrossRef]
- Sysolyatin, S.V.; Lobanova, A.A.; Chernikova, Y.T.; Sakovich, G.V. Methods of synthesis and properties of hexanitrohexaazaisowurtzitane. Russ. Chem. Rev. 2005, 74, 757–764. [Google Scholar] [CrossRef]
- Venkata Viswanath, J.; Venugopal, K.J.; Srinivasa Rao, N.V.; Venkataraman, A. An overview on importance, synthetic strategies and studies of 2,4,6,8,10,12-hexanitro-2,4,6,8,10,12-hexaazaisowurtzitane (HNIW). Def. Technol. 2016, 12, 401–418. [Google Scholar] [CrossRef]
- Nair, U.R.; Sivabalan, R.; Gore, G.M.; Geetha, M.; Asthana, S.N.; Singh, H. Hexanitrohexaazaisowurtzitane (CL-20) and CL-20-based formulations (review). Combust. Explos. Shock. Waves 2005, 41, 121–132. [Google Scholar] [CrossRef]
- Bumpus, J.A. A Theoretical Investigation of the Ring Strain Energy, Destabilization Energy, and Heat of Formation of CL-20. Adv. Phys. Chem. 2012, 2012, 175146. [Google Scholar] [CrossRef]
- Krause, H.H. New Energetic Materials. In Energetic Materials: Particle Processing and Characterization; Teipel, U., Ed.; Wiley-VCH: Weinheim, Germany, 2005; pp. 1–25. [Google Scholar]
- Mandal, A.K.; Pant, C.S.; Kasar, S.M.; Soman, T. Process Optimization for Synthesis of CL-20. J. Energ. Mater 2009, 27, 231–246. [Google Scholar] [CrossRef]
- Talawar, M.B.; Sivabalan, R.; Anniyappan, M.; Gore, G.M.; Asthana, S.N.; Gandhe, B.R. Emerging trends in advanced high energy materials. Combust. Explos. Shock Waves 2007, 43, 62–72. [Google Scholar] [CrossRef]
- Singh, H. Survey of New Energetic and Eco-friendly. Materials for Propulsion of Space Vehicles. In Chemical Rocket Propulsion. A Comprehensive Survey of Energetic Materials; De Luca, L.T., Shimada, T., Sinditskii, V.P., Calabro, M., Eds.; Springer: Dordrecht, The Netherlands, 2017; pp. 127–138. [Google Scholar]
- Aldoshin, S.M.; Lempert, D.B.; Goncharov, T.K.; Kazakov, A.I.; Soglasnova, S.I.; Dorofeenko, E.M.; Plishkin, N.A. Energetic potential of solid composite propellants based on CL-20-containing bimolecular crystals. Russ. Chem. Bull. 2016, 65, 2018–2024. [Google Scholar] [CrossRef]
- Wu, Z.; Liu, N.; Zheng, W.; Chen, J.; Song, X.; Wang, J.; Cui, C.; Zhang, D.; Zhao, F. Application and Properties of CL-20/HMX Cocrystal in Composite Modified Double Base Propellants. Propellants Explos. Pyrotech. 2020, 45, 92–100. [Google Scholar] [CrossRef]
- Wang, J.; Yang, L.; Zheng, W.; Zhang, J. Study on Comparative Performance of CL-20/RDX-based CMDB Propellants. Propellants Explos. Pyrotech. 2019, 44, 1175–1182. [Google Scholar] [CrossRef]
- Sergienko, A.V.; Popenko, E.M.; Slyusarsky, K.V.; Larionov, K.B.; Dzidziguri, E.L.; Kondratyeva, E.S.; Gromov, A.A. Burning Characteristics of the HMX/CL-20/AP/Polyvinyltetrazole Binder/Al Solid Propellants Loaded with Nanometals. Propellants Explos. Pyrotech. 2019, 44, 217–223. [Google Scholar] [CrossRef]
- Sinditskii, V.P.; Chernyi, A.N.; Egorshev, V.Y.; Dashko, D.V.; Goncharov, T.K.; Shisho, N.I. Combustion of CL-20 cocrystals. Combust. Flame 2019, 207, 51–62. [Google Scholar] [CrossRef]
- Zhou, S.; Wu, F.; Tang, G.; Wang, Y.; Pang, A. Effects of 2CL-20/HMX cocrystals on the thermal decomposition behavior and combustion properties of polyether solid propellants. Energetic Mater. Front. 2021, 2, 96–104. [Google Scholar] [CrossRef]
