
Molecular Dynamics Simulations of Amylose- and Cellulose-Based Selectors and Related Enantioseparations in Liquid Phase Chromatography




Abstract
:1. Introduction
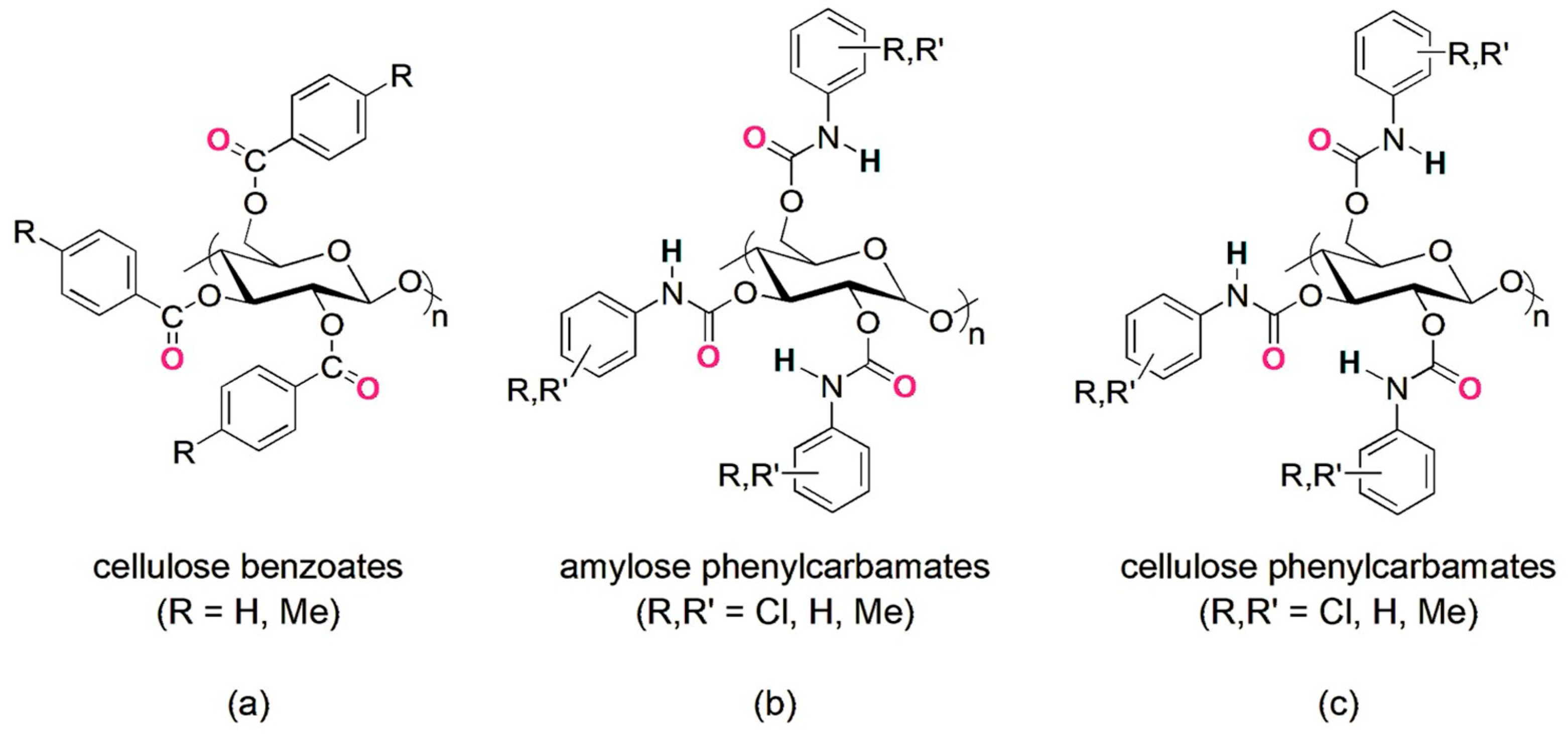
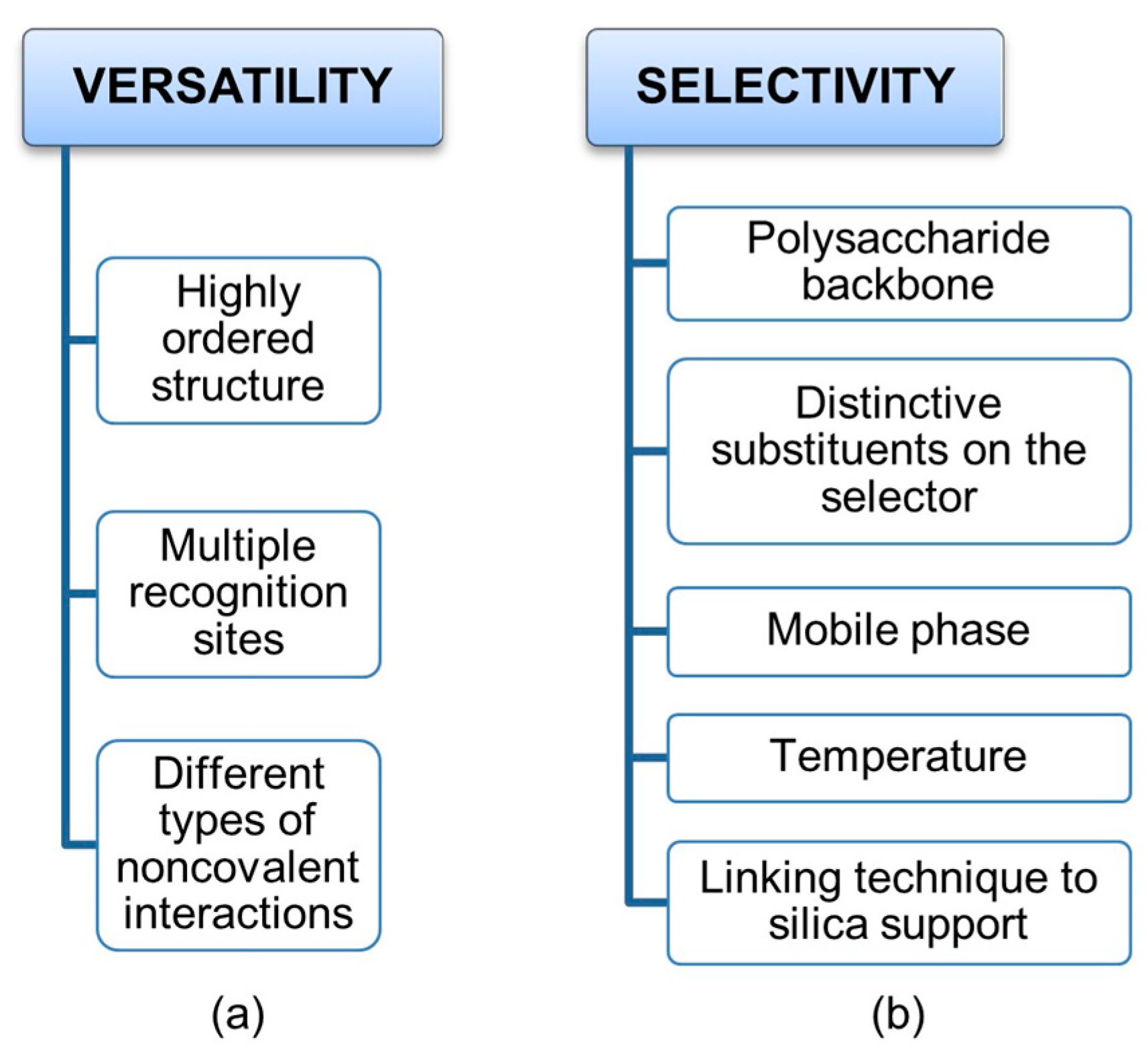
