
Double Attack to Oxidative Stress in Neurodegenerative Disorders: MAO-B and Nrf2 as Elected Targets
 , , , ,
, , , ,  , and
, and
Abstract
:
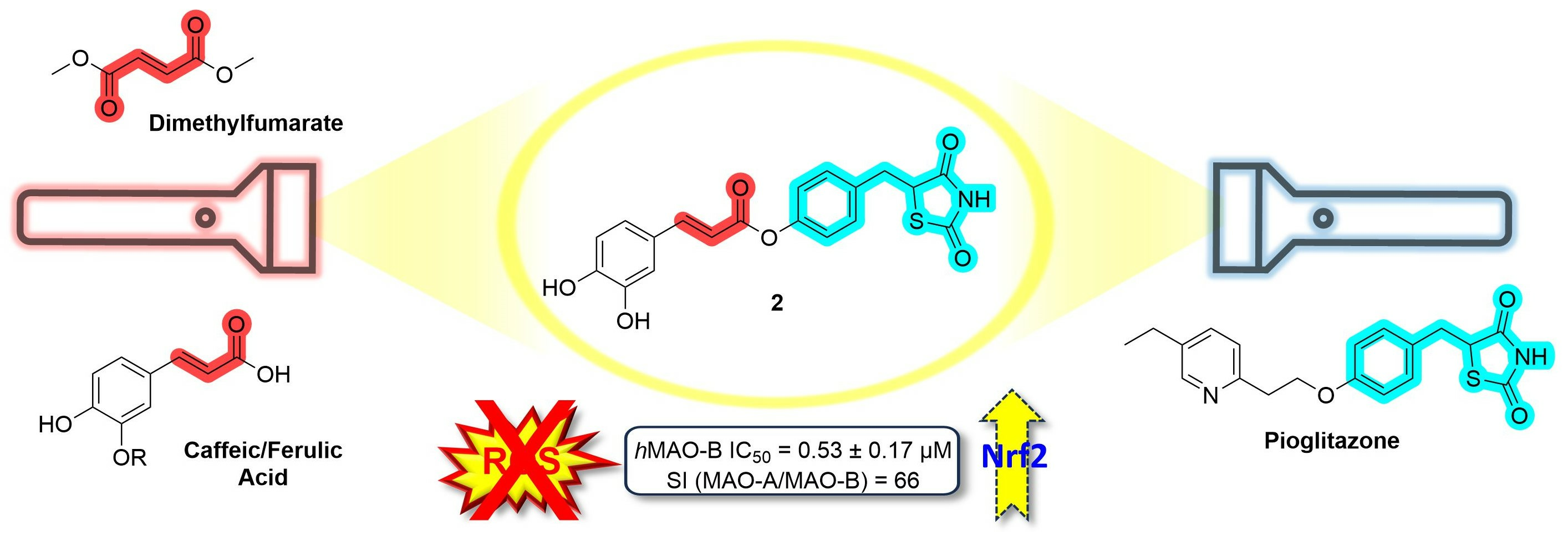
1. Introduction
2. Results and Discussion
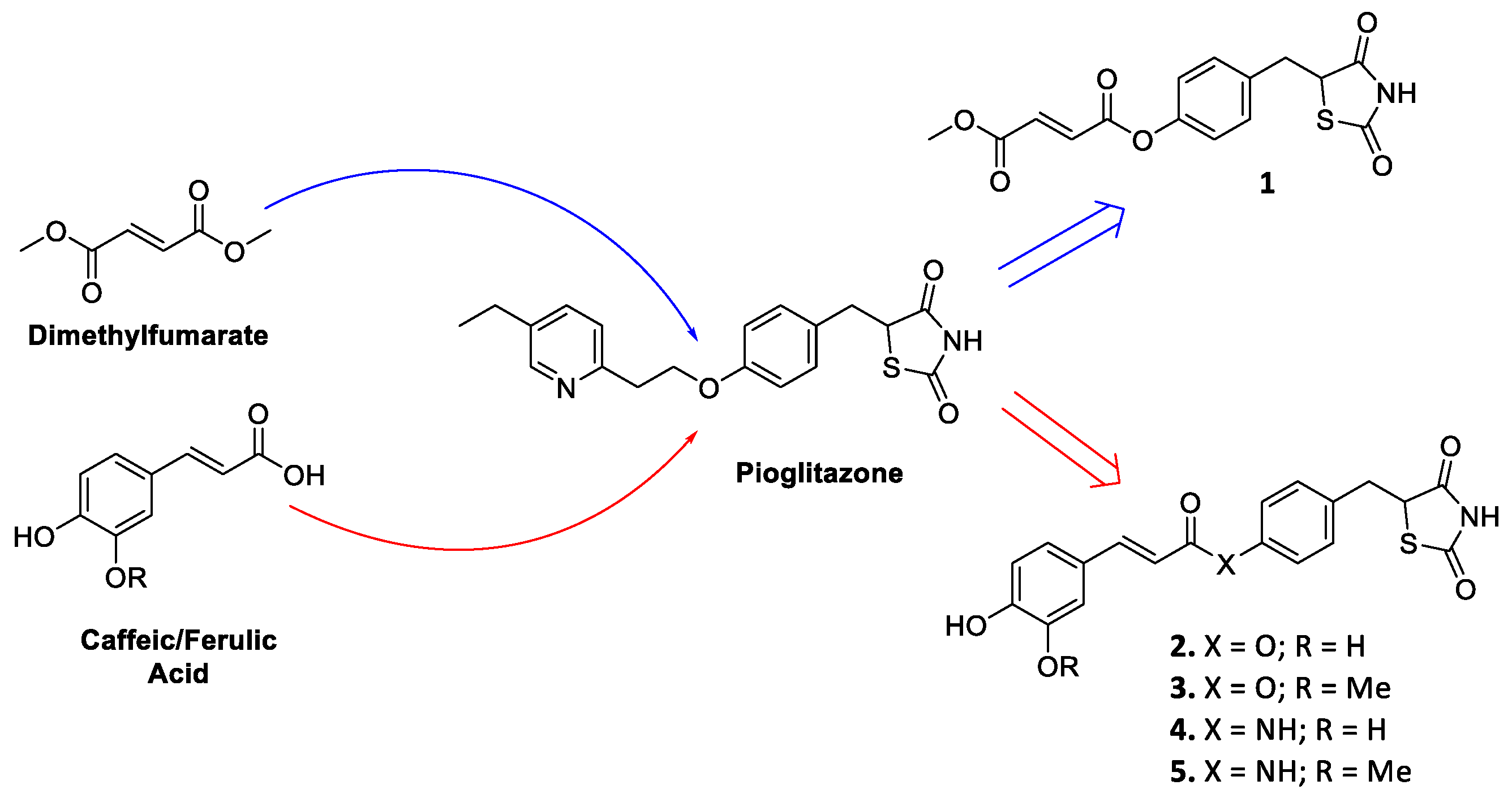
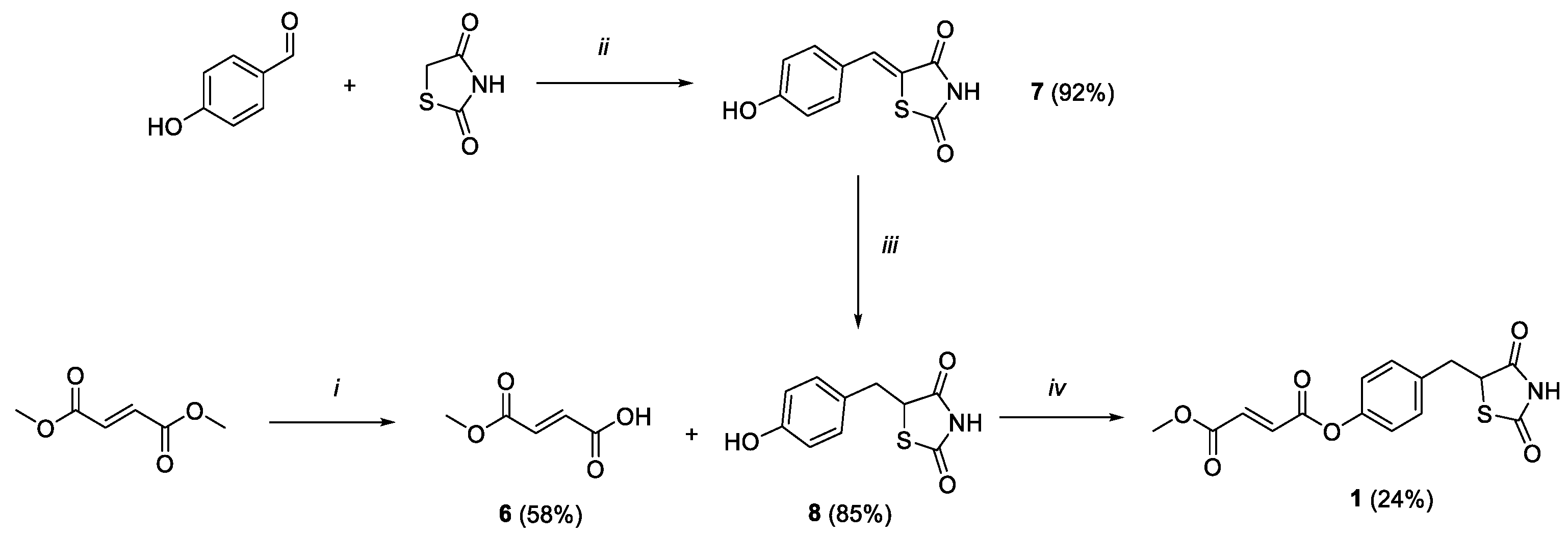
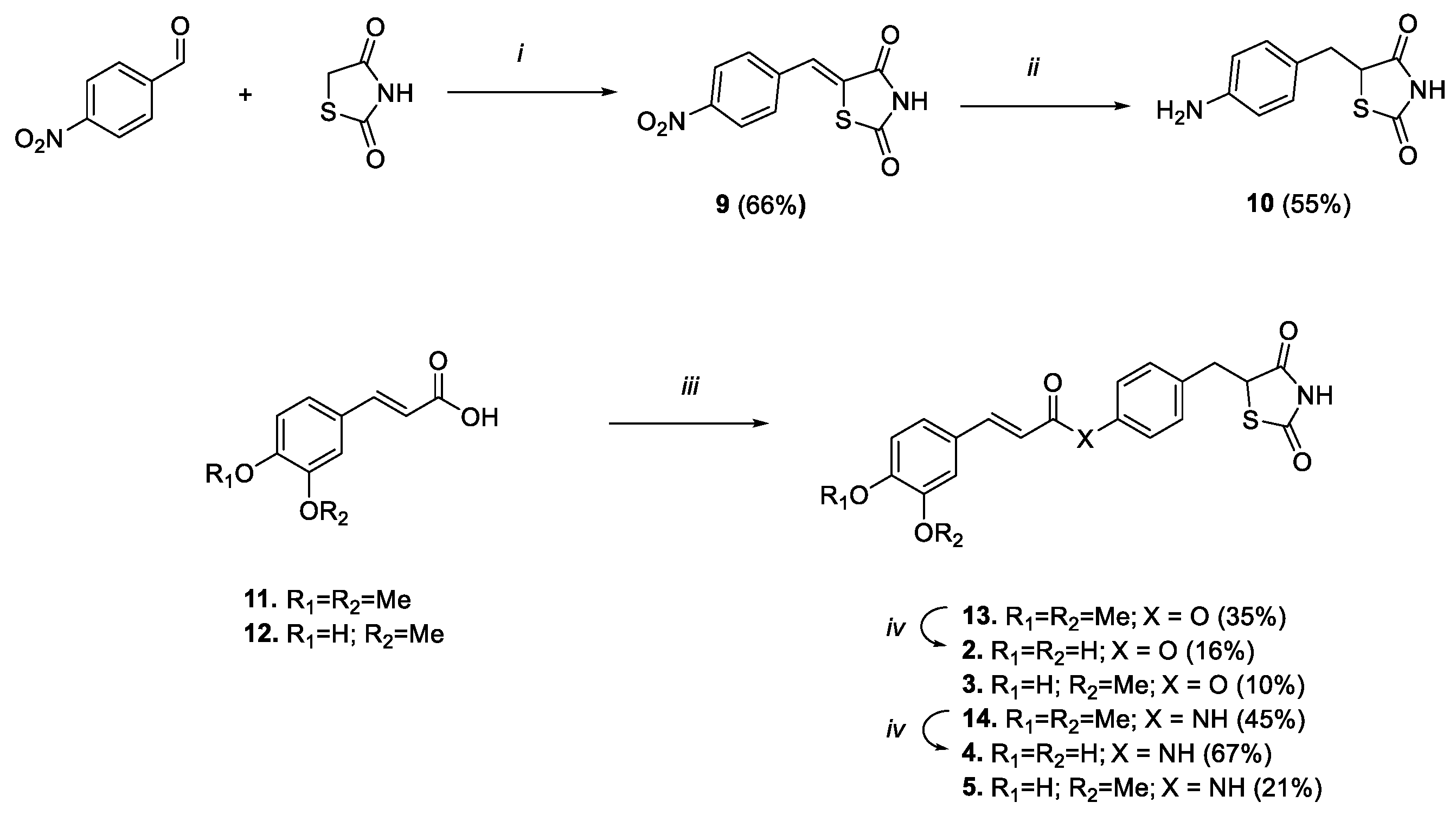
2.1. Chemistry
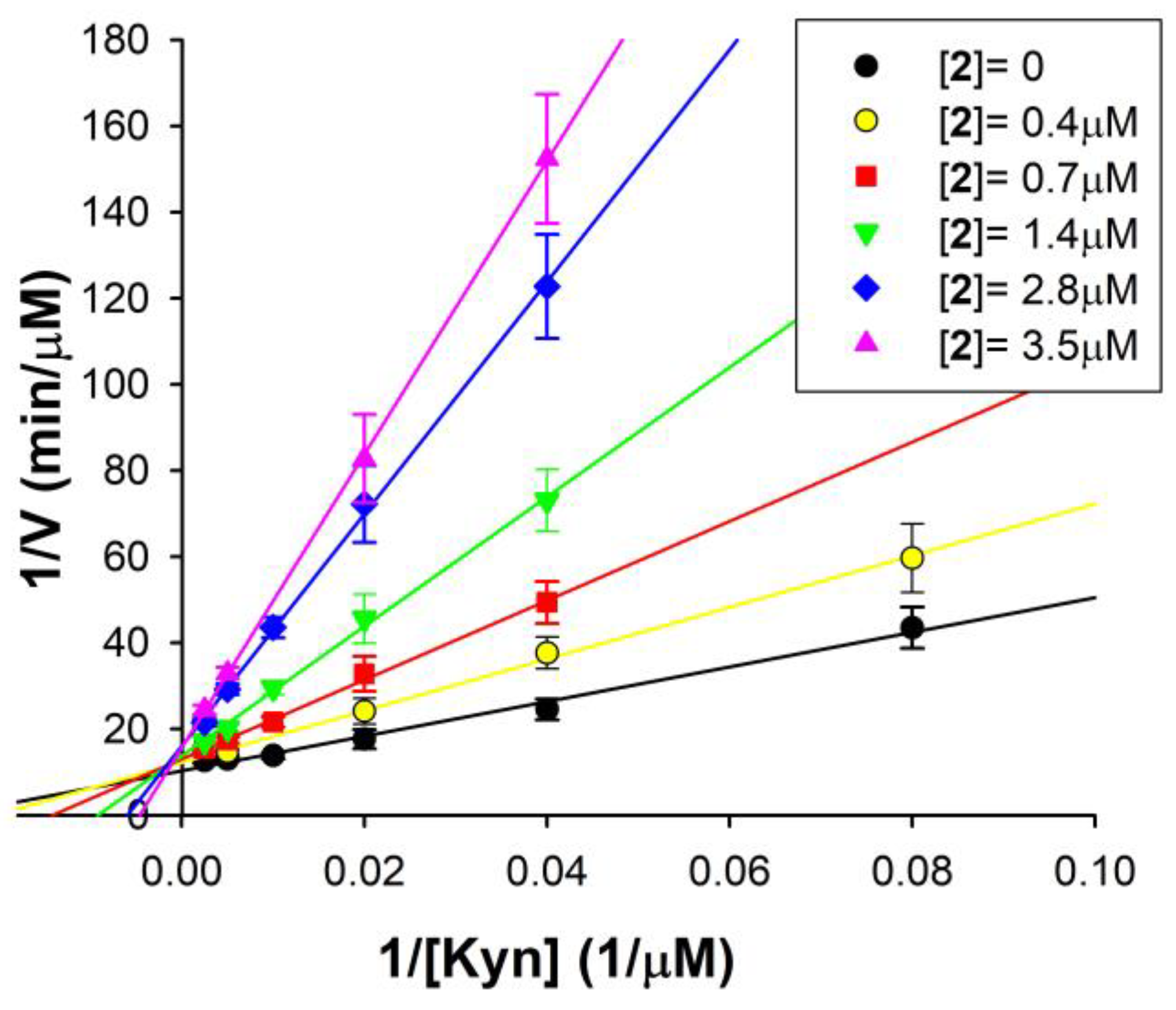
2.2. Effects and Kinetic Characterization of Compounds 1–5 on hMAOs
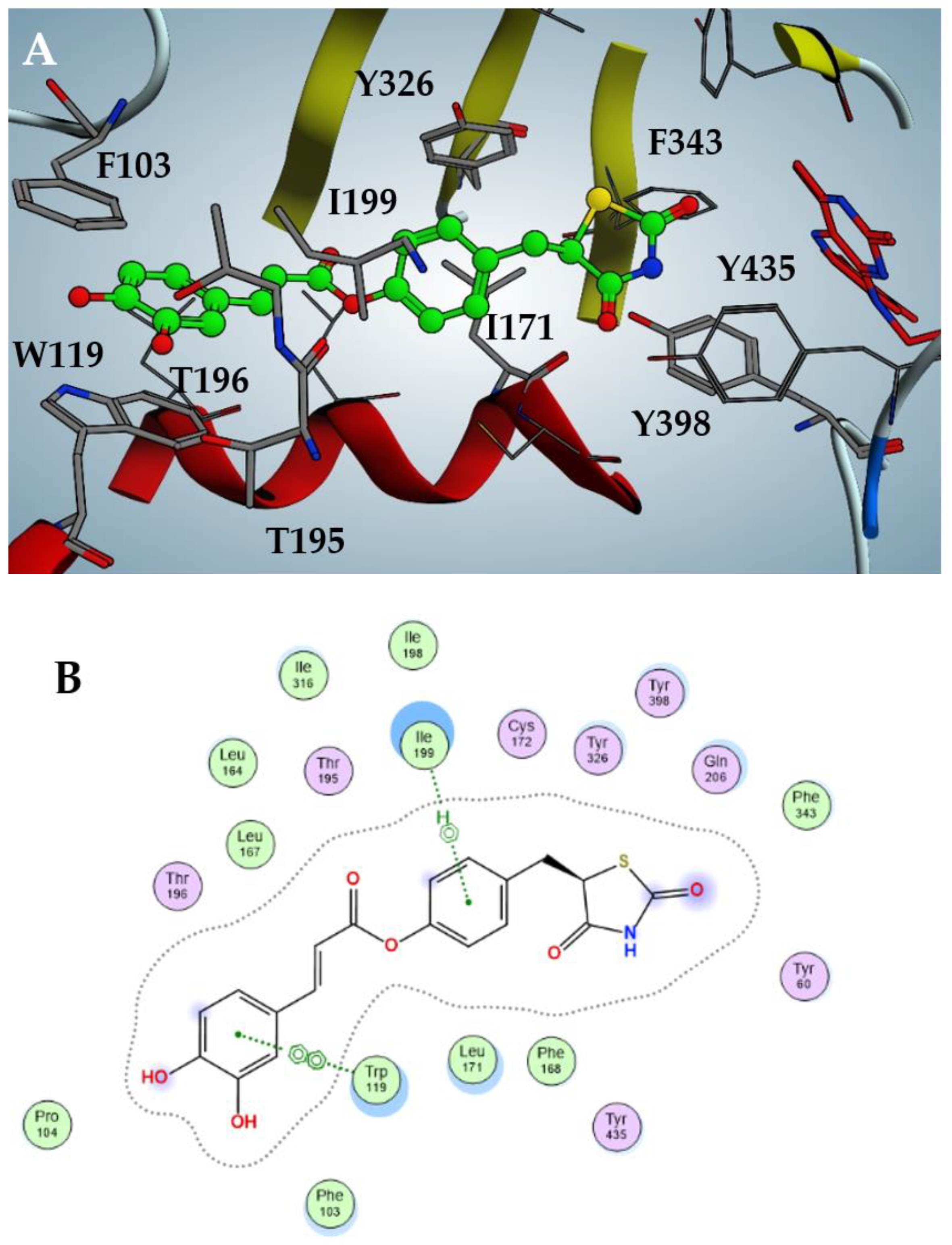
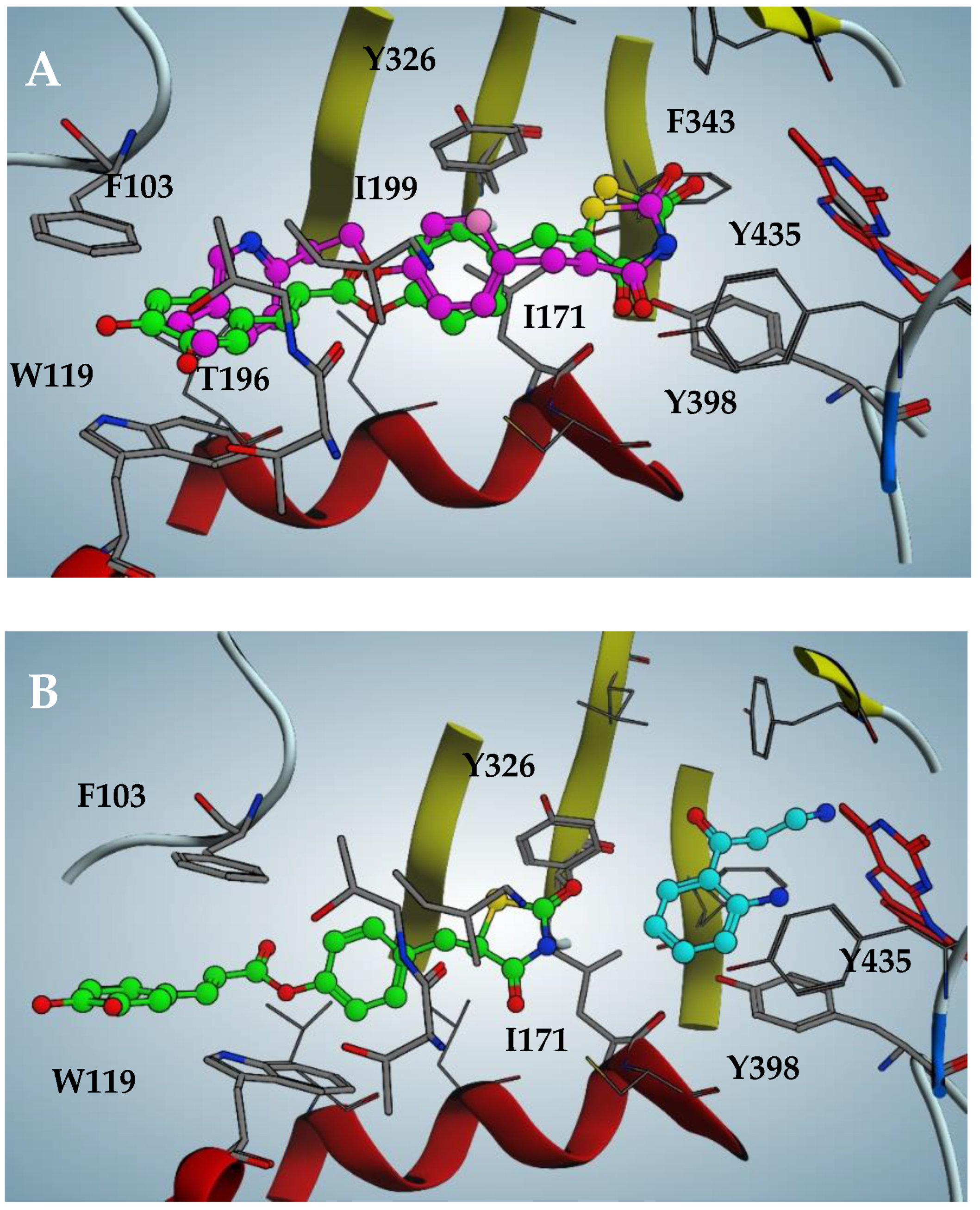
2.3. Docking Analysis of Compounds 2–5 against hMAO-B
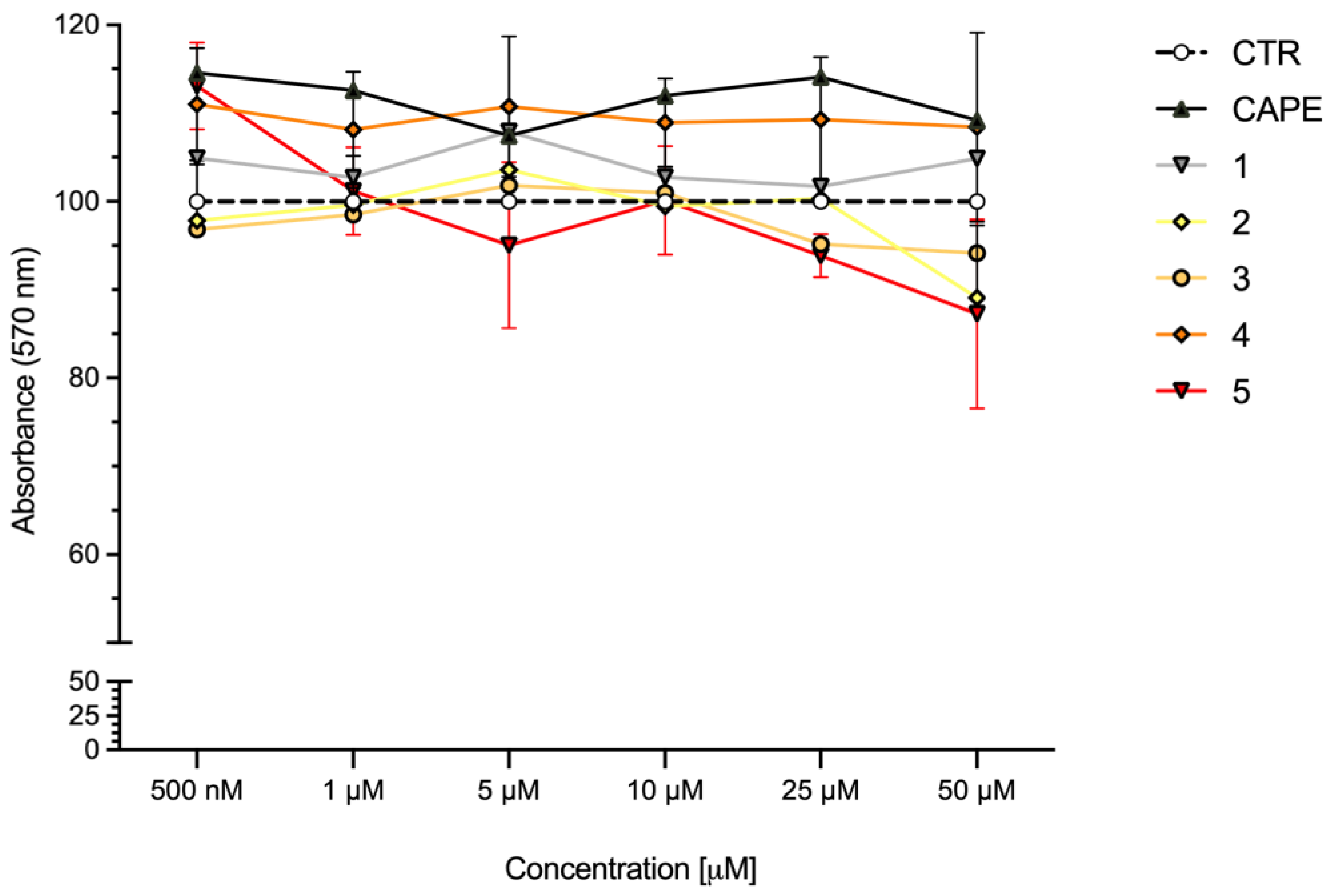
2.4. Cell Viability
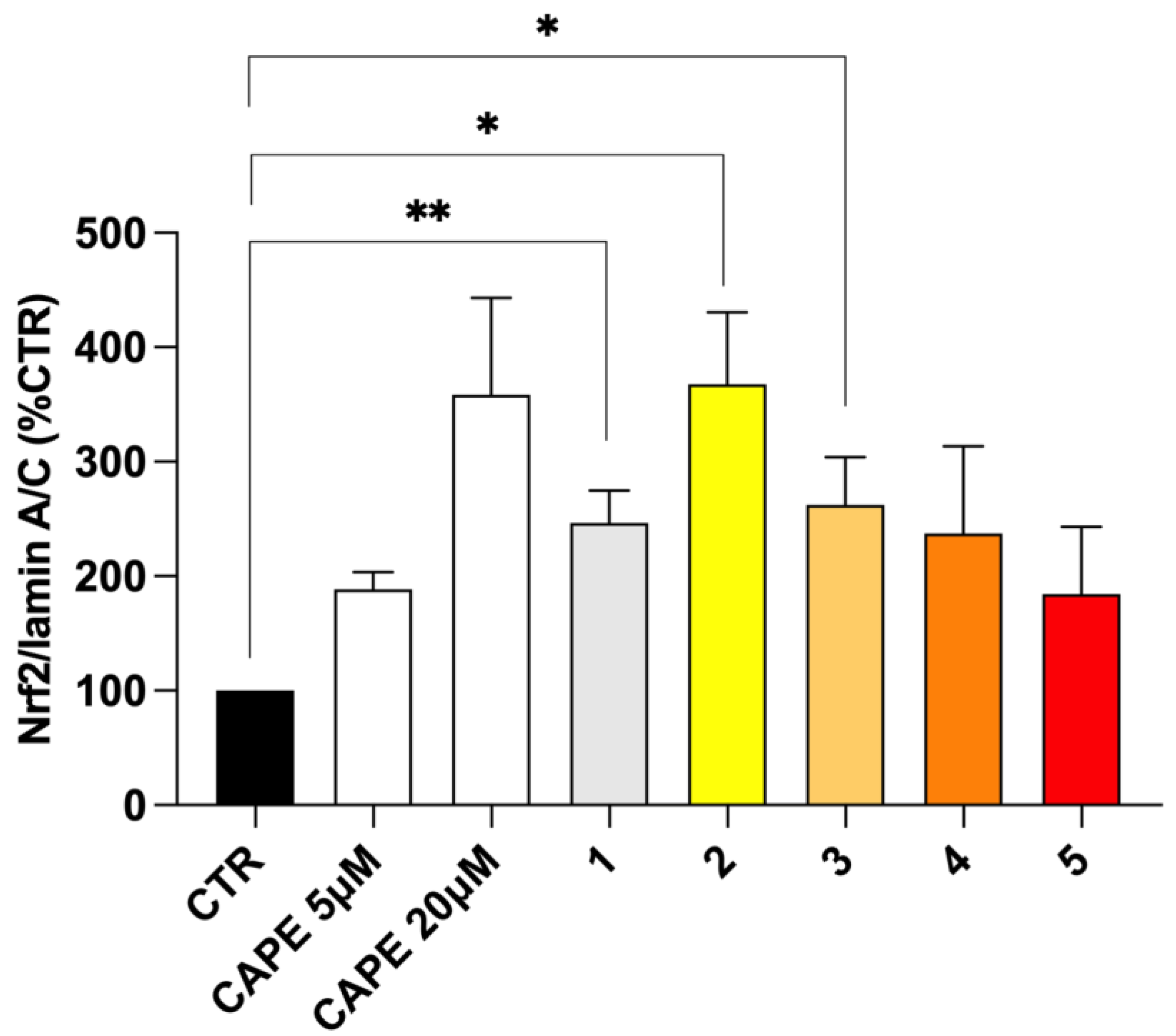
2.5. Nrf2 Nuclear Translocation
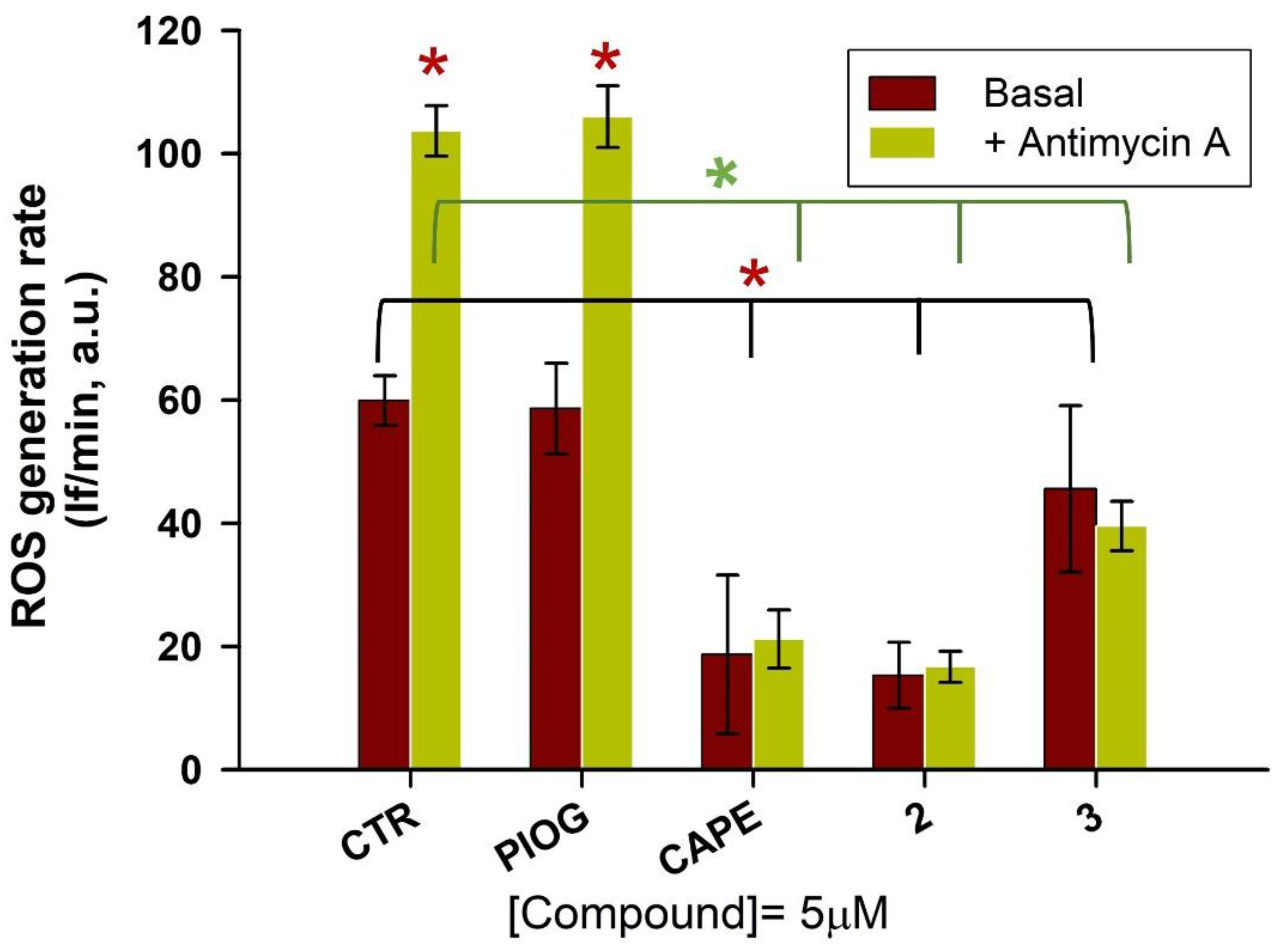
2.6. Antioxidant Efficacy in HepG2 Cells
3. Materials and Methods
3.1. Chemistry
3.1.1. (E)-4-Methoxy-4-oxobut-2-enoic Acid (6)
3.1.2. (Z)-5-(4-Hydroxybenzylidene)thiazolidine-2,4-dione (7)
3.1.3. 5-(4-Hydroxybenzyl)thiazolidine-2,4-dione (8)
3.1.4. 4-((2,4-Dioxothiazolidin-5-yl)methyl)phenyl Methyl Fumarate (1)
3.1.5. (Z)-5-(4-Nitrobenzylidene)thiazolidine-2,4-dione (9)
3.1.6. 5-(4-Aminobenzyl)thiazolidine-2,4-dione (10)
3.1.7. General Procedure for the Synthesis of Compounds 13, 14, 3 and 5
3.1.8. 4-((2,4-Dioxothiazolidin-5-yl)methyl)phenyl (E)-3-(3,4-Dimethoxyphenyl)acrylate (13)
3.1.9. (E)-3-(3,4-Dimethoxyphenyl)-N-(4-((2,4-dioxothiazolidin-5-yl)methyl)phenyl)acrylamide (14)
3.1.10. 4-((2,4-Dioxothiazolidin-5-yl)methyl)phenyl (E)-3-(4-hydroxy-3-methoxyphenyl)acrylate (3)
3.1.11. (E)-N-(4-((2,4-Dioxothiazolidin-5-yl)methyl)phenyl)-3-(4-hydroxy-3-methoxyphenyl)acrylamide (5)
3.1.12. General Procedure for the Synthesis of Compounds 2 and 4
3.1.13. 4-((2,4-Dioxothiazolidin-5-yl)methyl)phenyl (E)-3-(3,4-Dihydroxyphenyl)acrylate (2)
3.1.14. (E)-3-(3,4-Dihydroxyphenyl)-N-(4-((2,4-dioxothiazolidin-5-yl)methyl)phenyl)acrylamide (4)
3.2. hMAOs Activity Assays
3.3. Docking Analysis at hMAO-B and Keap1
3.4. Cell Viability in SH-SY5Y Cells
3.5. Nrf2 Nuclear Translocation