- Wang, Y.; Wen, J.; Yang, J.; Zhang, G.; Wang, N.; Wu, Y. Investigations on the thermal response of a solid rocket motor with complex charge structure using CL-20/GAP propellant. Case Stud. Therm. Eng. 2022, 37, 102257. [Google Scholar] [CrossRef]
- Li, M.; Hu, R.; Xu, M.; Wang, Q.; Yang, W. Burning characteristics of high density foamed GAP/CL-20 propellants. Def. Technol. 2021, 18, 1914–1921. [Google Scholar] [CrossRef]
- Yang, L.-F.; Shi, X.-R.; Li, C.-Z.; Wu, B.; Pei, C.-H. Microfluidic assisted 90% loading CL-20 spherical particles: Enhancing self-sustaining combustion performance. Def. Technol. 2023, 22, 176–184. [Google Scholar] [CrossRef]
- Shi, Y.; Bai, L.; Gong, J.; Ju, X. Theoretical calculation into the structures, stability, sensitivity, and mechanical properties of 2,4,6,8,10,12-hexanitro-2,4,6,8,10,12 hexaazaisowurtzitane (CL-20)/1-amino-3-methyl-1,2,3-triazoliumnitrate (1-AMTN) cocrystal and its mixture. Struct. Chem. 2020, 31, 647–655. [Google Scholar] [CrossRef]
- Zhu, Y.; Luo, J.; Lu, Y.; Li, H.; Gao, B.; Wang, D.; Zhang, X.; Guo, C. Emulsion synthesis of CL-20/DNA composite with excellent superfine spherical improved sensitivity performance via a combined ultrasonic–microwave irradiation approach. J. Mater. Sci. 2018, 53, 14231–14240. [Google Scholar] [CrossRef]
- Chen, T.; Zhang, Y.; Guo, S.-F.; Zhao, L.-m.; Chen, W.; Hao, G.-Z.; Xiao, L.; Ke, X.; Jiang, W. Preparation and property of CL-20/BAMO-THF energetic nanocomposites. Def. Technol. 2019, 15, 306–312. [Google Scholar] [CrossRef]
- Chapman, C.J.; Groven, L.J. Evaluation of a CL-20/TATB Energetic Co-crystal. Propellants Explos. Pyrotech. 2019, 44, 293–300. [Google Scholar] [CrossRef]
- Liu, N.; Duan, B.; Lu, X.; Mo, H.; Xu, M.; Zhanga, Q.; Wang, B. Preparation of CL-20/DNDAP cocrystals by a rapid and continuous spray drying method: An alternative to cocrystal formation. CrystEngComm 2018, 20, 2060–2067. [Google Scholar] [CrossRef]
- Herrmannsdorfer, D.; Gerber, P.; Heintz, T.; Herrmann, M.J.; Klapotke, T.M. Investigation of Crystallisation Conditions to Produce CL-20/HMX Cocrystal for Polymer-bonded Explosives. Propellants Explos. Pyrotech. 2019, 44, 668–678. [Google Scholar] [CrossRef]
- Tan, Y.; Yang, Z.; Wang, H.; Li, H.; Nie, F.; Liu, Y.; Yu, Y. High Energy Explosive with Low Sensitivity: A New Energetic Cocrystal Based on CL-20 and 1,4-DNI. Cryst. Growth Des. 2019, 19, 4476–4482. [Google Scholar] [CrossRef]
- Li, P.; Liu, K.; Ao, D.; Liu, X.; Xu, H.; Duan, X.; Pei, C. A Low-Sensitivity Nanocomposite of CL-20 and TATB. Cryst. Res. Technol. 2018, 53, 1800189. [Google Scholar] [CrossRef]
- Wu, C.-L.; Zhang, S.-H.; Gou, R.-J.; Ren, F.-D.; Han, G.; Zhu, S.-F. Theoretical insight into the effect of solvent polarity on the formation and morphology of 2,4,6,8,10,12-hexanitrohexaazaisowurtzitane (CL-20)/2,4,6-trinitro-toluene(TNT) cocrystal explosive. Comput. Theor. Chem. 2018, 1127, 22–30. [Google Scholar] [CrossRef]
- Vuppuluri, V.S.; Samuels, P.J.; Caflin, K.C.; Gunduz, I.E.; Son, S.F. Detonation Performance Characterization of a Novel CL-20 Cocrystal Using Microwave Interferometry. Propellants Explos. Pyrotech. 2018, 43, 38–47. [Google Scholar] [CrossRef]
