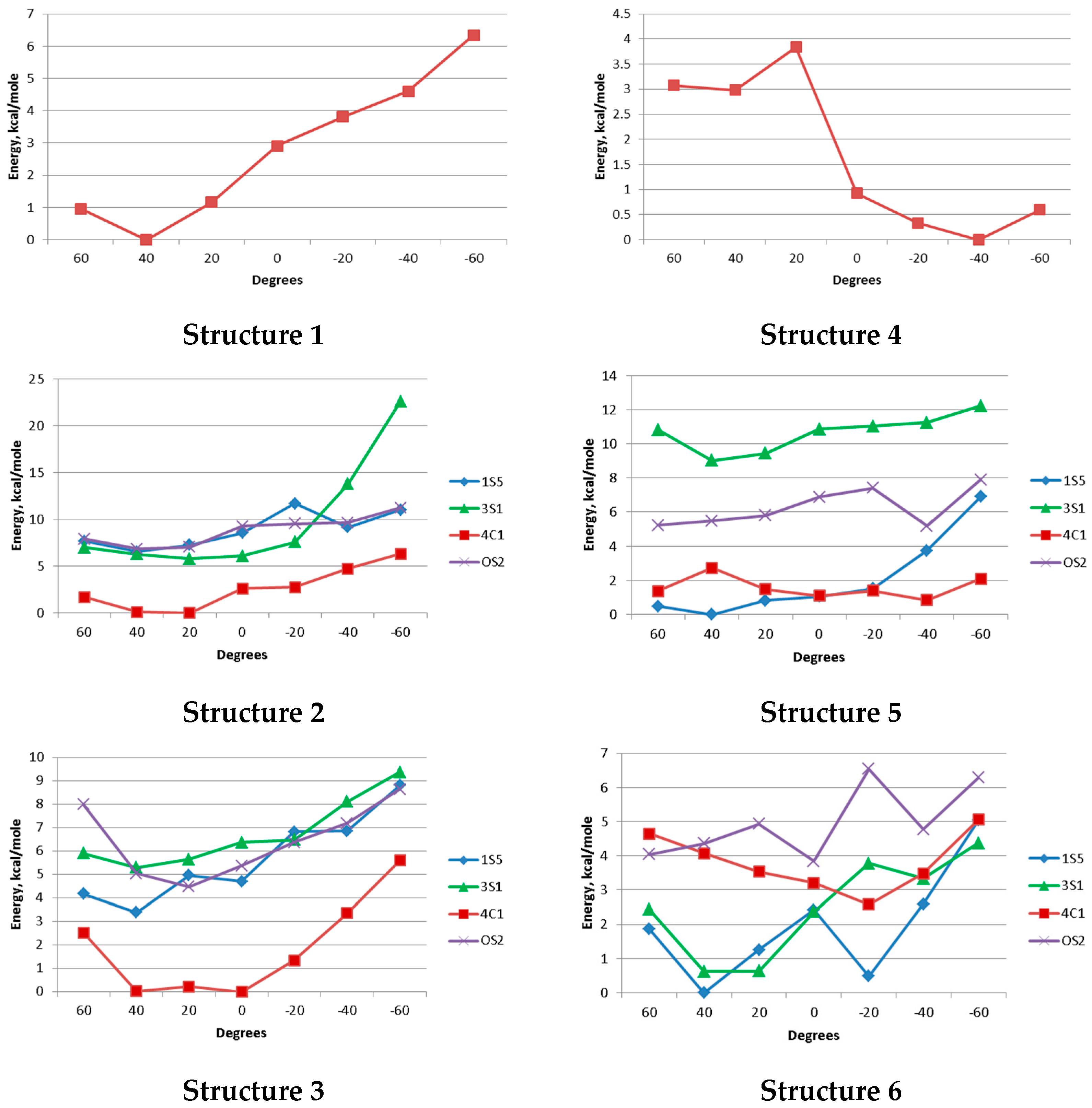
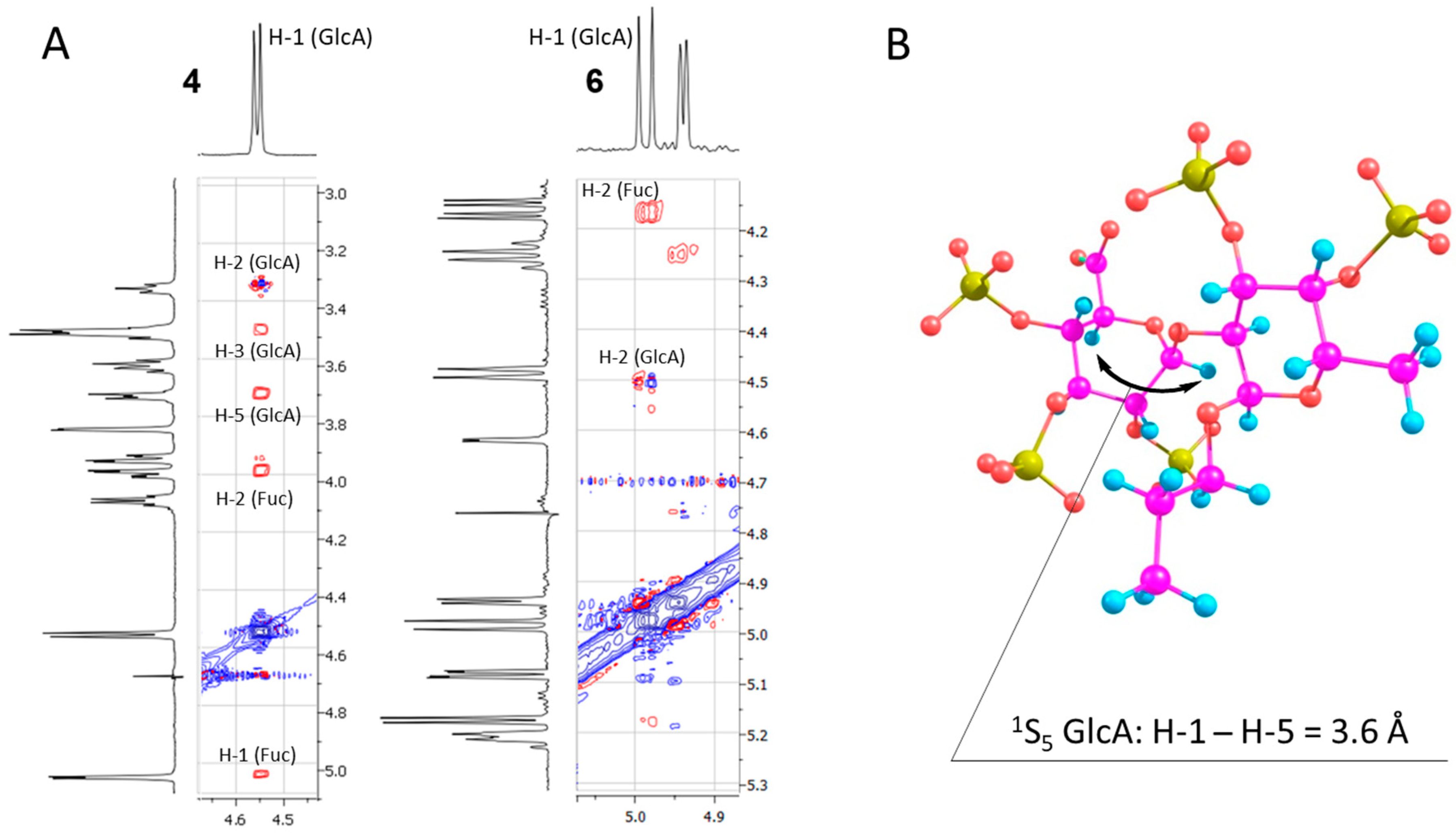
3.3. Synthesis of Compounds 9, 10, 11, 12, 13, 18, 19, 20, 21a, 21b, 1, 2, 3, 4, 5, and 6
3.3.1. (2-Methyl-5-tert-butylphenyl) 4,6-O-(4-methoxybenzylidene)-1-thio-β-d-glucopyranoside (9)
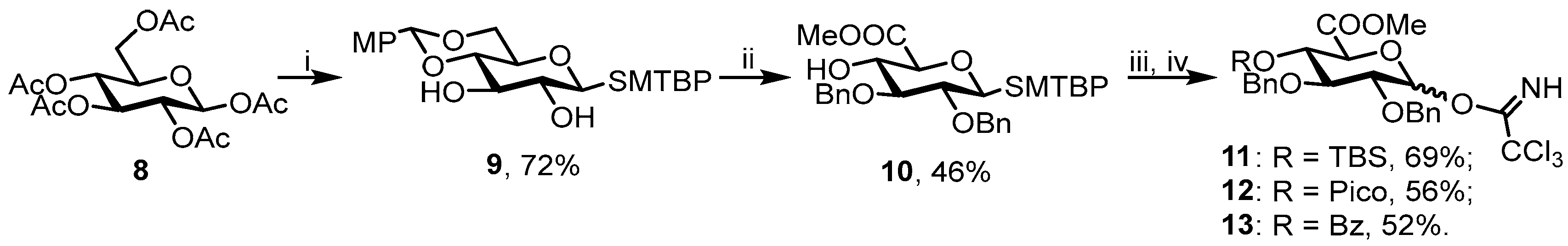
To a solution of glucose β-d-pentaacetate 8 [2 g, 5.1 mmol] in DCM [20 mL], MTBTP [1.32 mL, 1.4 eq.] was added. The mixture was cooled to 0 °C and BF3·Et2O [1.26 mL, 2 eq.] was added dropwise. The reaction mixture was allowed to warm to rt and stirred for 2 h. After 2 h, TLC [hexane/EtOAc = 1:1] showed full consumption of the starting material, and the reaction mixture was neutralized with Et3N, diluted with EtOAc [150 mL], and washed with H2O [2 × 100 mL]. The organic layer was dried (Na2SO4) and concentrated in vacuo. The crude thioglycoside was dissolved in MeOH [10 mL] and 1 M solution of MeONa in MeOH was added [1 mL]. After 20 min, TLC [EtOAc] showed formation of the final product and the reaction mixture was neutralized with ion exchange resin IR-120 in H+ form. The resin was filtered off and the filtrate was concentrated in vacuo. The crude residue was dissolved in CH3CN [15 mL] and CSA was added [150 mg, 10 mg/mL] followed by HC(OMe)3 [0.84 mL, 1.5 eq.] and 4-anisaldehyde [0.81 mL, 1.3 eq.]. After 30 min, TLC showed full consumption of the staring material, and the reaction mixture was diluted with EtOAc [150 mL] and washed with NaHCO3 [100 mL] and H2O [100 mL]. The organic layer was dried (Na2SO4) and concentrated in vacuo. The residue was purified by chromatography (silica gel, eluent: toluene/EtOA c = 10:1→3:1) to give 9 (1.68 g, 72%) as white crystals. Rf = 0.4 (toluene/EtOAc = 2:1). [α]D = −41.9° (c = 1, EtOAc). 1H NMR (300 MHz, CDCl3): δ 7.61 (d, 1H, J = 2.0 Hz, SMTBP), 7.41 (d, 2H, J = 8.7 Hz, o-MPh), 7.28–7.20 (m, 1H, SMTBP), 7.15 (d, 1H, J = 8.0 Hz, SMTBP), 6.88 (d, 2H, J = 8.8 Hz, m-MPh), 5.48 (s, 1H, CHMPh), 4.62 (d, 1H, J1–2 = 9.8 Hz, H-1), 4.38–4.27 (m, 1H, H-6), 3.87–3.69 (m, 5H, H-3. H-6′, OCH3), 3.58–3.43 (m, 3H, H-2, H-4, H-5), 3.08 (br s, 1H, 3-OH), 2.84 (s, 1H, 2-OH), 2.42 (s, 3H, Me (SMTBP)), 1.31 (s, 9H, tert-Bu (SMTBP)). 13C NMR (75 MHz, CDCl3): δ 160.39 (SMTBP), 149.75 (i-MBn), 137.15 (SMTBP), 130.90 (SMTBP), 130.38 (SMTBP), 130.23 (SMTBP), 129.46 (p-MPh), 127.72 (o-MPh), 125.56 (SMTBP), 113.84 (m-MPh), 101.99 (MPhCH), 88.83 (C-1), 80.31 (C-4), 74.79 (C-3), 73.02 (C-2), 70.52 (C-5), 68.71 (C-6), 55.42 (OMe), 31.42 (tert-Bu(SMTBP)), 20.59 (Me(SMTBP)). HRMS (ESI): Calcd. m/z for [M + H]+ C25H32O6S 461.1992 found 461.1991.