3.6. Measurement of Intracellular ROS Generation Rate
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, Y.C.; Wu, J.S.; Tsai, H.D.; Huang, C.Y.; Chen, J.J.; Sun, G.Y.; Lin, T.N. Peroxisome proliferator-activated receptor gamma (PPAR-γ) and neurodegenerative disorders. Mol. Neurobiol. 2012, 46, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Zolezzi, J.M.; Santos, M.J.; Bastías-Candia, S.; Pinto, C.; Godoy, J.A.; Inestrosa, N.C. PPARs in the central nervous system: Roles in neurodegeneration and neuroinflammation. Biol. Rev. Camb Philos. Soc. 2017, 92, 2046–2069. [Google Scholar] [CrossRef]
- Swanson, C.R.; Joers, V.; Bondarenko, V.; Brunner, K.; Simmons, H.A.; Ziegler, T.E.; Kemnitz, J.W.; Johnson, J.A.; Emborg, M.E. The PPAR-γ agonist pioglitazone modulates inflammation and induces neuroprotection in parkinsonian monkeys. J. Neuroinflamm. 2011, 8, 91. [Google Scholar] [CrossRef]
- Breidert, T.; Callebert, J.; Heneka, M.T.; Landreth, G.; Launay, J.M.; Hirsch, E.C. Protective action of the peroxisome proliferator-activated receptor-gamma agonist pioglitazone in a mouse model of Parkinson’s disease. J. Neurochem. 2002, 82, 615–624. [Google Scholar] [CrossRef]
- Quinn, L.P.; Crook, B.; Hows, M.E.; Vidgeon-Hart, M.; Chapman, H.; Upton, N.; Medhurst, A.D.; Virley, D.J. The PPARgamma agonist pioglitazone is effective in the MPTP mouse model of Parkinson’s disease through inhibition of monoamine oxidase B. Br. J. Pharmacol. 2008, 154, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Edmonson, D.E.; Binda, C. Monoamine Oxidases. In Membrane Protein Complexes: Structure and Function; Harris, J.R., Boekema, E.J., Eds.; Springer Nature: Singapore, 2018; pp. 117–140. [Google Scholar]
- Schedin-Weiss, S.; Inoue, M.; Hromadkova, L.; Teranishi, Y.; Yamamoto, N.G.; Wiehager, B.; Bogdanovic, N.; Winblad, B.; Sandebring-Matton, A.; Frykman, S.; et al. Monoamine oxidase B is elevated in Alzheimer disease neurons, is associated with γ-secretase and regulates neuronal amyloid β-peptide levels. Alzheimer’s Res. Ther. 2017, 9, 57. [Google Scholar] [CrossRef] [PubMed]