- Hai, L.; Yi, L.; Zhaoxia, M.; Zhixuan, Z.; Junling, L.; Yuanhang, H. Study on the Initial Decomposition Mechanism of Energetic Co-Crystal 2,4,6,8,10,12-Hexanitro-2,4,6,8,10,12-Hexaazaisowurtzitane (CL-20)/1,3,5,7-Tetranitro-1,3,5,7-Tetrazacyclooctane (HMX) under a Steady Shock Wave. Acta Phys.-Chim. Sin. 2019, 35, 858–867. [Google Scholar] [CrossRef]
- Sun, S.; Zhang, H.; Xu, J.; Wang, H.; Wang, S.; Yu, Z.; Zhua, C.; Suna, J. Design, preparation, characterization and formation mechanism of a novel kinetic CL-20-based cocrystal. Acta Cryst. 2019, B75, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Gou, R.-j.; Zhang, S.-h.; Chen, Y.-H.; Chen, M.-H.; Liu, Y.-B. Effect of solvent mixture on the formation of CL-20/HMX cocrystal explosives. J. Mol. Model. 2020, 26, 8. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Duan, B.; Lu, X.; Zhang, Q.; Xu, M.; Moa, H.; Wang, B. Preparation of CL-20/TFAZ cocrystals under aqueous conditions: Balancing high performance and low sensitivity. CrystEngComm 2019, 21, 7271–7279. [Google Scholar] [CrossRef]
- Viswanath, J.V.; Shanigaram, B.; Vijayadarshan, P.; Chowadary, T.V.; Gupta, A.; Bhanuprakash, K.; Niranjana, S.R.; Venkataraman, A. Studies and Theoretical Optimization of CL-20: RDX Cocrystal. Propellants Explos. Pyrotech. 2019, 44, 1570–1582. [Google Scholar] [CrossRef]
- Stepanov, V.; Patel, R.B.; Mudryy, R.; Qiu, H. Investigation of Nitramine-Based Amorphous Energetics. Propellants Explos. Pyrotech. 2016, 41, 142–147. [Google Scholar] [CrossRef]
- Chiquete, C.; Jackson, S.I. Detonation performance of the CL-20-based explosive LX-19. Proc. Combust. Inst. 2021, 38, 3661–3669. [Google Scholar] [CrossRef]
- Sysolyatin, S.V.; Chernikova, Y.T.; Lobanova, A.A.; Sakovich, G.V.; Surmachev, V.N.; Kadulin, V.V.; Kalashnikov, A.I.; Gudkova, N.I. Nitrolysis of tetraacetyl derivatives of hexaazaisowurtzitanes. Khimicheskaya Tekhnologiya 2005, 11, 12–15. [Google Scholar]
- Bayat, Y.; Hajighasemali, F. Synthesis of CL-20 by a Greener Method Using Nitroguanidine/HNO3. Propellants Explos. Pyrotech. 2016, 41, 20–23. [Google Scholar] [CrossRef]
- Koskin, A.P.; Simakova, I.L.; Parmon, V.N. Reductive debenzylation of hexabenzylhexaazaisowurtzitane—The key step of the synthesis of polycyclic nitramine hexanitrohexaazaisowurtzitane. Russ. Chem. Bull. 2007, 56, 2370–2375. [Google Scholar] [CrossRef]
- Zhao, W.; Liu, S.; Wang, H.; Yang, J.; Chen, X. Ultrasmall Pd Nanoparticles Supported on TiO2 for Catalytic Debenzylation via Hydrogenative C–N Bond Cleavage. ACS Appl. Nano Mater. 2020, 4, 159–166. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, D.; Dong, K.; Lv, P.; Pang, S.; Sun, C. Kinetics Study of a Complex Reaction: Nitration of Caged 2,6,8,12-Tetraacetyl-4,10-dinitro-2,4,6,8,10,12-hexaazaisowurtzitane. Org. Process Res. Dev. 2016, 20, 1911–1916. [Google Scholar] [CrossRef]
- Latypov, N.V.; Wellmar, U.; Goede, P.; Bellamy, A.J. Synthesis and Scale-Up of 2,4,6,8,10,12-Hexanitro-2,4,6,8,10,12-hexaazaisowurtzitane from 2,6,8,12-Tetraacetyl-4,10-dibenzyl-2,4,6,8,10,12-hexaazaisowurtzitane (HNIW, CL-20). Org. Process Res. Dev. 2000, 4, 156–158. [Google Scholar] [CrossRef]
- Dong, K.; Sun, C.H.; Song, J.W.; Wei, G.X.; Pang, S.P. Synthesis of 2,6,8,12-Tetraacetyl-2,4,6,8,10,12-hexaazaisowurtzitane (TAIW) from 2,6,8,12-Tetraacetyl-4,10-dibenzyl-2,4,6,8,10,12-hexaazaisowurtzitane (TADBIW) by Catalytic Hydrogenolysis Using a Continuous Flow Process. Org. Process Res. Dev. 2014, 18, 1321–1325. [Google Scholar] [CrossRef]