3.3.2. Methyl [(2-methyl-5-tert-butylphenyl) 2,3-di-O-benzyl-1-thio-β-d-glucopyranosyl] Uronate (10)
A solution of 9 [700 mg, 1.58 mmol] in DMF [7 mL] was cooled to 0 °C and NaH [250 mg, 4 eq.] was added portionwise. The reaction mixture was allowed to warm to rt and stirred for 30 min. Then, the mixture was cooled to 0 °C again and BnBr [0.57 mL, 3 eq.] was added dropwise. After 2 h, TLC [hexane/EtOAc = 2:1] showed formation of the final product and the reaction mixture was cooled to 0 °C and TFA [0.5 mL, 90% aq] was added. Reaction mixture was stirred at rt until TLC [hexane/EtOAc = 2:1] showed full consumption of the starting material. Then, the mixture was diluted with EtOAc [100 mL] and washed with NaHCO3 [100 mL] and H2O [100 mL]. The organic layer was dried (Na2SO4) and concentrated in vacuo. The crude residue was purified by chromatography (silica gel, eluent: hexane/EtOAc = 10:1→3:1) to give diol. The latter was dissolved in DCM [5 mL] and H2O [1 mL] and TEMPO [50 mg, 0.2 eq.] were added. The mixture was cooled to 0 °C and BAIB [1g, 2.5 eq.] was added portionwise. The reaction mixture was vigorously stirred for 3 h until TLC [hexane/EtOAc = 2:1] showed full consumption of the starting material. Then, the mixture was diluted with EtOAc [100 mL] and washed with Na2S2O3 [100 mL] and H2O [100 mL]. The organic layer was dried (Na2SO4) and concentrated in vacuo. The crude residue was dissolved in DMF [5 mL] and K2CO3 [200 mg, 1.5 eq.] and MeI [175 μL, 3 eq.] were added. The mixture was vigorously stirred for 3 h until TLC [hexane/EtOAc = 2:1] showed full consumption of the starting material. Then, the mixture was diluted with EtOAc [100 mL] and washed with H2O [2 × 100 mL]. The organic layer was dried (Na2SO4) and concentrated in vacuo. The residue was purified by chromatography (silica gel, eluent: hexane/EtOAc = 15:1→6:1) to give 10 (400 mg, 46%) as colorless oil. Rf = 0.55 (hexane/EtOAc = 2:1). [α]D = 7.2° (c = 1, EtOAc). 1H NMR (300 MHz, CDCl3): δ 7.63 (d, 1H, J = 1.9 Hz, SMTBP), 7.43–7.13 (m, 11H, SMTBP, 2xBn), 7.08 (d, 1H, J = 8.0 Hz, SMTBP), 4.90 (d, 1H, J = 10.3 Hz, CHH’Ph), 4.84 (s, 2H, CH2Ph), 4.75 (d, 1H, J = 10.3 Hz, CHH’Ph), 4.61 (d, 1H, J1–2 = 9.4 Hz, H-1), 3.97–3.86 (m, 1H, H-4), 3.79–3.75 (m, 4H, H-5, OMe), 3.61–3.44 (m, 2H, H-2, H-3), 2.97 (br s, 1H, 4-OH), 2.35 (s, 3H, Me(SMTBP)), 1.25 (s, 9H, tert-Bu(SMTBP)). 13C NMR (75 MHz, CDCl3): δ 169.67 (C-6), 149.76 (Bn), 138.01 (SMTBP), 136.43 (Bn), 132.90 (SMTBP), 129.99 (SMTBP), 129.61 (SMTBP), 128.64 (Bn), 128.51 (Bn), 128.35 (Bn), 128.11 (Bn), 127.99 (Bn), 124.97 (SMTBP), 89.03 (C-1), 85.38 (C-3), 80.20 (C-2), 77.45 (C-5), 75.83 (CH2Ph), 75.77 (CH2Ph), 71.99 (C-4), 52.78 (OMe), 31.35 (tert-Bu(SMTBP)), 20.45 (Me(SMTBP). HRMS (ESI): Calcd. m/z for [M + Na]+ C32H38O6S 573.2281 found 573.2287.
3.3.3. Methyl (2,3-di-O-benzyl-4-O-tert-butyldimethylsilyl-α,β-d-glucopyranosyl) Uronate Trichloroacetimidate (11)
NEt
3 [65 μL, 2 eq.] and TBSOTf [80 μL, 1.5 eq.] were added under Ar protection to a solution of
10 [125 mg, 0.23 mmol] in DCM [2 mL] and the resulting mixture was stirred at rt for 2 h. After TLC showed full consumption of the starting material, the mixture was diluted with EtOAc [50 mL] and washed with H
2O [2 × 50 mL]. The organic layer was dried (Na
2SO
4) and concentrated in vacuo. The crude product was treated according to the standard procedure A and after purification by chromatography (silica gel neutralized with NEt
3, eluent: hexane/EtOAc = 30:1→10:1) gave
11 (102 mg, 69%) as a mixture of α/β-isomers = 1:1.4. R
f = 0.9 (hexane/EtOAc = 2:1). Analytical data for
11 were in agreement with those previously reported [
25].
3.3.4. Methyl (2,3-di-O-benzyl-4-O-picoloyl-α,β-d-glucopyranosyl) Uronate Trichloroacetimidate (12)
PicoA [47 mg, 1.5 eq.], DCC [70 mg, 1.5 eq.], and DMAP [6 mg, 0.2 eq.] were added under Ar protection to a solution of 10 [124 mg, 0.23 mmol] in DCM [2 mL] and the resulting mixture was stirred at rt for 1 h. After TLC showed full consumption of the starting material, the mixture was diluted with EtOAc [50 mL] and washed with H2O [2 × 50 mL]. The organic layer was dried (Na2SO4) and concentrated in vacuo. The residue was purified by chromatography (silica gel, eluent: hexane/EtOAc = 8:1→2:1) to give fully protected thioglycoside. The latter was treated according to the standard procedure A and after purification by chromatography (silica gel neutralized with NEt3, eluent: hexane/EtOAc = 6:1→1:1) gave 12 (80 mg, 56%) as a mixture of α/β-isomers 1:7. Rf = 0.3 and 0.15 (hexane/EtOAc = 2:1). 1H NMR (400 MHz, CDCl3): δ 8.75 (d, 1H, J = 3.8 Hz, Pico), 8.66 (s, 1H, NH), 7.99 (d, J = 7.8 Hz, Pico), 7.79 (td, 1H, J = 7.7, J = 1.8 Hz, Pico), 7.46 (ddd, 1, J = 7.6 Hz, J = 4.8 Hz, J = 1.2 Hz, Pico), 7.34–6.96 (m, 10H, 2xBn), 6.58 (d, 1H, J1–2 = 3.5 Hz, H-1 (β)), 6.55 (d, 1H, J1–2 = 2.5 Hz, H-4 (α)) 6.19 (d, 1H, J4–5 = 2.8 Hz, H-4 (α)), 5.49–5.33 (m, 1H, H-4 (β)), 4.81 (d, 1H, J = 11.3 Hz, CHH’Ph), 4.76–4.64 (m, 3H, CHH’Ph, CH2Ph), 4.59 (d, 1H, J4–5 = 10.3 Hz, H-5 (β)), 4.43 (dd, 1H, J2–3 = 7.8 Hz, J3-4 = 2.8 Hz, H-3 (α)), 4.25 (t, 1H, J2–3, J3–4 = 9.5 Hz, H-3 (β)), 3.93 (dd, J2–3 = 7.7 Hz, J1–2 = 2.5 Hz, H-2 (α)), 3.85 (dd, 1H, J2–3 = 9.5 Hz, J1–2 = 3.6 Hz, H-2 (β)), 3.59 (s, 3H, OMe). 13C NMR (100 MHz, CDCl3): δ 167.91 (C-6), 164.21 (CO(Pico)), 161.01 (CCl3), 147.37 (Pico), 137.99 (i-Bn), 137.65 (i-Bn), 137.14 (Pico), 128.56-127.28 (Ar), 125.74 (Pico), 111.19 (C-1 (α)), 93.90 (C-1 (β)), 78.44 (C-2 (β)), 77.65 (C-3 (β)), 75.57 (CH2Ph), 73.42 (CH2Ph), 72.04 (C-4 (β)), 70.89 (C-5 (β)), 52.98 (OMe). HRMS (ESI): Calcd. m/z for [M + Na]+ C29H27Cl3N2O8 659.0725, found 659.0715.