- Santin, Y.; Resta, J.; Parini, A.; Mialet-Perez, J. Monoamine oxidases in age-associated diseases: New perspectives for old enzymes. Ageing Res. Rev. 2021, 66, 101256. [Google Scholar] [CrossRef]
- Binda, C.; Aldeco, M.; Geldenhuys, W.J.; Tortorici, M.; Mattevi, A.; Edmondson, D.E. Molecular Insights into Human Monoamine Oxidase B Inhibition by the Glitazone Anti-Diabetes Drugs. ACS Med. Chem. Lett. 2011, 3, 39–42. [Google Scholar] [CrossRef]
- Saunders, A.; Burns, D.; Gottschalk, W. Reassessment of Pioglitazone for Alzheimer’s Disease. Front. Neurosci. 2021, 15, 666958. [Google Scholar] [CrossRef]
- Galimberti, D.; Scarpini, E. Pioglitazone for the treatment of Alzheimer’s disease. Expert Opin. Investig. Drugs 2017, 26, 97–101. [Google Scholar] [CrossRef]
- Burns, D.; Alexander, R.; Welsh-Bohmer, K.; Culp, M.; Chiang, C.; O’Neil, J.; Evans, R.; Harrigan, P.; Plassman, B.; Burke, J.; et al. Safety and efficacy of pioglitazone for the delay of cognitive impairment in people at risk of Alzheimer’s disease (TOMMORROW): A prognostic biomarker study and a phase 3, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2021, 20, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Simuni, T.; Kieburtz, K.; Tilley, B.; Elm, J.; Ravina, B.; Babcock, D.; Emborg, M.; Hauser, R.; Kamp, C.; Morgan, J.; et al. Pioglitazone in early Parkinson’s disease: A phase 2, multicentre, double-blind, randomised trial. Lancet Neurol. 2015, 14, 795–803. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Kostov, R.V.; Kazantsev, A.G. The role of Nrf2 signaling in counteracting neurodegenerative diseases. FEBS J. 2018, 285, 3576–3590. [Google Scholar] [CrossRef]
- Bomprezzi, R. Dimethyl fumarate in the treatment of relapsing-remitting multiple sclerosis: An overview. Ther. Adv. Neurol. Disord. 2015, 8, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Moratilla-Rivera, I.; Sanchez, M.; Valdes-Gonzalez, J.; Gomez-Serranillos, M. Natural Products as Modulators of Nrf2 Signaling Pathway in Neuroprotection. Int. J. Mol. Sci. 2023, 24, 3748. [Google Scholar] [CrossRef]