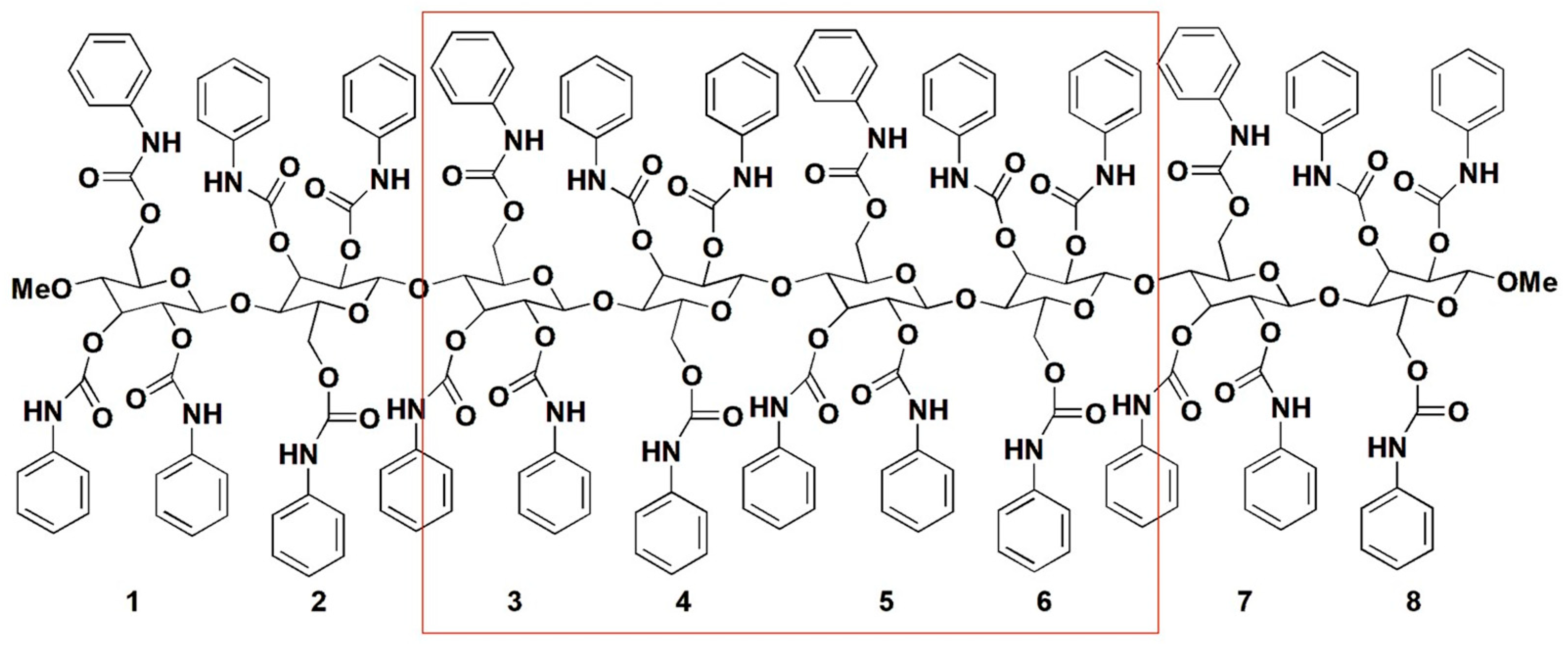
2. Polysaccharide-Based Selectors