- Bayat, Y.; Hajimirsadeghi, S.S.; Pourmortazavi, S.M. Statistical Optimization of Reaction Parameters for the Synthesis of 2,4,6,8,10,12-Hexanitro-2,4,6,8,10,12-hexaazaisowurtzitane. Org. Process Res. Dev. 2011, 15, 810–816. [Google Scholar] [CrossRef]
- Pang, S.-P.; Yu, Y.-Z.; Zhao, X.-Q. A Novel Synthetic Route to Hexanitrohexaazaisowurtzitane. Propellants Explos. Pyrotech. 2005, 30, 442–444. [Google Scholar] [CrossRef]
- Surmachev, V.N.; Kubasova, V.A.; Zimin, D.E. A Study on Nitration of 4,10-Dibenzyl-2,6,8,12-Tetraacetyl-2,4,6,8,10,12-Hexaazaisowurtzitane. Propellants Explos. Pyrotech. 2020, 45, 1841–1844. [Google Scholar] [CrossRef]
- Kalashnikov, A.I.; Sysolyatin, S.V.; Sakovich, G.V.; Dubkov, A.S.; Kulagina, D.A. Nitrolysis of 2,6,8,12-tetraacetyl-4,10-dibenzyl-2,4,6,8,10,12-hexaazatetracyclo[5.5.0.03,11.05,9]dodecane. Russ. Chem. Bull. 2017, 66, 531–536. [Google Scholar] [CrossRef]
- Chapman, R.D.; Hollins, R.A. Benzylamine-Free, Heavy-Metal-Free Synthesis of CL-20 via Hexa(1-propenyl)hexaazaisowurtzitane. J. Energ. Mater. 2008, 26, 246–273. [Google Scholar] [CrossRef]
- Lou, D.; Wang, H.; Liu, S.; Li, L.; Zhao, W.; Chen, X.; Yang, J. PdFe bimetallic catalysts for debenzylation of hexabenzylhexaazaisowurtzitane (HBIW) and tetraacetyldibenzylhexaazaisowurtzitane (TADBIW). Catal. Commun. 2018, 109, 28–32. [Google Scholar] [CrossRef]
- Qian, H.; Ye, Z.-W.; Lv, C.-X. An Efficient and Facile Synthesis of Hexanitrohexaazaisowurtzitane (HNIW). Lett. Org. Chem. 2007, 4, 482–485. [Google Scholar] [CrossRef]
- Kai, W.; Dong, B.; Yang, C.; Qian, H. Acidic ionic liquids and green and recyclable catalysts in the clean nitration of TAIW to CL-20 using HNO3 electrolyte. Can. J. Chem. 2017, 95, 190–193. [Google Scholar] [CrossRef]
- Chapman, R.D.; Hollins, R.A. Processes for Preparing Certain Hexaazaisowurtzitanes and Their Use in Preparing Hexanitrohexaazaisowurtzitane. U.S. Patent 8268991 B1, 18 September 2012. [Google Scholar]
- Wright, M.E. Three-Step Synthesis of CL-20. U.S. Patent 9056868 B1, 16 June 2015. [Google Scholar]
- Wardle, R.B.; Hinshaw, J.C. Polycyclic, Polyamides as Precursors for Energetic Polycyclic Polynitramine Oxidizers. U.S. Patent 7129348 B1, 31 October 2006. [Google Scholar]
- Lu, Y.; Lei, Q.; Ren, X.; Ye, D.; Guo, Y.; Ding, N.; He, J. A Kind of Method that Three-Step Reaction Prepares CL 20. CN Patent 107353293 A, 16 August 2019. [Google Scholar]
- Cagnon, G.; Eck, G.; Herve, G.; Jacob, G. Process for the Synthesis of Hexanitrohexaazaisowurtzitane in 2 Steps from a Primary Amine. EP Patent 1479683 A1, 26 October 2005. [Google Scholar]
- Wang, J.; Li, Y.; Xue, M.; Chen, L.; Cao, D.; Li, Y. Method for Preparing CL-20 through Two-Step Method. CN Patent 110117289 B, 4 January 2022. [Google Scholar]
- Kalashnikov, A.I.; Sysoljatin, S.V.; Lapina, J.T.; Lobanova, A.A.; Kadulin, V.V. Method for Preparation of 2,4,6,8,10,12-hexanitro-2,4,6,8,10,12-hexaazatetracyclo[5,5,0,03,11,05,9]dodecane. RU Patent 2360916 C1, 10 July 2009. [Google Scholar]
- Jin, Z.; Wang, M.; Li, J.; Tie, Z. Method for Efficiently Synthesizing CL-20 High-Energy Cage Compound Based on Monatomic Catalyst. CN Patent 115611905 A, 2023. Available online: https://patents.google.com/patent/CN115611905A/en?oq=CN+Patent+115611905+A (accessed on 12 October 2023).