- Scapagnini, G.; Vasto, S.; Abraham, N.; Calogero, C.; Zella, D.; Galvano, F. Modulation of Nrf2/ARE Pathway by Food Polyphenols: A Nutritional Neuroprotective Strategy for Cognitive and Neurodegenerative Disorders. Mol. Neurobiol. 2011, 44, 192–201. [Google Scholar] [CrossRef]
- Zhou, Y.; Jiang, Z.; Lu, H.; Xu, Z.; Tong, R.; Shi, J.; Jia, G. Recent Advances of Natural Polyphenols Activators for Keap1-Nrf2 Signaling Pathway. Chem. Biodivers. 2019, 16, e1900400. [Google Scholar] [CrossRef]
- Basagni, F.; Lanni, C.; Minarini, A.; Rosini, M. Lights and shadows of electrophile signaling: Focus on the Nrf2-Keap1 pathway. Futur. Med. Chem. 2019, 11, 707–721. [Google Scholar] [CrossRef]
- Geldenhuys, W.J.; Darvesh, A.S.; Funk, M.O.; Van der Schyf, C.J.; Carroll, R.T. Identification of novel monoamine oxidase B inhibitors by structure-based virtual screening. Bioorg. Med. Chem. Lett. 2010, 20, 5295–5298. [Google Scholar] [CrossRef]
- Satoh, T.; Lipton, S. Recent advances in understanding NRF2 as a druggable target: Development of pro-electrophilic and non-covalent NRF2 activators to overcome systemic side effects of electrophilic drugs like dimethyl fumarate. F1000Res 2017, 6, 2138. [Google Scholar] [CrossRef]
- Lategan, T.; Wang, L.; Sprague, T.; Rousseau, F. Pharmacokinetics and Bioavailability of Monomethyl Fumarate Following a Single Oral Dose of Bafiertam (TM) (Monomethyl Fumarate) or Tecfidera(R) (Dimethyl Fumarate). CNS Drugs 2021, 35, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Jamali, B.; Bjørnsdottir, I.; Nordfang, O.; Hansen, S.H. Investigation of racemisation of the enantiomers of glitazone drug compounds at different pH using chiral HPLC and chiral CE. J. Pharm. Biomed. Anal. 2008, 46, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.A. Isolation and structure elucidation of the new fungal metabolite (-)-xylariamide A. J. Nat. Prod. 2005, 68, 769–772. [Google Scholar] [CrossRef] [PubMed]