3.3.5. Methyl (2,3-di-O-benzyl-4-O-benzoyl-α,β-d-glucopyranosyl) Uronate Trichloroacetimidate (13)
NEt3 [100 μL, 3 eq.] and BzCl [40 μL, 1.5 eq.] were added to a solution of 10 [129 mg, 0.23 mmol] in DCM [2 mL] and the resulting mixture was stirred overnight. After TLC showed full consumption of the starting material, an excess of MeOH was added and the mixture was kept for 30 min and concentrated in vacuo. The residue was purified by chromatography (silica gel, eluent: hexane/EtOAc = 30:1→12:1) to give fully protected thioglycoside. The latter was treated according to the standard procedure A and after purification by chromatography (silica gel neutralized with NEt3, eluent: hexane/EtOAc = 30:1→4:1) gave 13 (77 mg, 52%) as a mixture of α/β-isomers 1:2.1. Rf = 0.8 (hexane/EtOAc = 2:1). 1H NMR (400 MHz, CDCl3): δ 8.73 (s, 1H, NH(β)), 8.66 (s, 1H, NH(α)), 7.97–7.87 (m, 4H, o-Bz(α), o-Bz(β)), 7.56–7.52 (m, 2H, p-Bz(α), p-Bz(β)), 7.43–7.35 (m, 4H, m-Bz(α), m-Bz(β)), 7.29–7.06 (m, 20H, 2xBn(α), 2xBn(β)), 6.55 (d, 1H, J1–2 = 3.4 Hz, H-1 (α)), 5.91 (d, 1H, J1–2 = 7.3 Hz, H-1 (β)), 5.48 (t, 1H, J3–4, J4–5 = 9.2 Hz, H-4 (β)), 5.37 (t, 1H, J3–4, J4–5 = 9.8 Hz, H-4 (α)), 4.90 (d, 1H, J = 10.9 Hz, CHH’Ph(β)), 4.77–4.63 (m, 7H, CHH’Ph(β), CH2Ph(β), 2xCH2Ph(α)), 4.46 (d, 1H, J4–5 = 10.3 Hz, H-5 (α)), 4.22 (d, 1H, J4–5 = 9.6 Hz, H-5 (β)), 4.14 (t, 1H, J2–3, J3–4 = 9.4 Hz, H-3 (α)), 3.94–3.84 (m, 3H, H-2 (α), H-2 (β), H-3 (β)), 3.58 (s, 3H, OMe (β)), 3.56 (s, 3H, OMe (α)). 13C NMR (100 MHz, CDCl3): δ 168.04 (C-6 (α)), 167.57 (C-6 (β)), 165.47 (COPh (α)), 165.37 (COPh (β)), 161.15 (CCl3 (β)), 160.85 (CCl3 (α)), 137.67 (i-Bn), 133.51 (p-Bz (α), p-Bz (β)), 129.95 (o-Bz(α)), 129.90 (o-Bz(β)), 128.60–127.81 (Ar), 97.72 (C-1 (β)), 93.86 (C-1(α)), 80.73 (C-3 (β)), 80.15 (C-2 (β)), 78.48 (C-2 (α)), 77.49 (C-3 (α)), 75.47 (CH2Ph (α)), 75.24 (CH2Ph (β)), 75.10 (CH2Ph (β)), 73.45 (C-5 (β)), 73.40 (CH2Ph (α)), 71.34 (C-4 (β)), 71.14 (C-4 (α), C-5 (α)), 52.96 (OMe (α)), 52.92 (OMe (β)). HRMS(ESI): Calcd. m/z for [M + Na]+ C30H28Cl3NO8 658.0773 found 658.0769.
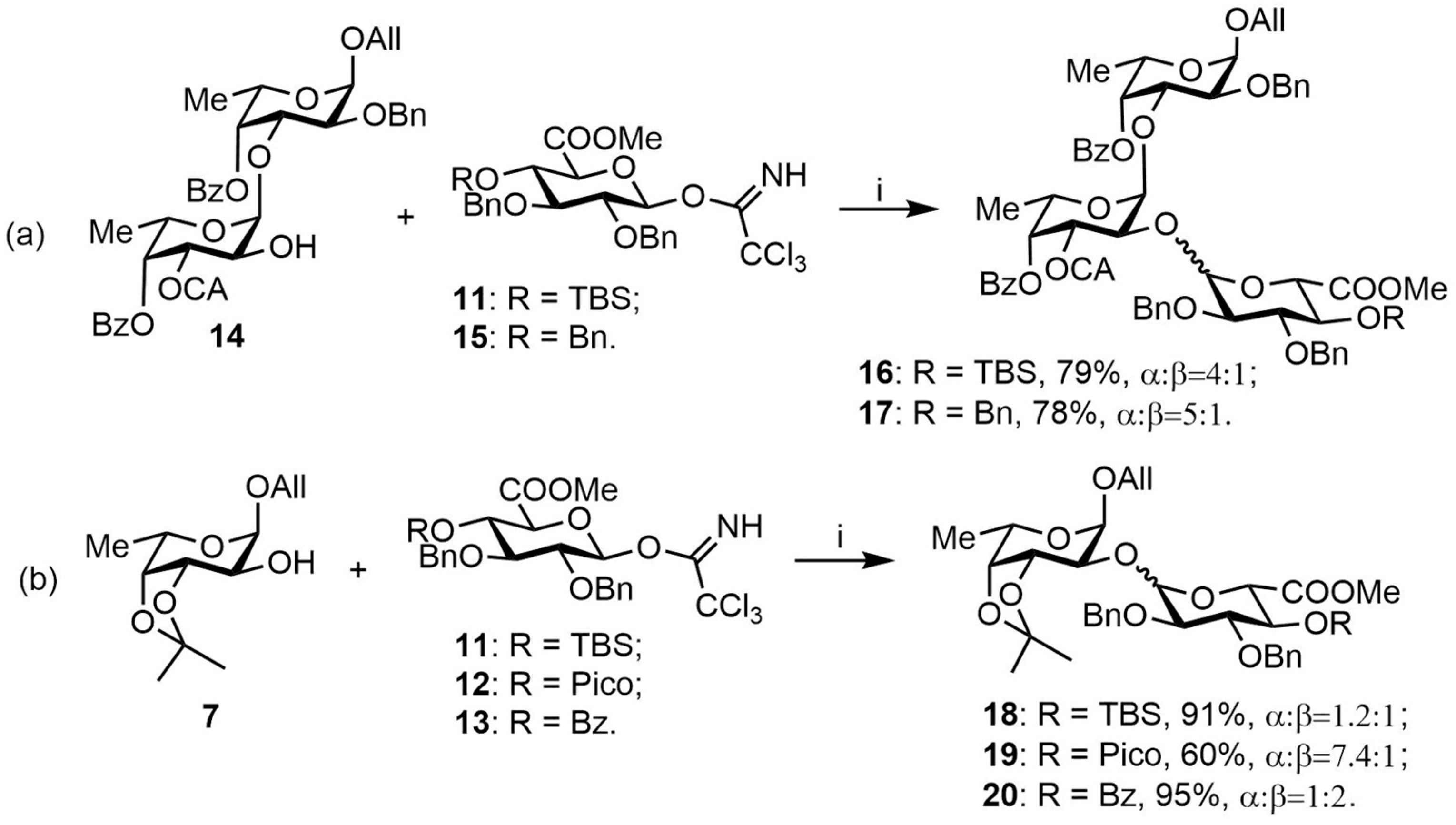
3.3.6. Allyl 3,4-O-isopropylidene-2-O-(methyl 2,3-di-O-benzyl-4-O-tert-butyldimethylsilyl-α,β-d-glucopyranosyluronate)-α-L-fucopyranoside (18)
The glycosylation was performed according to the standard procedure B with 7 [40 mg, 0.16 mmol] and 11 [102 mg, 0.16 mmol]. Purification by chromatography (silica gel, eluent: hexane/EtOAc = 20:1→8:1) gave 18 (109 mg, 91%) as a mixture of α/β-isomers 1.2:1. Rf = 0.8 (hexane/EtOAc = 2:1). 1H NMR (600 MHz, CDCl3): δ 7.36–7.21 (m, 20H, 2xBn (α), 2xBn (β)), 6.01 (ddd, 1H, J = 22.7 Hz, J = 10.8 Hz, J = 5.6 Hz, CH2CH=CH2 (α)), 5.80 (ddd, 1H, J = 22.5 Hz, J = 10.8 Hz, J = 5.6 Hz, CH2CH=CH2 (β)), 5.41 (dd, 1H, J = 17.2, J = 1.6 Hz, CH2CH=CHH’ (α)), 5.28–5.22 (m, 3H, H-1 (GlcA, α) CH2CH=CHH’ (α), CH2CH=CHH’ (β)), 5.13–5.08 (m, 2H, CH2CH=CHH’ (β), CHH’Ph (α)), 5.04–5.02 (m, 2H, 2xCHH’Ph (β)), 4.97 (d, 1H, J1–2 = 3.3 Hz, H-1 (Fuc, β)), 4.96 (d, 1H, J1–2 = 3.6 Hz, H-1(Fuc, α)), 4.76 (d, 1H, J = 11.3 Hz, CHH’Ph (α)), 4.72–4.68 (m, 2H, CHH’Ph (α), CHH’Ph (β)), 4.63–4.61 (m, 2H, H-1 (GlcA (β), CHH’Ph (β)), 4.58 (d, 1H, J = 11.3 Hz, CHH’Ph (α)), 4.45 (dd, 1H, J2–3 = 8.1 Hz, J3–4 = 5.5 Hz, H-3 (Fuc, α)), 4.40 (dd, 1H, J2–3 = 7.8 Hz, J3–4 = 5.5 Hz, H-3 (Fuc, β)), 4.28 (d, 1H, J4–5 = 9.5 Hz, H-5 (GlcA, α)), 4.26–4.08 (m, 7H, H-4 (Fuc, α), H-4 (Fuc, β), H-5 (Fuc, α), H-5 (Fuc, β), OCH2 (α), OCHH’ (β)), 4.04–3.98 (m, 2H, H-2 (Fuc, β), H-4 (GlcA, β), 3.94 (dd, 1H, J = 12.9 Hz, J = 6.0 Hz, CHH’ (β)), 3.87 (t, 1H, J4–5 = 9.1 Hz, H-4 (GlcA, α)), 3.83–3.80 (m, 2H, H-3 (GlcA, α), H-5 (GlcA, β)), 3.77–3.72 (m, 7H, H-2 (Fuc, α), OMe (α), OMe (β)), 3.64–3.59 (m, 2H, H-2 (GlcA, α), H-2 (GlcA, β)), 3.48 (t, 1H, J2–3, J3–4 = 8.7 Hz, H-3 (Glc, β)), 1.55 (s, 3H, CMe (α)), 1.50 (s, 3H, CMe (β)), 1.40–1.34 (m, 12H, H-6 (Fuc, α), H-6 (Fuc, β), CMe (α), CMe (β)), 0.85 (s, 9H, tert-BuSi (α)), 0.83 (s, 9H, tert-BuSi (β)), 0.01 (s, 3H, SiMe (α)), −0.02 (s, 3H, SiMe (β)), −0.02 (s, 3H, SiMe (α)), −0.03 (s, 3H, SiMe (β)). 