- Badard, O.; Renouard, J.; Marc, S.; Tenaglia, A. Procede de Synthese de l’Hexanitrohexaazaisowurtzitane a Partir de l’Hexaallylhexaazaisowurtzitane; Intermediaires. FR Patent 2997697 A1, 17 June 2016. [Google Scholar]
- Parakhin, V.V.; Pokhvisneva, G.V.; Ternikova, T.V.; Nikitin, S.V.; Smirnov, G.A.; Kon’kova, A.S.; Lempert, D.B.; Pivkina, A.N. Energetic alkylnitramine-functionalized pentanitro hexaazaisowurtzitanes: Towards advanced less sensitive CL-20 analogues. J. Mater. Chem. A 2022, 10, 818–828. [Google Scholar] [CrossRef]
- Luk’yanov, O.A.; Shlykova, N.I. Pentanitro- and pentanitronitroso-2,4,6,8,10,12-hexaazaisowurtzitanes. Russ. Chem. Bull. 2004, 53, 566–568. [Google Scholar] [CrossRef]
- Bellamy, A.J.; MacCuish, A.; Golding, P.; Mahon, M.F. The Use of trifluoroacetyl as an N- and O-protecting group during the synthesis of energetic compounds containing nitramine and/or nitrate ester groups. Propellants Explos. Pyrotech. 2007, 32, 20–31. [Google Scholar] [CrossRef]
- Sun, C.H.; Zhao, X.Q.; Li, Y.C.; Pang, S.P. Synthesis of two new cage molecules containing trinitromethyl group. Chin. Chem. Lett. 2010, 21, 572–575. [Google Scholar] [CrossRef]
- Zhang, Y.; Hu, J.; Li, Y.; Xu, C.; Chen, S.; Ge, Z.; Sun, C.; Pang, S. Energetic properties, thermal behavior and thermal safety of 4-(2,2,2-trinitroethyl)-2,6,8,10,12-pentanitro-2,4,6,8,10,12-hexaazaisowurtzitane. J. Anal. Appl. Pyrolysis 2020, 152, 104924. [Google Scholar] [CrossRef]
- Yukai, W.; Yuxiang, O.; Jinquan, L.; Lihua, L.; Boren, C.; Zhiguo, L. Synthesis, crystal structure and theoretical study of tetranitrodiazidoacetylhexaazaisowurtzitane (TNDAIW). Propellants Explos. Pyrotech. 2004, 29, 155–159. [Google Scholar] [CrossRef]
- Duddu, R.; Dave, P.R.; Damavarapu, R.; Surapaneni, R.; Gilardi, R.; Parrish, D. Synthesis of Azido Heterocycles. Synth. Commun. 2008, 38, 767–774. [Google Scholar] [CrossRef]
- Meng, Z.; Ou, Y.; Liu, J.; Wang, Y. Synthesis mechanism of tetranitrodiazidopropionylhexaazaisowurtzitane. Chin. J. Explos. Propellants 2006, 29, 65–67. [Google Scholar]
- Arabian, R.; Ramazani, A.; Mohtat, B.; Azizkhani, V.; Joo, S.W.; Rouhani, M. A Convenient and Efficient Protocol for the Synthesis of HBIW Catalyzed by Silica Nanoparticles under Ultrasound Irradiation. J. Energ. Mater. 2014, 32, 300–305. [Google Scholar] [CrossRef]
- Azizkhani, V.; Montazeri, F.; Molashahi, E.; Ramazani, A. Magnetically Recyclable CuFe2O4 Nanoparticles as an Efficient and Reusable Catalyst for the Green Synthesis of 2,4,6,8,10,12-Hexabenzyl-2,4,6,8,10,12-hexaazaisowurtzitane as CL-20 Explosive Precursor. J. Energ. Mater 2016, 35, 314–320. [Google Scholar] [CrossRef]
- Wang, L.; Yu, Z. Method for Efficiently Preparing Hexabenzyl Hexaazaisowurtzitanes. CN Patent 115594685 A, 2023. Available online: https://patents.google.com/patent/CN115594685A/en?oq=CN+Patent+115594685+A (accessed on 12 October 2023).