- Goel, A.; Parihar, A.; Mishra, P.; Varshney, S.; Nag, P.; Beg, M.; Gaikwad, A.; Rath, S.K. Design and synthesis of novel pyranone-based insulin sensitizers exhibiting in vivo hepatoprotective activity. Med. Chem. Commun. 2013, 4, 1532–1536. [Google Scholar] [CrossRef]
- Scapagnini, G.; Foresti, R.; Calabrese, V.; Stella, A.; Green, C.; Motterlini, R. Caffeic acid phenethyl ester and curcumin: A novel class of heme oxygenase-1 inducers. Mol. Pharmacol. 2002, 61, 554–561. [Google Scholar] [CrossRef]
- Morroni, F.; Sita, G.; Graziosi, A.; Turrini, E.; Fimognari, C.; Tarozzi, A.; Hrelia, P. Neuroprotective Effect of Caffeic Acid Phenethyl Ester in A Mouse Model of Alzheimer’s Disease Involves Nrf2/HO-1 Pathway. Aging Dis. 2018, 9, 605–622. [Google Scholar] [CrossRef] [PubMed]
- Dhiman, P.; Malik, N.; Khatkar, A. Hybrid caffeic acid derivatives as monoamine oxidases inhibitors: Synthesis, radical scavenging activity, molecular docking studies and in silico ADMET analysis. Chem. Cent. J. 2018, 12, 112. [Google Scholar] [CrossRef]
- Chavarria, D.; Benfeito, S.; Soares, P.; Lima, C.; Garrido, J.; Serra, P.; Soares-da-Silva, P.; Remia, F.; Oliveira, P.; Borges, F. Boosting caffeic acid performance as antioxidant and monoamine oxidase B/catechol-O-methyltransferase inhibitor. Eur. J. Med. Chem. 2022, 243, 114740. [Google Scholar] [CrossRef]
- Park, J.H.; Ju, Y.H.; Choi, J.W.; Song, H.J.; Jang, B.K.; Woo, J.; Chun, H.; Kim, H.J.; Shin, S.J.; Yarishkin, O.; et al. Newly developed reversible MAO-B inhibitor circumvents the shortcomings of irreversible inhibitors in Alzheimer’s disease. Sci. Adv. 2019, 5, eaav0316. [Google Scholar] [CrossRef]
- Deeks, E. Safinamide: First Global Approval. Drugs 2015, 75, 705–711. [Google Scholar] [CrossRef]
- Parvez, S.; Long, M.J.C.; Poganik, J.R.; Aye, Y. Redox Signaling by Reactive Electrophiles and Oxidants. Chem. Rev. 2018, 118, 8798–8888. [Google Scholar] [CrossRef] [PubMed]
- Simoni, E.; Serafini, M.M.; Caporaso, R.; Marchetti, C.; Racchi, M.; Minarini, A.; Bartolini, M.; Lanni, C.; Rosini, M. Targeting the Nrf2/Amyloid-Beta Liaison in Alzheimer’s Disease: A Rational Approach. ACS Chem. Neurosci. 2017, 8, 1618–1627. [Google Scholar] [CrossRef]