13C NMR (150 MHz, CDCl3): δ 170.08 (C-6 (GlcA, β)), 168.63 (C-6 (GlcA, α)), 139.26 (i-Bn (α)), 138.89 (i-Bn (β)), 138.48 (i-Bn (β)), 138.06 (i-Bn (α)), 133.94 (CH2CH=CH2 (α)), 133.87 (CH2CH=CH2 (β)), 128.50–127.04 (Ar), 117.72 (CH2CH=CH2 (α)), 117.38 (CH2CH=CH2 (β)), 109.05 (CMe3 (β)), 108.92 (CMe3 (α)), 102.13 (C-1 (GlcA, β)), 99.17 (C-1 (GlcA, α)), 97.59 (C-1 (Fuc, α)), 95.45 (C-1 (Fuc, β)), 84.10 (C-3 (Glc, β)), 81.67 (C-2 (GlcA, β)), 80.83 (C-3 (GlcA, α)), 79.51 (C-2 (GlcA, α)), 78.78 (C-2 (Fuc, α)), 76.58 (C-5 (Glc, β)), 76.44 (C-4 (Fuc, α)), 76.34 (C-4 (Fuc, β)), 75.38 (C-2 (Fuc, β)), 75.21 (C-3 (Fuc, α)), 75.00 (CH2Ph, α), 74.84 (CH2Ph, β), 74.27 (C-3 (Fuc, β), CH2Ph, β), 72.61 (C-5 (GlcA, α)), 72.27 (C-4 (GlcA, β)), 72.07 (C-4 (GlcA, β)), 71.98 (OCH2Ph, α), 68.77 (OCH2, α), 68.62 (OCH2, β), 64.07 (C-5 (Fuc, α)), 63.25 (C-5 (Fuc, α)), 52.30 (OMe, α), 52.16 (OMe, β), 28.65 (CMe, α), 27.82 (CMe, β), 26.62 (CMe, α), 26.43 (CMe, β), 25.86 (tert-BuSi, β), 25.81 (tert-BuSi, α), 16.41 (C-6 (Fuc, α), C-6 (Fuc, β)), −3.83 (SiMe, α), −3.93 (SiMe, β), −5.20 (SiMe, β), −5.23 (SiMe, α). HRMS (ESI): Calcd. m/z for [M + NH4]+ C39H56O11Si 746.3930, found 746.3933.
3.3.7. Allyl 3,4-O-isopropylidene-2-O-(methyl 2,3-di-O-benzyl-4-O-picoloyl-α,β-d-glucopyranosyluronate)-α-L-fucopyranoside (19)
The glycosylation was performed according to the standard procedure B with 7 [30 mg, 0.12 mmol] and 12 [80 mg, 0.12 mmol]. Purification by chromatography (silica gel, eluent: hexane/EtOAc = 4:1→1:1) gave 19 (53 mg, 60%) as a mixture of α/β-isomers 7.4:1. Rf = 0.2 (hexane/EtOAc = 2:1). 1H NMR (600 MHz, CDCl3): δ 8.77 (d, 1H, J = 4.2 Hz, 1-Pico, α), 8.74 (d, 1H, J = 4.1 Hz, 1-Pico, β), 8.02 (d, 1H, J = 7.9 Hz, 4-Pico, β), 8.00 (d, 1H, J = 7.8 Hz, 4-Pico, α), 7.82–7.79 (m, 2H, 3-Pico (α), 3-Pico (β)), 7.50–7.44 (m, 2H, 2-Pico (α), 2-Pico (β)), 7.41–7.06 (m, 20H, 2xBn (α), 2xBn (β)), 5.93 (ddd, 1H, J = 22.8 Hz, J = 10.8 Hz, J = 5.7 Hz, CH2CH=CH2 (α)), 5.82 (ddd, 1H, J = 22.4 Hz, J = 10.7 Hz, J = 5.6 Hz, CH2CH=CH2 (β)), 5.52 (t, 1H, J3–4, J4–5 = 9.7 Hz, H-4 (GlcA, β)), 5.37 (t, 1H, J3–4, J4–5 = 9.8 Hz, H-4 (GlcA, α), 5.31 (dd, 1H, J = 17.2 Hz, J = 1.6 Hz, CH2CH=CHH’ (α)), 5.28–5.24 (m, 2H, H-1 (GlcA, α), CH2CH=CHH’ (β)), 5.16 (dd, 1H, J = 10.4 Hz, J = 1.2 Hz, CH2CH=CHH’ (α)), 5.12 (dd, 1H, J = 10.4 Hz, J = 1.3 Hz, CH2CH=CHH’ (β)), 5.03 (d, 1H, J = 11.4 Hz, CHH’Ph, β), 4.98 (d, 1H, J1–2 = 3.3 Hz, H-1 (Fuc, β)), 4.91 (d, 1H, J1–2 = 3.5 Hz, H-1 (Fuc, α)), 4.87 (d, 1H, J = 11.3 Hz, CHH’Ph, α), 4.78–4.71 (m, 6H, CHH’Ph (α), CHH’Ph (β), CH2Ph (α), CH2Ph (β)), 4.66–4.63 (m, 2H, H-1 (GlcA, β), H-5 (GlcA, α)), 4.42–4.40 (m, 2H, H-3 (Fuc, α), H-3 (Fuc, β)), 4.27 (t, 1H, J2–3, J3–4 = 9.4 Hz, H-3 (GlcA, α)), 4.18–4.08 (m, 7H, H-5 (GlcA, β), H-5 (Fuc, α), H-5 (Fuc, β), H-4 (Fuc, α), H-4 (Fuc, β), OCHH’ (α), OCHH’ (β)), 4.02 (dd, 1H, J2–3 = 7.9 Hz, J1–2 = 3.3 Hz, H-2 (Fuc, β)), 3.98–3.93 (m, 2H,OCHH’ (α), OCHH’ (β)), 3.90 (t, 1H, J2–3, J3–4 = 9.2 Hz, H-3 (GlcA, β)), 3.75 (dd, 1H, J2–3 = 8.0 Hz, J1–2 = 3.6 Hz, H-2 (Fuc, α)), 3.70–3.67 (m, 2H, H-2 (GlcA, α), H-2 (GlcA, β)), 3.63 (s, 3H, OMe, β), 3.61 (s, 3H, OMe, α), 1.55 (s, 3H, CMe, α), 1.52 (s, 3H, CMe, β), 1.38 (s, 3H, CMe’, α), 1.35–1.33 (m, 9H, H-6 (Fuc, α), H-6 (Fuc, β), CMe’ (β)). 13C NMR (150 MHz, CDCl3): δ 168.84 (C-6 (GlcA, α), 164.16 (CO (Pico, α), 150.06 (1-Pico, α), 147.70 (1-Pico, β), 138.51 (i-Bn), 138.19 (i-Bn), 137.00 (3-Pico α, β), 133.97 (CH2CH=CHH’, α, β), 128.48–127.06 (2xBn α, 2xBn β, 2 × 2-Pico α, β) 125.66 (4-Pico, β), 125.50 (4-Pico, α), 117.56 (CH2CH=CH2, α, β), 101.91 (C-1 (GlcA, β)), 98.96 (C-1 (GlcA, α)), 97.37 (C-1 (Fuc, α)), 95.39 (C-1 (Fuc, β)), 81.24 (C-3 (GlcA, β)), 81.10 (C-2 (GlcA, β)), 79.19 (C-2 (Fuc, α)), 78.66 (C-2 (GlcA, α)), 78.43 (C-3 (GlcA, α)), 76.36 (C-4 (Fuc, α)), 76.28 (C-4 (Fuc, β)), 75.95 (C-2 (Fuc, β)), 75.58 (CH2Ph, α), 75.39 (CH2Ph, β), 74.98 (C-3 (Fuc, α)), 74.69 (C-3 (Fuc, β), 74.28 (CH2Ph, β)), 72.71 (C-4 (GlcA, α), CH2Ph, α), 72.64 (C-5 (GlcA, β)), 72.26 (C-4 (GlcA, β)), 68.98 (C-5 (GlcA, α)), 68.75 (OCH2, α), 68.64 (OCH2, β), 64.08 (C-5 (Fuc, β)), 63.37 (C-5 (Fuc, α)), 52.68 (OMe, α, β), 28.60 (CMe, α), 28.00 (CMe, β), 26.52 (CMe’, α), 26.40 (CMe’, β), 16.41 (C-6 (Fuc, β), 16.35 (C-6 (Fuc, α). HRMS (ESI): Calcd. m/z for [M + Na]+ C39H45NO12 742.2834 found 742.2839.