3. Molecular Dynamics: A Short Description of the Technique
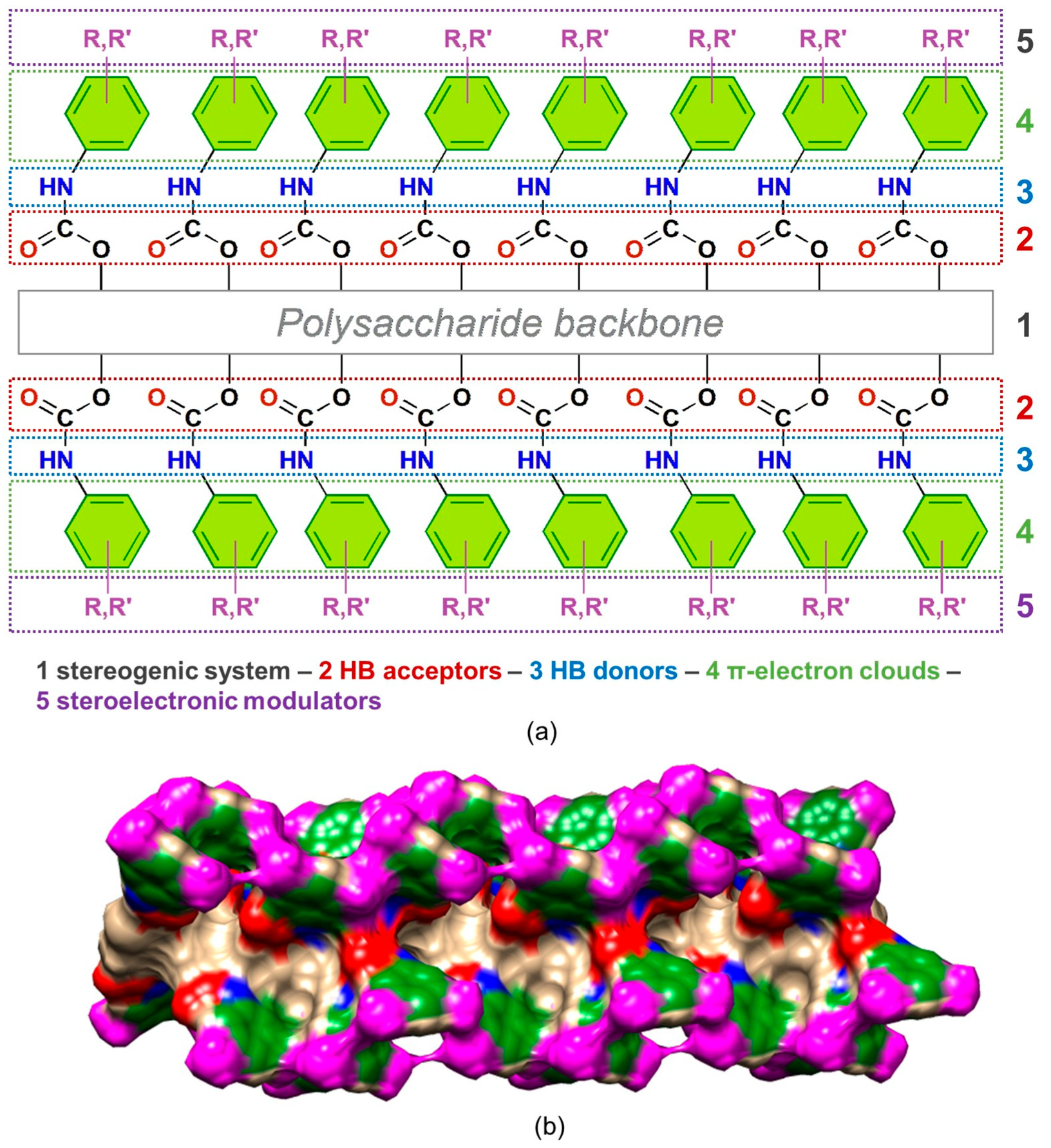