- Wu, Y.; Shi, Y.; Zheng, X.; Dang, Y.; Zhu, C.; Zhang, R.; Fu, Y.; Zhou, T.; Li, J. Lipophilic ferulic acid derivatives protect PC12 cells against oxidative damage via modulating beta-amyloid aggregation and activating Nrf2 enzymes. Food Funct. 2020, 11, 4707–4718. [Google Scholar] [CrossRef] [PubMed]
- Martin-Camara, O.; Arribas, M.; Wells, G.; Morales-Tenorio, M.; Martin-Requero, A.; Porras, G.; Martinez, A.; Giorgi, G.; Lopez-Alvarado, P.; Lastres-Becker, I.; et al. Multitarget Hybrid Fasudil Derivatives as a New Approach to the Potential Treatment of Amyotrophic Lateral Sclerosis. J. Med. Chem. 2022, 65, 1867–1882. [Google Scholar] [CrossRef] [PubMed]
- Rosini, M.; Simoni, E.; Caporaso, R.; Basagni, F.; Catanzaro, M.; Abu, I.F.; Fagiani, F.; Fusco, F.; Masuzzo, S.; Albani, D.; et al. Merging memantine and ferulic acid to probe connections between NMDA receptors, oxidative stress and amyloid-β peptide in Alzheimer’s disease. Eur. J. Med. Chem. 2019, 180, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kim, W.; Yum, S.; Hong, S.; Oh, J.; Lee, J.; Kwak, M.; Park, E.; Na, D.; Jung, Y. Caffeic acid phenethyl ester activation of Nrf2 pathway is enhanced under oxidative state: Structural analysis and potential as a pathologically targeted therapeutic agent in treatment of colonic inflammation. Free Radic. Biol. Med. 2013, 65, 552–562. [Google Scholar] [CrossRef]
- Pagnotta, S.; Tramutola, A.; Barone, E.; Di Domenico, F.; Pittalà, V.; Salerno, L.; Folgiero, V.; Caforio, M.; Locatelli, F.; Petrini, S.; et al. CAPE and its synthetic derivative VP961 restore BACH1/NRF2 axis in Down Syndrome. Free Radic. Biol. Med. 2022, 183, 1–13. [Google Scholar] [CrossRef]
- Zakaria, A.; Rady, M.; Mahran, L.; Abou-Aisha, K. Pioglitazone Attenuates Lipopolysaccharide-Induced Oxidative Stress, Dopaminergic Neuronal Loss and Neurobehavioral Impairment by Activating Nrf2/ARE/HO-1. Neurochem. Res. 2019, 44, 2856–2868. [Google Scholar] [CrossRef]