3.3.8. Allyl 3,4-O-isopropylidene-2-O-(methyl 2,3-di-O-benzyl-4-O-benzoyl-α,β-d-glucopyranosyluronate)-α-L-fucopyranoside (20)
The glycosylation was performed according to the standard procedure B with 7 [30 mg, 0.12 mmol] and 13 [77 mg, 0.12 mmol]. Purification by chromatography (silica gel, eluent: toluene/EtOAc = 20:1→10:1) gave 20 (84 mg, 95%) as a mixture of α/β-isomers 1:2. Rf = 0.7 (hexane/EtOAc = 2:1). 1H NMR (600 MHz, CDCl3): δ 7.97 (d, 2H, J = 7.1 Hz, o-Bz, α), 7.94 (d, 2H, J = 7.1 Hz, o-Bz, β), 7.60–7.54 (m, 2H, p-Bz, α, β), 7.46–7.39 (m, 8H, m-Bz, α, β, Bn, β), 7.36–7.26 (m, 13H, Bn, α, β), 7.18–7.07 (m, 15H, Bn, α, β), 5.93 (ddd, 1H, J = 22.7 Hz, J = 10.8 Hz, J = 5.6 Hz, CH2CH=CH2, α), 5.81 (ddd, 1H, J = 22.2 Hz, J = 10.8, J = 5.6 Hz, CH2CH=CH2, β), 5.44 (t, 1H, J3–4, J4–5 = 9.6 Hz, H-4 (GlcA, β)), 5.36–5.25 (m, 3H, H-4 (GlcA, α), CH2CH=CHH’, α, CH2CH=CHH’, β), 5.22 (d, 1H, J1–2 = 3.7 Hz, H-1 (GlcA, α)), 5.18 (dd, 1H, J = 10.4 Hz, J = 1.4 Hz, CH2CH=CHH’, α), 5.13 (dd, 1H, J = 10.4 Hz, J = 1.4 Hz, CH2CH=CHH’, β), 5.05 (d, 1H, J = 11.3 Hz, CHH’Ph, β), 4.99 (d, 1H, J1–2 = 3.3 Hz, H-1 (Fuc, β)), 4.92 (d, 1H, J1–2 = 3.6 Hz, H-1 (Fuc, α)), 4.88 (d, 1H, J = 11.2 Hz, CHH’, α), 4.79–4.73 (m, 4H, CH2Ph, α, CHH’Ph, β, CHH’Ph, β), 4.69 (d, 1H, J = 11.2 Hz, CHH’Ph, α), 4.66–4.63 (m, 2H, H-1 (GlcA, β), CHH’Ph, β), 4.51 (d, 1H, J4–5 = 10.2 Hz, H-5 (GlcA, α)), 4.45–4.42 (m, 2H, H-3 (Fuc, α), H-3 (Fuc, β)), 4.22–4.12 (m, 5H, H-3 (GlcA, α), H-5 (Fuc, α), H-5 (Fuc, β), OCHH’, α, OCHH’, β), 4.12–4.09 (m, 2H, H-4 (Fuc, α), H-4 (Fuc, β)), 4.03–3.99 (m, 2H, H-2 (Fuc, β), H-5 (GlcA, β)), 3.98–3.91 (m, 2H, OCHH’, α, OCHH’, β), 3.81 (t, 1H, J2–3, J3–4 = 9.2 Hz, H-3 (GlcA, β)), 3.74 (dd, 1H, J2–3 = 8.0 Hz, J1–2 = 3.6 Hz, H-2 (Fuc, α)), 3.71–3.68 (m, 2H, H-2 (GlcA, α), H-2 (GlcA, β)), 3.62 (s, 3H, OMe, β), 3.58 (s, 3H, OMe, α), 1.57 (s, 3H, CMe, α), 1.52 (s, 3H, CMe, β), 1.41 (s, 3H, CMe’, α), 1.37–1.33 (m, 9H, H-6 (Fuc, α, H-6 (Fuc, β, CMe’, β). 13C NMR (150 MHz, CDCl3): δ 168.93 (C-6 (GlcA, α)), 167.66 (C-6 (GlcA, β)), 165.51 (CO (Bz, α)), 165.34 (CO, (Bz, β)), 138.35 (i-Bn), 137.83 (i-Bn), 133.76 (CH2CH=CH2, β), 133.72 (CH2CH=CH2, α), 133.37 (p-Bz, α, β), 129.84 (o-Bz, β), 129.82 (o-Bz, α), 128.54–127.65 (m-Bz, α, β, Bn, α, β), 117.64 (CH2CH=CH2, α), 117.51 (CH2CH=CH2, β), 109.16 (CMe2, α, β), 101.79 (C-1 (GlcA, β)), 99.05 (C-1 (GlcA, α)), 97.19 (C-1 (Fuc, α)), 95.30 (C-1 (Fuc, β)), 81.03 (C-2 (GlcA, β)), 80.95 (C-3 (GlcA, β)), 79.17 (C-2 (Fuc, α)), 78.53 (C-2 (GlcA, α)), 78.38 (C-3 (GlcA, α)), 76.30 (C-4 (Fuc, α)), 76.24 (C-4 (Fuc, β)), 75.95 (C-2 (Fuc, β)), 75.22 (CH2Ph, β), 74.84 (CH2Ph, α), 74.70 (CH2Ph, β), 74.18 (C-3 (Fuc, α), C-3 (Fuc, β)), 72.87 (CH2Ph, α), 72.82 (C-5 (GlcA, β)), 71.79 (C-4 (GlcA, α)), 71.48 (C-4 (GlcA, β)), 69.03 (C-5 (GlcA, α)), 68.58 (OCH2, β), 68.56 (OCH2, α), 63.98 (C-5 (Fuc, β)), 63.28 (C-5 (Fuc, α)), 52.73 (OMe, α), 52.65 (OMe, β), 28.63 (CMe, α), 28.01 (CMe, β), 26.54 (CMe’, α), 26.42 (CMe’, β), 16.41 (C-6 (Fuc, β)), 16.36 (C-6 (Fuc, α)). HRMS (ESI): Calcd. m/z for [M + NH4]+ C40H46O12 736.3328 found 736.3326.
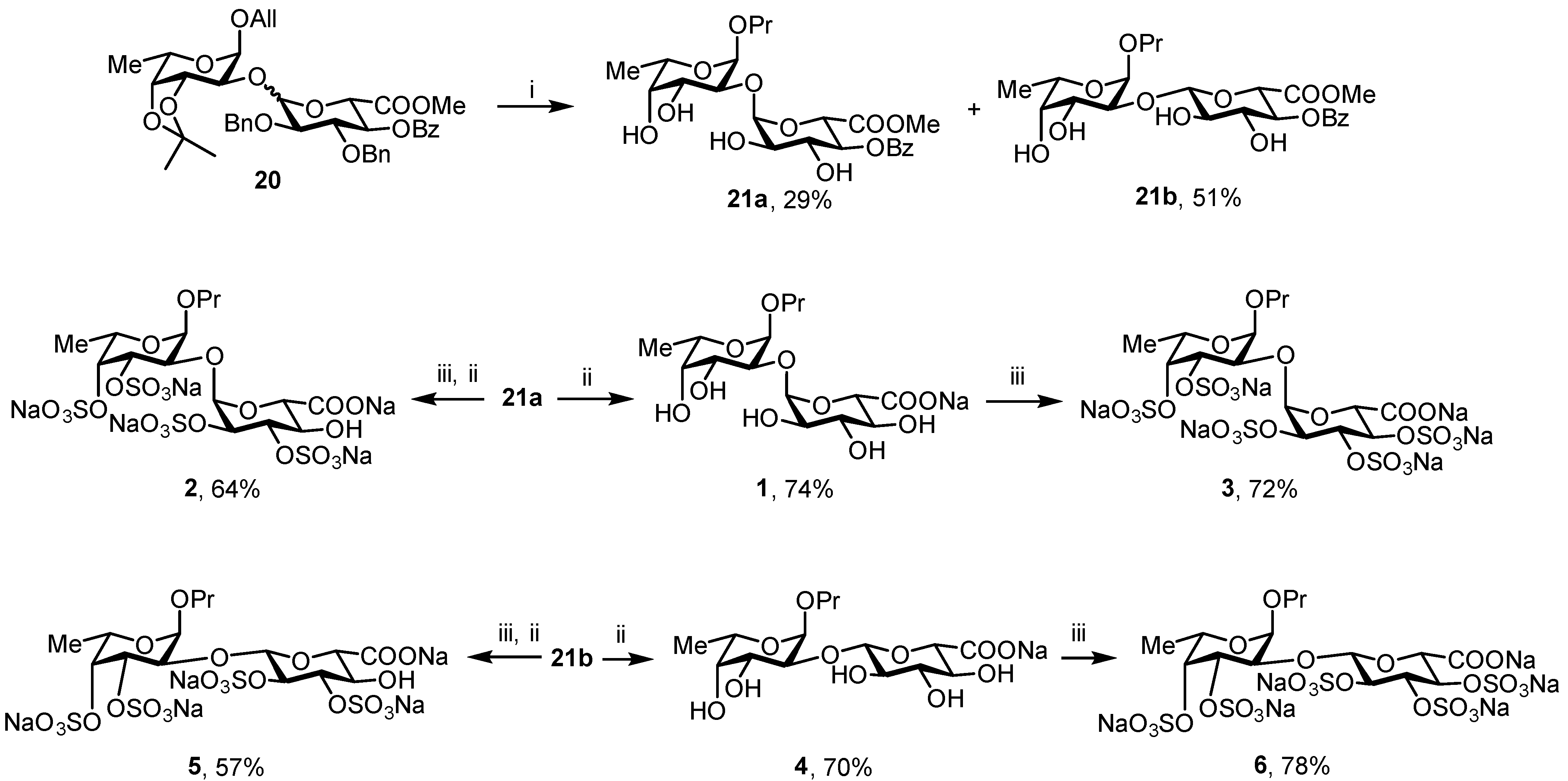
3.3.9. Propyl 2-O-(methyl 4-O-benzoyl-α-d-glucopyranosyluronate)-α-L-fucopyranoside (21a) and propyl 2-O-(methyl 4-O-benzoyl-β-d-glucopyranosyluronate)-α-L-fucopyranoside (21b)
TFA [150 μL, 90% aq.] was added to a solution of 20 [84 mg, 0.11 mmol] in DCM [1.5 mL]. The reaction mixture was kept at 40 °C for 30 min. After TLC [hexane/EtOAc = 1:1] showed full consumption of the starting material, the mixture was neutralized with NEt3 and concentrated in vacuo. The residue was purified by chromatography (silica gel, eluent: hexane/EtOAc = 3:1→1:2) to give diol. A mixture of the resulting product and the catalyst 10% Pd/C [25 mg] in EtOAc–MeOH (1:1) [2 mL] was stirred under H2 at rt for 2 h. After TLC (toluene:EtOAc = 1:1) showed formation of the final product, the reaction mixture was filtered through a nylon membrane syringe filter (0.45 μm). The filtrate was concentrated in vacuo and the residue was purified by chromatography (silica gel, eluent: DCM/MeOH = 20:1→10:1) to give 21a (17 mg, 29%) and 21b (30 mg, 51%). Rf = 0.25 and 0.1 (toluene/EtOAc = 1:1).