- Liu, W.; She, C.C.; Chao, H.; Wang, N.; Chen, S.S.; Jin, S.H.; Wang, J.F.; Chen, K. Role of the Bromide on the Hydrodebenzylation of 2,4,6,8,10,12-Hexabenzyl-2,4,6,8,10,12-hexaazaisowurtzitane (HBIW). ChemistrySelect 2022, 7, e202104216. [Google Scholar] [CrossRef]
- Yang, J.; Liu, S. Application of the Pd Radicel Duplex Metal Catalyst in HBIW Catalytic Hydrogenolytic Cleavages. CN Patent 106946894A, 15 March 2019. [Google Scholar]
- Kozlov, A.I.; Zbarskij, V.L.; Grunskij, V.N.; Judin, N.V.; Kuznetsov, L.A.; Merkin, A.A.; Komarov, A.A.; Kozlov, I.A.; Rybin, V.E.; Mikhajlov, J.M.; et al. Method of Producing Substituted Hexaazaisowurtzitanes. RU Patent 2451020 C1, 20 May 2012. [Google Scholar]
- Koskin, A.P.; Simakova, I.L.; Parmon, V.N. Study of palladium catalyst deactivation in synthesis of 4,10-diformyl-2,6,8,12-tetraacetyl-2,4,6,8,10,12-hexaazaisowurtzitane. React. Kinet. Catal. Lett. 2007, 92, 293–302. [Google Scholar] [CrossRef]
- Tang, X.; Lian, P.; Zhang, Y.; Zhu, J.; Jang, S.-R.; Wang, X.; Li, W.; Chen, S. Method for Synthesizing TADB by Continuous Hydrogenolysis and Debenzylation of HBIW through Acoustic Resonance Enhancement. CN Patent 114634513 A, 12 September 2023. [Google Scholar]
- Zhang, Q.; Wang, M.; Qian, J.; Lou, S.; Jin, J.; Li, B.; Lu, C.; Feng, F.; Lv, J.; Wang, Q.; et al. Deactivation and Regeneration of Palladium Catalysts for Hydrogenation Debenzylation of 2,4,6,8,10,12-Hexabenzyl-2,4,6,8,10,12-Hexaazaisowurtzitane (HBIW). Catalysts 2022, 12, 1547. [Google Scholar] [CrossRef]
- Fotouhi-Far, F.; Bashiri, H.; Hamadanian, M. Study of Deactivation of Pd(OH)2/C Catalyst in Reductive Debenzylation of Hexabenzylhexaazaisowurtzitane. Propellants Explos. Pyrotech. 2016, 42, 213–219. [Google Scholar] [CrossRef]
- Aravindu, P.; Rani, K.D.; Shaik, A.M.; Kommu, N.; Rao, V.K. Synthesis of Novel Hexaazaisowurtzitane Cages to Access CL-20. Asian J. Org. Chem. 2022, 11, 333–337. [Google Scholar] [CrossRef]
- Shang, F.; Liu, R.; Lv, M.; Ma, Y.; Liu, J.; Zhou, P.; Zhang, C.; Han, K.L. Unraveling the Key Role of the Benzyl Group in the Synthesis of CL-20 Precursor HBIW. ACS Omega 2022, 7, 21912–21924. [Google Scholar] [CrossRef] [PubMed]
- Paromov, A.; Shchurova, I.; Rogova, A.; Bagryanskaya, I.; Polovyanenko, D. Acid-Catalyzed Condensation of Benzamide with Glyoxal, and Reaction Features. Molecules 2022, 27, 1094. [Google Scholar] [CrossRef] [PubMed]
- Paromov, A.E.; Sysolyatin, S.V.; Gatilov, Y.V. An acid-catalyzed cascade synthesis of oxaazatetracyclo[5.5.0.03,11.05,9]dodecane Derivatives. J. Energ. Mater. 2017, 35, 363–373. [Google Scholar] [CrossRef]