- Zhao, Y.; Lutzen, U.; Gohlke, P.; Jiang, P.; Herdegen, T.; Culman, J. Neuroprotective and antioxidative effects of pioglitazone in brain tissue adjacent to the ischemic core are mediated by PI3K/Akt and Nrf2/ARE pathways. J. Mol. Med.-Jmm 2021, 99, 1073–1083. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Z.; Liu, J.; Hu, J.; Chen, H.; Li, W.; Hai, C. Double antioxidant activities of rosiglitazone against high glucose-induced oxidative stress in hepatocyte. Toxicol. Vitr. 2011, 25, 839–847. [Google Scholar] [CrossRef]
- Shahidi, F.; Chandrasekara, A. Hydroxycinnamates and their in vitro and in vivo antioxidant activities. Phytochem. Rev. 2010, 9, 147–170. [Google Scholar] [CrossRef]
- Zhang, H.; Yin, M.; Huang, L.; Wang, J.; Gong, L.; Liu, J.; Sun, B. Evaluation of the Cellular and Animal Models for the Study of Antioxidant Activity: A Review. J. Food Sci. 2017, 82, 278–288. [Google Scholar] [CrossRef] [PubMed]
- Bartolomei, M.; Bollati, C.; Bellumori, M.; Cecchi, L.; Cruz-Chamorro, I.; Santos-Sanchez, G.; Ranaldi, G.; Ferruzza, S.; Sambuy, Y.; Arnoldi, A.; et al. Extra Virgin Olive Oil Phenolic Extract on Human Hepatic HepG2 and Intestinal Caco-2 Cells: Assessment of the Antioxidant Activity and Intestinal Trans-Epithelial Transport. Antioxidants 2021, 10, 118. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, N.; Bennis, K.; Vivier, D.; Pereira, V.; Chatelain, F.; Chapuy, E.; Deokar, H.; Busserolles, J.; Lesage, F.; Eschalier, A.; et al. Synthesis and structure-activity relationship study of substituted caffeate esters as antinociceptive agents modulating the TREK-1 channel. Eur. J. Med. Chem. 2014, 75, 391–402. [Google Scholar] [CrossRef] [PubMed]
- Herraiz, T.; Flores, A.; Fernández, L. Analysis of monoamine oxidase (MAO) enzymatic activity by high-performance liquid chromatography-diode array detection combined with an assay of oxidation with a peroxidase and its application to MAO inhibitors from foods and plants. J. Chromatogr. B-Anal. Technol. Biomed. Life Sci. 2018, 1073, 136–144. [Google Scholar] [CrossRef]