For 21a: [α]D = −98.5° (c = 1, MeOH). 1H NMR (600 MHz, CD3OD3): 8.05 (d, 2H, J = 7.1 Hz, o-Bz)), 7.66 (t, 1H, J = 7.4 Hz, p-Bz), 7.52 (t, 2H, J = 7.8 Hz, m-Bz), 5.13–5.12 (m, 2H, H-1 (GlcA), H-4 (GlcA)), 4.91 (d, 1H, J1–2 = 3.7 Hz, H-1 (Fuc)), 4.66 (d, 1H, J4–5 = 10.3 Hz, H-5 (GlcA)), 4.07 (t, 1H, J2–3, J3–4= 9.5 Hz, H-3 (GlcA)), 4.02 (dd, 1H, J2–3 = 10.0 Hz, J3–4 = 3.3 Hz, H-3 (Fuc)), 3.95 (q, 1H, J5–6 = 6.5 Hz, H-5 (Fuc)), 3.81 (dd, 1H, J2–3 = 10.0 Hz, J1–2 = 3.7 Hz, H-2 (Fuc)), 3.75 (d, 1H, J3–4 = 3.4 Hz, H-4 (Fuc)), 3.67–3.60 (m, 2H, H-2 (GlcA), OCHH’), 3.22 (dt, 1H, J = 9.3 Hz, J = 6.6 Hz, OCHH’), 1.66 (dq, 2H, J = 14.2 Hz, J = 7.1 Hz, OCH2CH2CH3), 1.23 (d, 3H, J5–6 = 6.6 Hz, H-6 (Fuc)), 0.99 (t, 3H, J = 7.4 Hz, OCH2CH2CH3). 13C NMR (150 MHz, CD3OD): δ 13C NMR (151 MHz, MeOD) δ 170.84 (C-6 (GlcA)), 167.20 (COPh), 134.53 (p-Bz), 130.66 (o-Bz), 129.59 (m-Bz), 103.26 (C-1 (GlcA)), 99.35 (C-1 (Fuc)), 81.39 (C-2 (Fuc)), 74.02 (C-4 (GlcA)), 73.43 (C-2 (GlcA), C-4 (Fuc)), 72.14 (C-3 (GlcA)), 70.39 (C-5 (GlcA)), 70.10 (C-3 (Fuc), OCH2), 67.19 (C-5 (Fuc)), 53.04 (OMe), 23.87 (OCH2CH2CH3), 16.50 (C-6 (Fuc)), 11.25 (OCH2CH2CH3). HRMS (ESI): Calcd. m/z for [M + Na]+ C23H32O12 523.1786 found 523.1786.
For 21b: [α]D = −17.6° (c = 1, MeOH). 1H NMR (600 MHz, CD3OD3): δ 8.07 (d, 2H, J = 7.9 Hz, o-Bz), 7.65 (t, 1H, J = 7.4 Hz, p-Bz), 7.51 (t, 2H, J = 7.8 Hz, m-Bz), 5.12 (t, 1H, J3–4, J4–5 = 9.7 Hz, H-4 (GlcA)), 4.98 (d, 1H, J1–2 = 3.7 Hz, H-1 (Fuc)), 4.66 (d, 1H, J1–2 = 7.9 Hz, H-1 (GlcA)), 4.30 (d, 1H, J4–5 = 10.0 Hz, H-5 (GlcA)), 4.04 (dd, 1H, J2–3 = 10.2 Hz, J1–2 = 3.7 Hz, H-2 (Fuc)), 4.00 (q, 1H, J5–6 = 6.6 Hz, H-5 (Fuc)), 3.94 (dd, 1H, J2–3 = 10.2 Hz, J3–4 = 3.3 Hz, H-3 (Fuc)), 3.87 (t, 1H, J2–3, J3–4 = 9.3 Hz, H-3 (GlcA)), 3.76 (d, 1H, J3–4 = 3.3 Hz, H-4 (Fuc)), 3.66–3.60 (m, 1H, OCHH’), 3.57 (s, 3H, OMe), 3.54–3.50 (m, 1H, H-2 (GlcA)), 3.49–3.46 (m, 1H, OCHH’), 1.71–1.63 (m,2H, CH2CH2CH3), 1.25 (d, 3H, J5–6 = 5.6 Hz, H-6 (Fuc)), 0.97 (t, 3H, J = 7.4 Hz, CH2CH2CH3). 13C NMR (150 MHz, CD3OD): δ 170.09 (C-6 (GlcA)), 167.32 (COPh), 134.46 (p-Bz), 130.69 (o-Bz), 129.52 (m-Bz), 102.78 (C-1 (GlcA)), 98.24 (C-1 (Fuc)), 77.11 (C-2 (Fuc)), 74.71 (C-3 (GlcA)), 74.08 (C-2 (GlcA)), 73.64 (C-4 (GlcA)), 73.39 (C-5 (GlcA)), 73.14 (C-4 (Fuc)), 70.85 (OCH2), 69.74 (C-3 (Fuc)), 67.22 (C-5 (Fuc)), 53.10 (OMe), 23.70 (CH2CH2CH3), 16.53 (C-6 (Fuc)), 11.03 (CH2CH2CH3). HRMS (ESI): Calcd. m/z for [M + Na]+ C23H32O12 523.1786 found 523.1786.
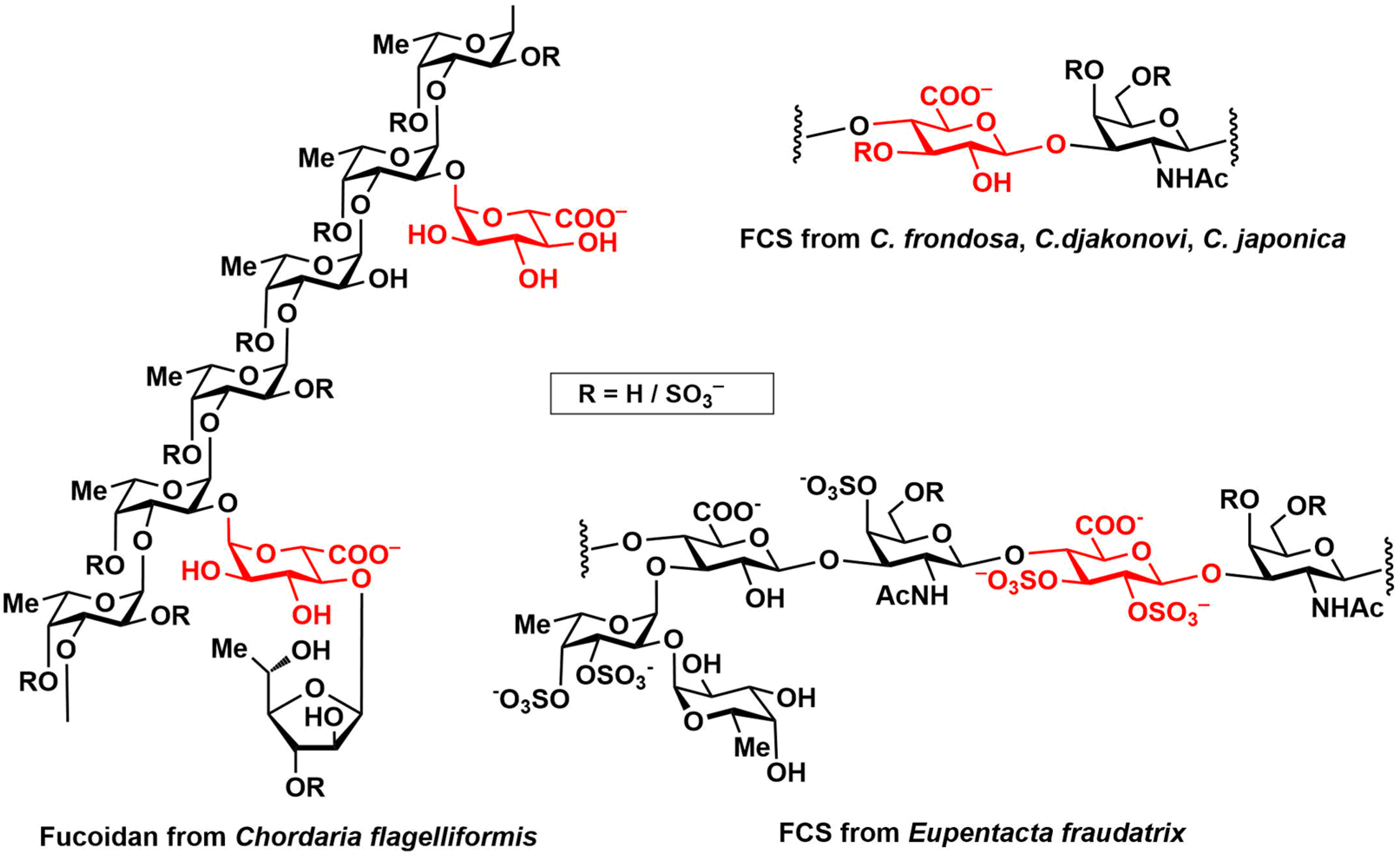
3.3.10. Propyl 2-O-(α-d-glucopyranosyluronic acid)-α-L-fucopyranoside Sodium Salt (1)
Final deprotection of
21a [11.5 mg, 0.024 eq.] was performed according to the standard procedure C. Purification by gel chromatography followed by lyophilization gave
1 (7 mg, 74%). Analytical data for
1 were in agreement with those previously reported [
36]. [α]
D = −21° (
c = 1, H
2O).