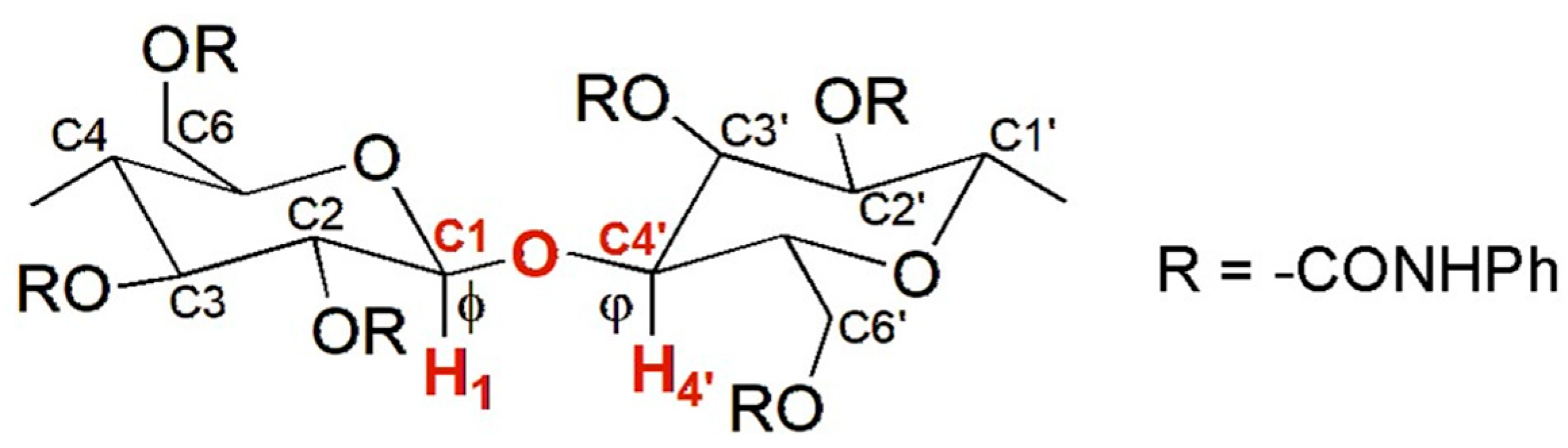
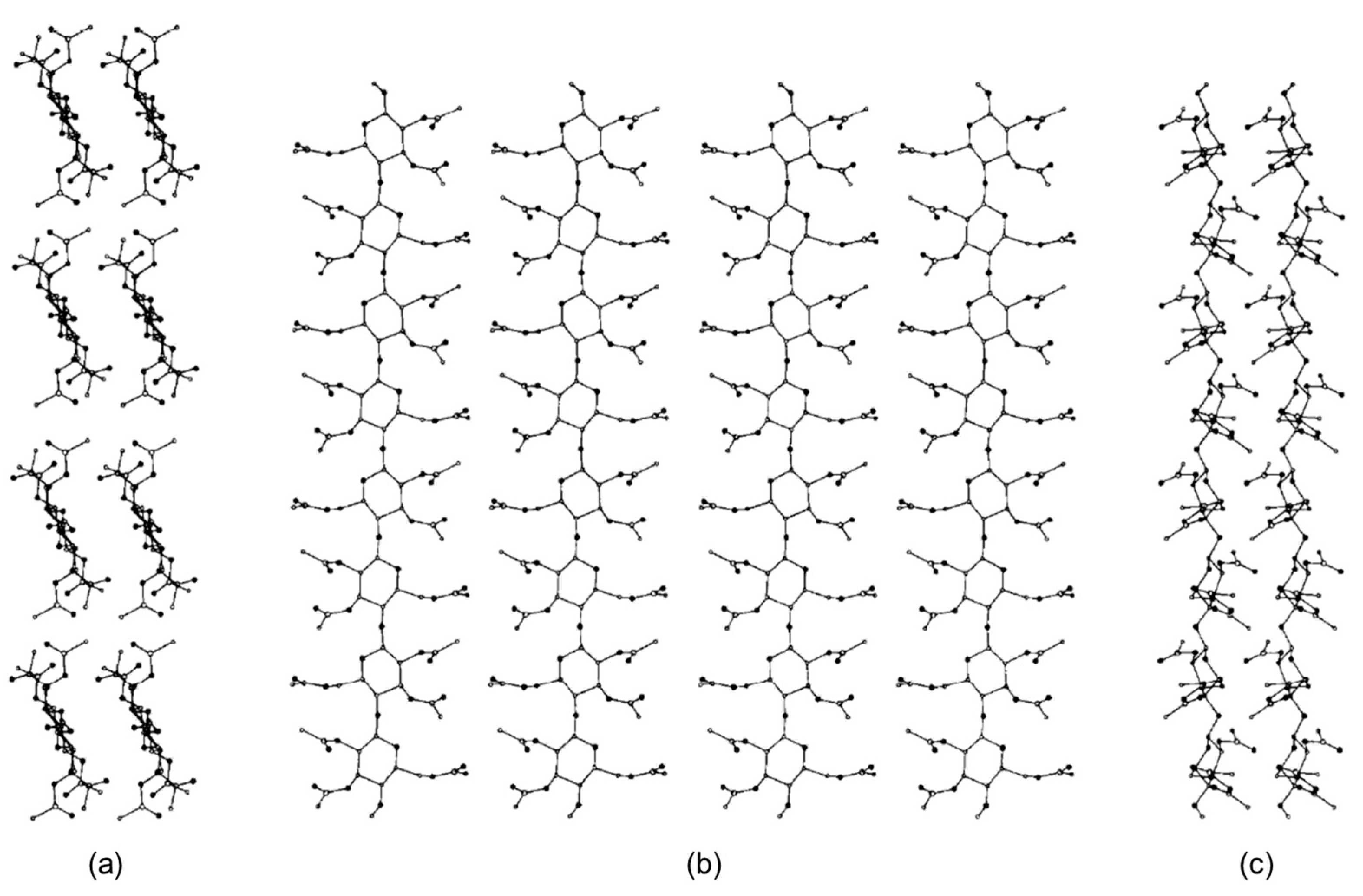