- Santillo, M.F.; Liu, Y.; Ferguson, M.; Vohra, S.N.; Wiesenfeld, P.L. Inhibition of monoamine oxidase (MAO) by β-carbolines and their interactions in live neuronal (PC12) and liver (HuH-7 and MH1C1) cells. Toxicol. Vitr. 2014, 28, 403–410. [Google Scholar] [CrossRef]
- Di Paolo, M.L.; Cozza, G.; Milelli, A.; Zonta, F.; Sarno, S.; Minniti, E.; Ursini, F.; Rosini, M.; Minarini, A. Benextramine and derivatives as novel human monoamine oxidases inhibitors: An integrated approach. FEBS J. 2019, 286, 4995–5015. [Google Scholar] [CrossRef]
- Copeland, R.A. Evaluation of Enzyme Inhibitors in Drug Discovery: A Guide for Medicinal Chemists and Pharmacologists; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013; pp. 57–121. [Google Scholar]
- Holdgate, G.; Meek, T.; Grimley, R. Mechanistic enzymology in drug discovery: A fresh perspective. Nat. Rev. Drug Discov. 2018, 17, 115–132. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cpd. | hMAO-B Ki (µM) | hMAO-A Ki (µM) | SI a |
|---|---|---|---|
| Pioglitazone | 0.061 ± 0.018 | >>100 | >>1000 |
| CAPE | 1.8 ± 0.2 | 12 ± 2 | 6.7 |
| 1 | 3.14 ± 0.20 | 25 ± 4 | 8.0 |
| 2 | 0.53 ± 0.17 | 35 ± 4 | 66 |
| 3 | 0.99 ± 0.15 | 32 ± 2 | 32.3 |
| 4 | 2.5 ± 1.0 | 39 ± 5 | 15.6 |
| 5 | 3.3 ± 1.0 | 47 ± 4 | 14.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Basagni, F.; Di Paolo, M.L.; Cozza, G.; Dalla Via, L.; Fagiani, F.; Lanni, C.; Rosini, M.; Minarini, A. Double Attack to Oxidative Stress in Neurodegenerative Disorders: MAO-B and Nrf2 as Elected Targets. Molecules 2023, 28, 7424. https://doi.org/10.3390/molecules28217424
Basagni F, Di Paolo ML, Cozza G, Dalla Via L, Fagiani F, Lanni C, Rosini M, Minarini A. Double Attack to Oxidative Stress in Neurodegenerative Disorders: MAO-B and Nrf2 as Elected Targets. Molecules. 2023; 28(21):7424. https://doi.org/10.3390/molecules28217424
Chicago/Turabian StyleBasagni, Filippo, Maria Luisa Di Paolo, Giorgio Cozza, Lisa Dalla Via, Francesca Fagiani, Cristina Lanni, Michela Rosini, and Anna Minarini. 2023. "Double Attack to Oxidative Stress in Neurodegenerative Disorders: MAO-B and Nrf2 as Elected Targets" Molecules 28, no. 21: 7424. https://doi.org/10.3390/molecules28217424