1H NMR (600 MHz, D
2O): δ 5.10 (d, 1H,
J1–2 = 3.9 Hz, H-1 (GlcA)), 5.02 (d, 1H,
J1–2 = 3.8 Hz, H-1 (Fuc)), 4.07 (q, 1H,
J5–6 = 6.5 Hz, H-5 (Fuc)), 4.03–3.98 (m, 2H, H-5 (GlcA), H-3 (Fuc)), 3.84 (d, 1H,
J3–4 = 3.0 Hz, H-4 (Fuc)), 3.78 (dd, 1H,
J2–3 = 10.3 Hz,
J1–2 = 3.8 Hz, H-2 (Fuc)), 3.74 (t, 1H,
J2–3,
J3–4 = 9.5 Hz, H-3 (GlcA)), 3.65–3.60 (m, 1H, OCHH’), 3.58 (dd, 1H,
J2–3 = 10.0 Hz,
J1–2 = 3.9 Hz, H-2 (GlcA)), 3.48 (t, 1H,
J3–4,
J4–5 = 9.6 Hz, H-4 (GlcA)), 3.43–3.39 (m, 1H, OCHH’), 1.66–1.58 (m, 2H, CH
2CH
2CH
3), 1.21 (d, 3H,
J5–6 = 6.6 Hz, H-6 (Fuc)), 0.91 (t,
J = 7.4 Hz, CH
2CH
2CH
3).
13C NMR (150 MHz, D
2O): δ 101.66 (C-1 (GlcA)), 98.39 (C-1 (Fuc)), 78.64 (C-2 (Fuc)), 73.27 (C-3 (GlcA)), 72.42 (C-4 (Fuc)), 72.37 (C-4 (GlcA)), 72.24 (C-5 (GlcA)), 72.02 (C-2 (GlcA)), 70.76 (OCH
2), 69.02 (C-3 (Fuc)), 66.80 (C-5 (Fuc)), 22.66 (CH
2CH
2CH
3), 15.68 (C-6 (Fuc)), 10.51 (CH
2CH
2CH
3). HRMS (ESI): Calcd.
m/
z for [M − H]
− C
15H
26O
11 381.1402 found 381.1393.
3.3.11. Propyl 2-O-(2,3-di-O-sulfo-α-d-glucopyranosyluronic acid)-3,4-di-O-sulfo-α-L-fucopyranoside Sodium Salt (2)
According to the standard procedure D, 21a [5.5 mg, 0.011 eq.] was treated followed by chromatography (silica gel, eluent: DCM/MeOH = 5:1→2:1). Product of sulfation was treated according to standard procedure C. Purification by gel chromatography followed by lyophilization gave 2 (3.5 mg, 64%). [α]D =−44° (c = 1, H2O). 1H NMR (600 MHz, D2O): δ 5.39 (d, 1H, J1–2 = 3.7 Hz, H-1 (GlcA)), 5.06 (d, 1H, J1–2 = 3.7 Hz, H-1 (Fuc)), 4.97 (d, 1H, J3–4 = 2.9 Hz, H-4 (Fuc)), 4.72 (dd, 1H, J2–3 = 10.6 Hz, J3–4 = 3.0 Hz, H-3 (Fuc)), 4.62 (t, 1H, J2–3, J3–4 = 9.5 Hz, H-3 (GlcA)), 4.29 (dd, 1H, J2–3 = 10.0 Hz, J1–2 = 3.8 Hz, H-2 (GlcA)), 4.22 (q, 1H, J5–6 = 6.5 Hz, H-5 (Fuc)), 4.11 (d, 1H, J4–5 = 10.2 Hz, H-5 (GlcA)), 4.01 (dd, 1H, J2–3 = 10.6 Hz, J1–2 = 3.7 Hz, H-2 (Fuc)), 3.71 (dd, 1H, J4–5 = 10.0 Hz, J3–4 = 9.0 Hz, H-4 (GlcA)), 3.66–3.61 (m, 1H, OCHH’), 3.47 (dt, 1H, J = 9.5 Hz, J = 6.6 Hz, OCHH’), 1.70–1.61 (m, 2H, CH2CH2CH3), 1.27 (d, 3H, J5–6 = 6.5 Hz, H-6 (Fuc)), 0.93 (t, 3H, J = 7.4 Hz, CH2CH2CH3). 13C NMR (150 MHz, D2O): δ 176.27 (C-6 (GlcA)), 99.35 (C-1 (GlcA)), 98.29 (C-1 (Fuc)), 79.74 (C-4 (Fuc)), 78.87 (C-3 (GlcA)), 76.18 (C-2 (Fuc)), 75.26 (C-2 (GlcA)), 73.94 (C-3 (Fuc)), 72.78 (C-5 (GlcA)), 71.79 (C-4 (GlcA)), 66.41 (C-5 (Fuc)), 22.80 (CH2CH2CH3), 16.36 (C-6 (Fuc)), 10.64 (CH2CH2CH3). HRMS (ESI): Calcd. m/z for [M − 2Na + H]− C15H21Na5O23S4 766.9133 found 766.9118.
3.3.12. Propyl 2-O-(2,3,4-tri-O-sulfo-α-d-glucopyranosyluronic acid)-3,4-di-O-sulfo-α-L-fucopyranoside Sodium Salt (3)
Exhaustive sulfation of
1 [3.2 mg, 0.008 eq.] was performed according to the standard procedure D. Purification by gel chromatography followed by lyophilization gave
3 (5 mg, 72%). Analytical data for
3 were in agreement with those previously reported [
37]. [α]
D = −13° (
c = 1, H
2O).
1H NMR (600 MHz, D
2O): δ 5.44 (d, 1H,
J1–2 = 2.6 Hz, H-1 (GlcA)), 5.07 (d, 1H,
J1–2 = 3.8 Hz, H-1 (Fuc)), 4.95–4.93 (m, 2H, H-3 (GlcA), H-4 (Fuc)), 4.76–4.71 (m, 2H, H-3 (Fuc), H-4 (GlcA)), 4.52 (dd, 1H,
J2–3 = 6.4 Hz,
J1–2 = 2.6 Hz, H-2 (GlcA)), 4.39 (d, 1H,
J4–5 = 5.4 Hz, H-5 (GlcA)), 4.24–4.19 (m, 2H, H-2 (Fuc), H-5 (Fuc)), 3.66–3.57 (m, 2H, OCH
2), 1.73–1.61 (m, 2H, CH
2CH
2CH
3), 1.28 (d, 3H,
J5–6 = 6.6 Hz, H-6 (Fuc)), 0.93 (t, 3H,
J = 7.4 Hz, CH
2CH
2CH
3).
13C NMR (150 MHz, D
2O): δ 98.53 (C-1 (Fuc)), 96.94 (C-1 (GlcA)), 79.75 (C-4 (Fuc)), 75.61 (C-5 (GlcA)), 74.86 (C-2 (Fuc)), 74.81 (C-4 (GlcA)), 74.55 (C-3 (GlcA)), 74.44 (C-3 (Fuc)), 73.35 (C-2 (GlcA)), 71.13 (OCH
2), 66.42 (C-5 (Fuc)), 22.67 (CH
2CH
2CH
3), 16.39 (C-6 (Fuc)), 10.56 (CH
2CH
2CH
3). HRMS (ESI): Calcd.
m/
z for [M − 2Na + H]
− C
15H
20Na
6O
26S
5 868.8515 found 868.8501.
3.3.13. Propyl 2-O-(β-d-glucopyranosyluronic acid)-α-L-fucopyranoside Sodium Salt (4)
Final deprotection of 21b [8.5 mg, 0.016 eq.] was performed according to the standard procedure C. Purification by gel chromatography followed by lyophilization gave 4 (4.8 mg, 70%). [α]D = −83° (c = 1, H2O). 1H NMR (600 MHz, D2O): δ 5.05 (d, 1H, J1–2 = 3.4 Hz, H-1 (Fuc)), 4.56 (d, 1H, J1–2 = 7.9 Hz, H-1 (GlcA)), 4.09 (q, 1H, J5–6 = 6.6 Hz, H-5 (Fuc)), 4.00 (dd, 1H, J2–3 = 10.4 Hz, J1–2 = 3.6 Hz, H-2 (Fuc)), 3.95 (dd, 1H, J2–3 = 10.3 Hz, J1–2 = 3.2 Hz, H-3 (Fuc)), 3.84 (d, 1H, J3–4 = 2.9 Hz, H-4 (Fuc)), 3.76–3.71 (m, 1H, H-5 (GlcA)), 3.63 (dt, 1H, J = 14.4 Hz, J = 7.2 Hz, OCHH’), 3.54–3.48 (m, 3H, H-3 (GlcA), H-4 (GlcA), OCHH’), 3.36 (t, 1H, J1–2, J2–3 = 8.0 Hz, H-2 (GlcA)), 1.62 (dd, 2H, J = 14.3 Hz, J = 7.1 Hz, CH2CH2CH3), 1.22 (d, 3H, J5–6 = 6.6 Hz, H-6 (Fuc)), 0.91 (t, 3H, J = 7.4 Hz, CH2CH2CH3). 13C NMR (150 MHz, D2O): δ 101.56 (C-1 (GlcA)), 96.73 (C-1 (Fuc)), 76.38 (C-5 (GlcA)), 76.14 (C-2 (Fuc)), 76.08 (C-3 (GlcA)), 73.39 (C-2 (GlcA)), 72.32 (C-4 (GlcA)), 72.25 (C-4 (Fuc)), 70.72 (OCH2), 68.76 (C-3 (Fuc)), 66.96 (C-5 (Fuc)), 22.59 (CH2CH2CH3), 15.78 (C-6 (Fuc)), 10.49 (CH2CH2CH3). HRMS (ESI): Calcd. m/z for [M − H]− C15H26O11 381.1402 found 381.1405.