- Paromov, A.E.; Sysolyatin, S.V. Synthesis of new N-polysubstituted oxaazaisowurtzitanes by acid-catalyzed condensation of sulfonamides with glyoxal. Russ. J. Org. Chem. 2017, 53, 1717–1725. [Google Scholar] [CrossRef]
- Paromov, A.E.; Sysolyatin, S.V.; Shchurova, I.A.; Rogova, A.I.; Malykhin, V.V.; Gatilov, Y.V. Synthesis of oxaazaisowurtzitanes by condensation of 4-dimethylaminobenzenesulfonamide with glyoxal. Tetrahedron 2020, 76, 131298. [Google Scholar] [CrossRef]
- Paromov, A.E.; Sysolyatin, S.V.; Shchurova, I.A. Condensation of 4-Tert-butyl-2,6-dimethylbenzenesulfonamide with Glyoxal and Reaction Features: A New Process for Symmetric and Asymmetric Aromatic Sulfones. Molecules 2022, 27, 7793. [Google Scholar] [CrossRef] [PubMed]
- Kovalevsky, R.A.; Vasechkin, K.V.; Kucherenko, A.S.; Zlotin, S.G. Enantioselective Catalytic Synthesis of α-Stereogenic Chromen-4-one Amino Derivatives. Adv. Synth. Catal. 2023, 365, 3162–3166. [Google Scholar] [CrossRef]
Figure 1.
Structural formulas of HMX and CL-20.

Scheme 1.
Synthesis of CL-20 via transfunctionalization of HBIW.

Scheme 2.
The formation of compounds 3–5.

Scheme 3.
Formation of compound 2.

Scheme 4.
The formation of compound 7.

Scheme 5.
The formation of compound 8.

Scheme 6.
The formation of compound 9.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Condensation products formed within 3 h from carbamate 1 and glyoxal in a range of polar protic and aprotic organic solvents when catalyzed by H2SO4 of varied concentrations.
Table 1.
Condensation products formed within 3 h from carbamate 1 and glyoxal in a range of polar protic and aprotic organic solvents when catalyzed by H2SO4 of varied concentrations.
| Entry | Solvent | ω(H2SO4 1),% 2 | Composition of Principal Reaction Products (HPLC),% | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 2 | 3 | 4 | 5 | 7 | 8 | 9 | ||||
| 1 | H2O | 40 | residue (0.339 g) 3: | 3.4 | 8.5 | 9.7 | 0.2 | - | 0.3 | - |
| 2 | 44 | residue (0.360 g) 3: | 0.3 | 4.3 | 18.3 | 1.8 | - | 4.1 | - | |
| 3 | 48 | residue (0.372 g) 3: | 0.2 | 4.2 | 18.5 | 2.7 | - | 7.3 | - | |
| 4 | 51 | residue (0.331 g) 3: | - | 2.5 | 12.9 | 3.0 | - | 15.0 | - | |
| 5 | 54 | residue (0.322 g) 3: | - | 1.4 | 3.6 | 3.0 | - | 25.1 | - | |
Note: 1 In the pure form. 2 Mass content in the mixture. 3 Compound 1 was taken as 0.36 g.
Table 2.
Condensation products formed within 20 h from carbamate 1 and glyoxal in a range of polar protic and aprotic organic solvents over H2SO4 of varied concentrations.
Table 2.
Condensation products formed within 20 h from carbamate 1 and glyoxal in a range of polar protic and aprotic organic solvents over H2SO4 of varied concentrations.