3.3.14. Propyl 2-O-(2,3-di-O-sulfo-β-d-glucopyranosyluronic acid)-3,4-di-O-sulfo- α-L-fucopyranoside Sodium Salt (5)
According to the standard procedure D, 21b [10.5 mg, 0.021 eq.] was treated followed by chromatography (silica gel, eluent: DCM/MeOH = 4:1→1:1). Product of sulfation was treated according to standard procedure C. Purification by gel chromatography followed by lyophilization gave 5 (6 mg, 57%). [α]D = −23° (c = 1, H2O). 1H NMR (600 MHz, D2O): δ 5.23 (d, 1H, J1–2 = 3.7 Hz, H-1 (Fuc)), 4.95 (d, 1H, J3–4 = 2.6 Hz, H-4 (Fuc)), 4.92 (d, 1H, J1–2 = 7.1 Hz, H-1 (GlcA)), 4.75 (dd, 1H, J2–3 = 6.3 Hz, J3–4 = 2.5 Hz, H-3 (Fuc)), 4.48 (t, 1H, J2–3, J3–4 = 7.8 Hz, H-3 (GlcA)), 4.28 (t, 1H, J1–2, J2–3 = 7.3 Hz, H-2 (Fuc)), 4.24 (q, 1H, J5–6 = 6.4 Hz, H-5 (Fuc)), 4.16 (dd, 1H, J2–3 = 10.5 Hz, J1–2 = 3.7 Hz, H-2 (Fuc)), 3.94–3.88 (m, 2H, H-4 (GlcA), H-5 (GlcA)), 3.64–3.57 (m, 2H, OCH2), 1.64 (dq, 2H, J = 14.5 Hz, J = 7.3 Hz, CH2CH2CH3), 1.27 (d, 3H, J5–6 = 6.6 Hz, H-6 (Fuc)), 0.91 (t, 3H, J = 7.4 Hz, CH2CH2CH3). 13C NMR (150 MHz, D2O): δ 101.41 (C-1 (GlcA)), 97.24 (C-1 (Fuc)), 81.63 (C-3 (GlcA)), 79.70 (C-4 (Fuc)), 77.99 (C-2 (GlcA)), 77.12 (C-5 (GlcA)), 75.44 (C-2 (Fuc)), 74.86 (C-3 (Fuc)), 71.33 (C-4 (GlcA)), 71.11 (OCH2), 66.52 (C-5 (Fuc)), 22.67 (CH2CH2CH3), 16.33 (C-6 (Fuc)), 10.48 (CH2CH2CH3). HRMS (ESI): Calcd. m/z for [M − 3Na + 2H]− C15H21Na5O23S4 744.9314 found 744.9217.
3.3.15. Propyl 2-O-(2,3,4-tri-O-sulfo-β-d-glucopyranosyluronic acid)-3,4-di-O-sulfo- α-L-fucopyranoside Sodium Salt (6)
Exhaustive sulfation of 4 [3.8 mg, 0.09 eq.] was performed according to the standard procedure D. Purification by gel chromatography followed by lyophilization gave 6 (6.8 mg, 78%). [α]D = −47° (c = 1, H2O). 1H NMR (400 MHz, D2O): δ 5.21 (dt, 1H, J3–4 = 4.1 Hz, J4–5 = 1.3 Hz, H-4 (GlcA)), 5.17 (d, 1H, J1–2 = 3.7 Hz, H-1 (Fuc)), 5.08 (dd, 1H, J3–4 = 4.1 Hz, J2–3 = 1.2 Hz, H-3 (GlcA)), 4.98 (d, 1H, J1–2 = 6.8 Hz, H-1 (GlcA)), 4.94 (d, 1H, J3–4 = 3.0 Hz, H-4 (Fuc)), 4.74 (dd, 1H, J2–3 = 10.6 Hz, J3–4 = 3.0 Hz, H-3 (Fuc)), 4.62 (d, 1H, J4–5 = 1.6 Hz, H-5 (GlcA)), 4.48 (d, 1H, J1–2 = 6.7 Hz, H-2 (GlcA)), 4.25 (q, 1H, J5–6 = 6.5 Hz, H-5 (Fuc)), 4.16 (dd, 1H, J2–3 = 10.6 Hz, J2–3 = 3.7 Hz, H-2 (Fuc)), 3.62 (t, 2H, J = 6.9 Hz, OCH2), 1.65 (h, 2H, J = 7.2 Hz, CH2CH2CH3), 1.27 (d, 3H, J5–6 = 6.6 Hz, H-6 (Fuc)), 0.91 (t, 3H, J = 7.4 Hz, CH2CH2CH3). 13C NMR (100 MHz, D2O): δ 99.91 (C-1 (GlcA)), 97.02 (C-1 (Fuc)), 79.70 (C-4 (Fuc)), 78.90 (C-5 (GlcA)), 78.60 (C-2 (GlcA)), 75.89 (C-3 (GlcA)), 75.28 (C-4 (GlcA)), 74.83 (C-2 (Fuc)), 74.33 (C-3 (Fuc)), 71.42 (OCH2), 66.56 (C-5 (Fuc)), 22.69 (CH2CH2CH3), 16.37 (C-6 (Fuc)), 10.51 (CH2CH2CH3). HRMS (ESI): Calcd. m/z for [M − 3Na + 2H]− C15H20Na6O26S5 846.8701 found 846.8489.



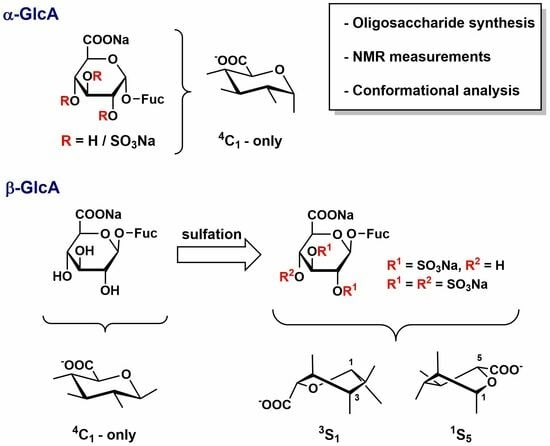
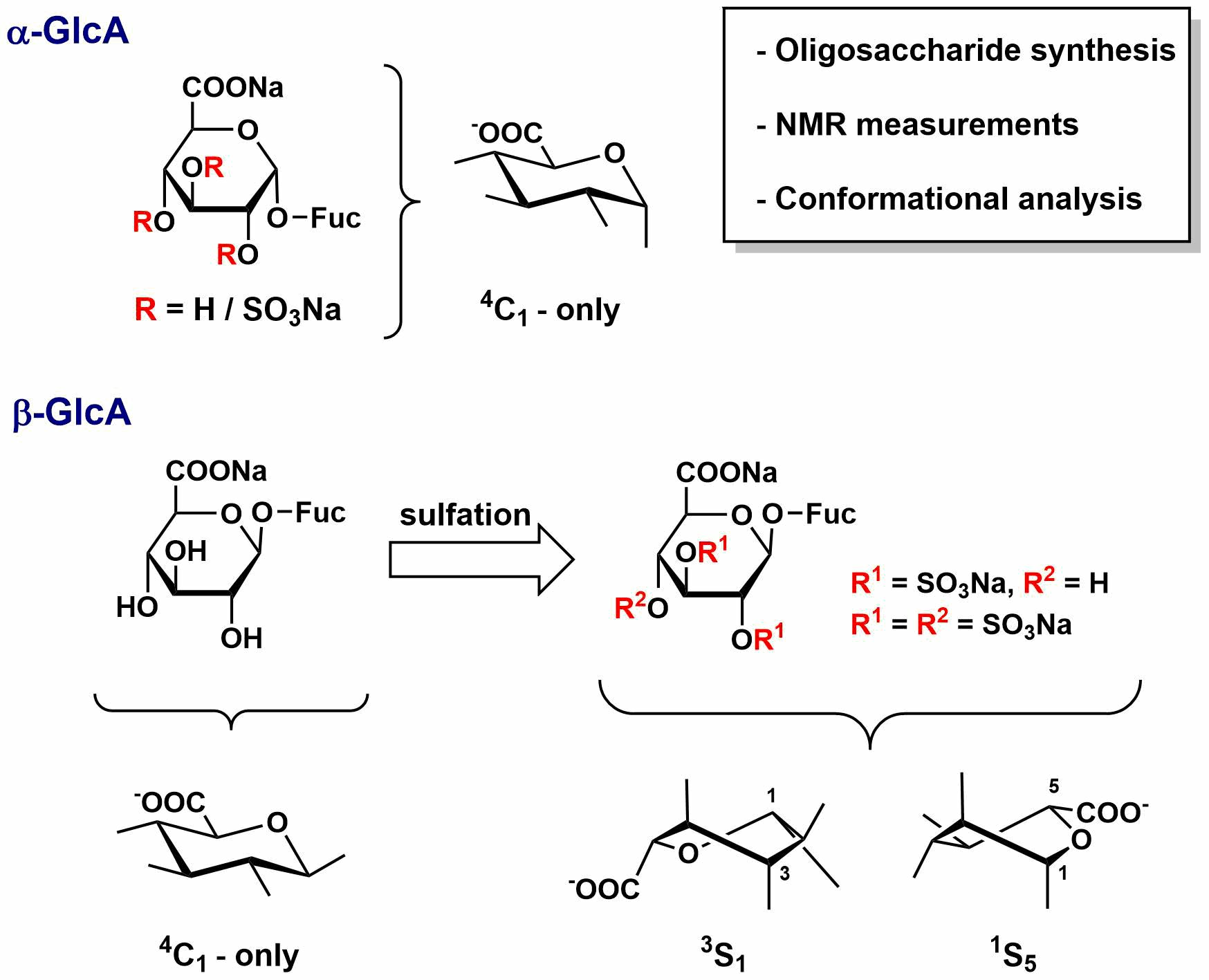
 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}