| Entry | Solvent | ω(H2SO4 1),% 2 | Composition of Principal Reaction Products (HPLC),% | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 2 | 3 | 4 | 5 | 7 | 8 | 9 | ||||
| 1 | Et2O | 0.3 | residue (0.033 g) 3: | 41.8 | 36.3 | 2.2 | - | traces | - | - |
| filtrate: | 3.0 | 12.1 | 1.5 | - | 0.2 | - | 2.7 | |||
| 2 | 2 | residue (0.630 g) 3: | - | 2.0 | 70.2 | - | 11.0 | 0.7 | - | |
| filtrate: | - | 4.4 | 5.3 | - | - | 0.6 | - | |||
| 3 | THF | 0.3 | reaction mass: | 6.7 | 20.6 | 3.8 | - | - | - | - |
| 4 | 2 | reaction mass: | 1.7 | 11.4 | 21.6 | traces | - | 0.4 | - | |
| 5 | 7 | residue (0.081 g) 3: | - | - | 10.4 | 82.1 | - | - | - | |
| filtrate: | 0.5 | 5.1 | 9.0 | - | - | 0.7 | - | |||
| 6 | AcOEt | 0.3 | residue (0.004 g) 3: | - | - | 1.6 | 43.4 | 44.9 | - | - |
| filtrate: | - | 2.8 | 1.8 | traces | 14.0 | - | 0.6 | |||
| 7 | 2 | residue (0.255 g) 3: | - | - | 1.6 | 17.1 | 61.1 | - | - | |
| filtrate: | - | 4.0 | 2.4 | 2.7 | 14.8 | 1.3 | - | |||
| 8 | (CH3)2CO | 0.3 | reaction mass: | 3.0 | 12.0 | 4.8 | - | - | - | - |
| 9 | 2 | residue (0.030 g) 3: | - | - | 15.4 | 72.5 | - | - | - | |
| filtrate: | 1.4 | 8.5 | 12.3 | traces | - | - | - | |||
| 10 | CH3CN | 0.3 | residue (0.131 g) 3: | - | - | 35.7 | 55.5 | - | - | - |
| filtrate: | 1.2 | 8.7 | 12.6 | - | - | - | - | |||
| 11 | 2 | residue (0.233 g) 3: | - | - | 14.5 | 75.2 | - | - | - | |
| filtrate: | 0.6 | 2.4 | 3.1 | - | - | - | - | |||
| 12 | 7 | residue (0.162 g) 3: | - | - | 4.5 | 74.0 | - | - | - | |
| filtrate: | - | - | - | - | - | - | - | |||
| 13 | CH2Cl2 | 0.3 | reaction mass: | 3.2 | 6.2 | 1.3 | 0.8 | - | - | - |
| 14 | 2 | residue (0.340 g) 3: | - | - | 7.7 | 81.0 | - | 2.8 | - | |
| filtrate: | - | 2.3 | 6.1 | - | - | 10.7 | - | |||
| 15 | DMSO | 0.3 | reaction mass: | traces | 1.9 | - | - | - | - | - |
| 16 | 2 | reaction mass: | 2.4 | 7.8 | 0.3 | - | - | - | - | |
| 17 | 7 | reaction mass: | 4.4 | 23.7 | 2.6 | 0.3 | - | - | - | |
| 18 | FoOH | - | residue (0.038 g) 3: | - | - | 15.5 | 67.0 | - | - | - |
| filtrate: | - | 0.7 | 5.2 | - | - | - | 23.0 | |||
| 19 | 2 | residue (0.034 g) 3: | - | - | 3.0 | 82.2 | - | - | - | |
| filtrate: | - | - | - | - | - | - | 74.9 | |||
| 20 | 7 | reaction mass: | - | - | - | - | - | - | 87.0 | |
| 21 | AcOH | - | residue (0.057 g) 3: | 70.0 | 16.0 | - | - | - | - | - |
| filtrate: | 0.7 | 6.1 | traces | - | - | - | - | |||
| 22 | 2 | residue (0.158 g) 3: | - | - | 9.9 | 72.5 | - | - | - | |
| filtrate: | - | - | 2.1 | - | - | - | - | |||
| 23 | 7 | residue (0.211 g) 3: | - | - | 5.8 | 81.8 | - | - | - | |
| filtrate: | - | - | - | - | - | - | - | |||
Note: 1 In the pure form. 2 Mass content in the mixture. 3 Compound 1 was taken as 0.72 g.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Paromov, A.E. Condensation of Benzyl Carbamate with Glyoxal in Polar Protic and Aprotic Solvents. Molecules 2023, 28, 7648. https://doi.org/10.3390/molecules28227648
AMA Style
Paromov AE. Condensation of Benzyl Carbamate with Glyoxal in Polar Protic and Aprotic Solvents. Molecules. 2023; 28(22):7648. https://doi.org/10.3390/molecules28227648
Chicago/Turabian StyleParomov, Artyom E. 2023. "Condensation of Benzyl Carbamate with Glyoxal in Polar Protic and Aprotic Solvents" Molecules 28, no. 22: 7648. https://doi.org/10.3390/molecules28227648