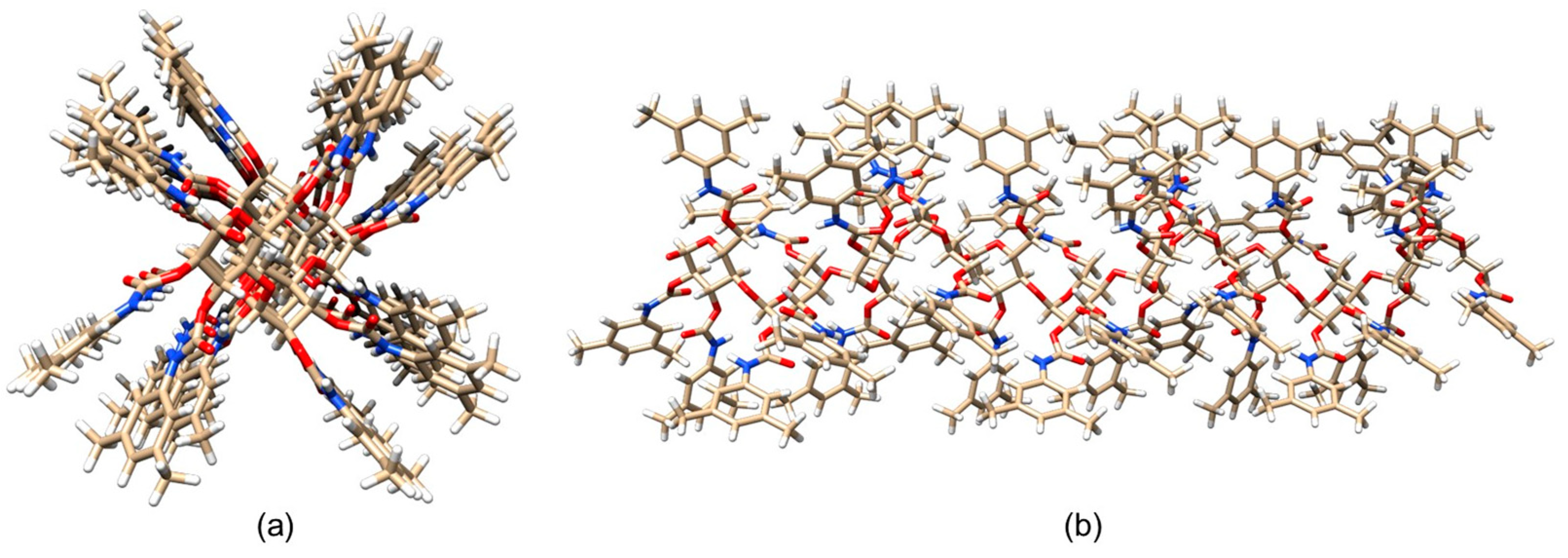
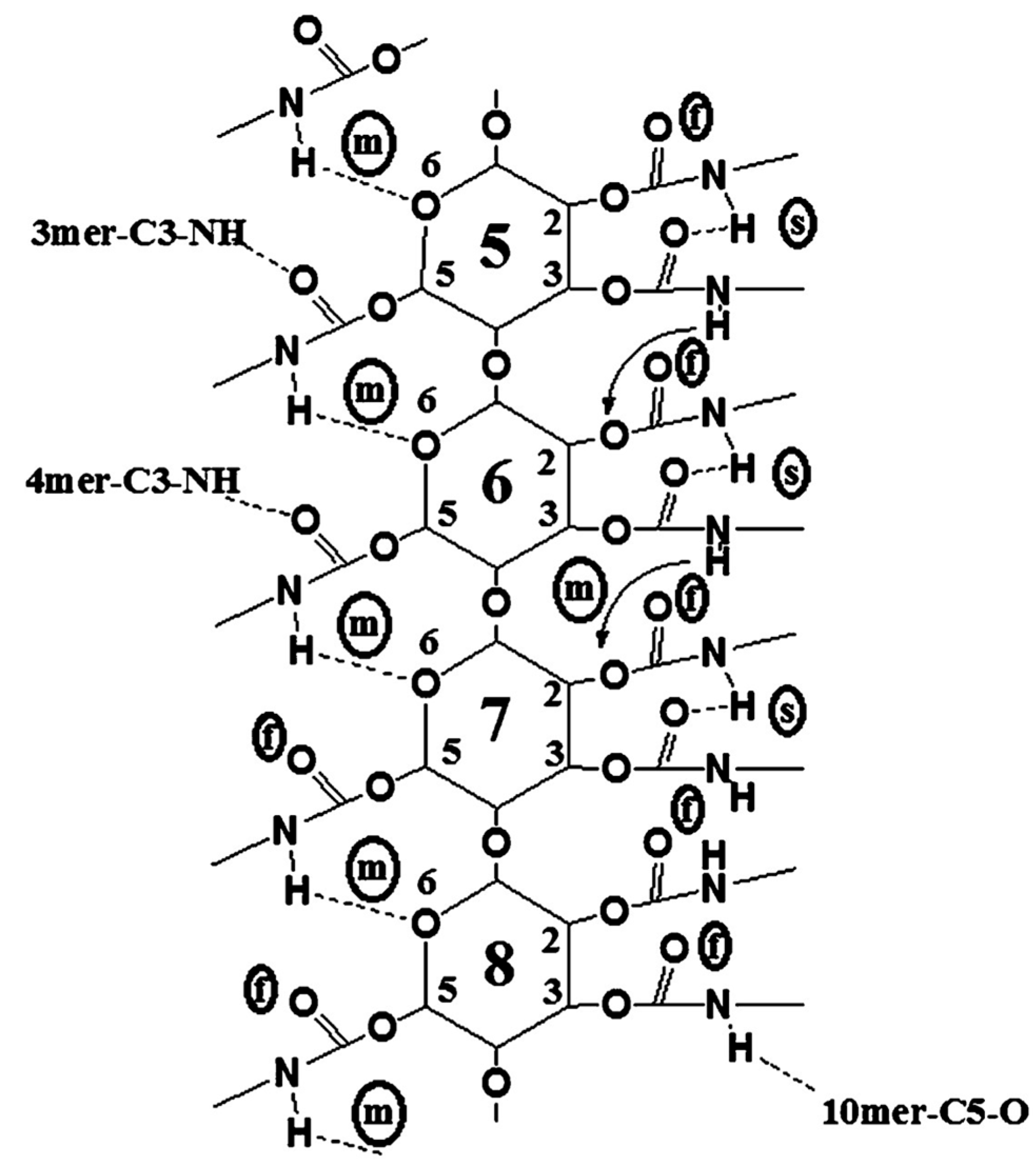
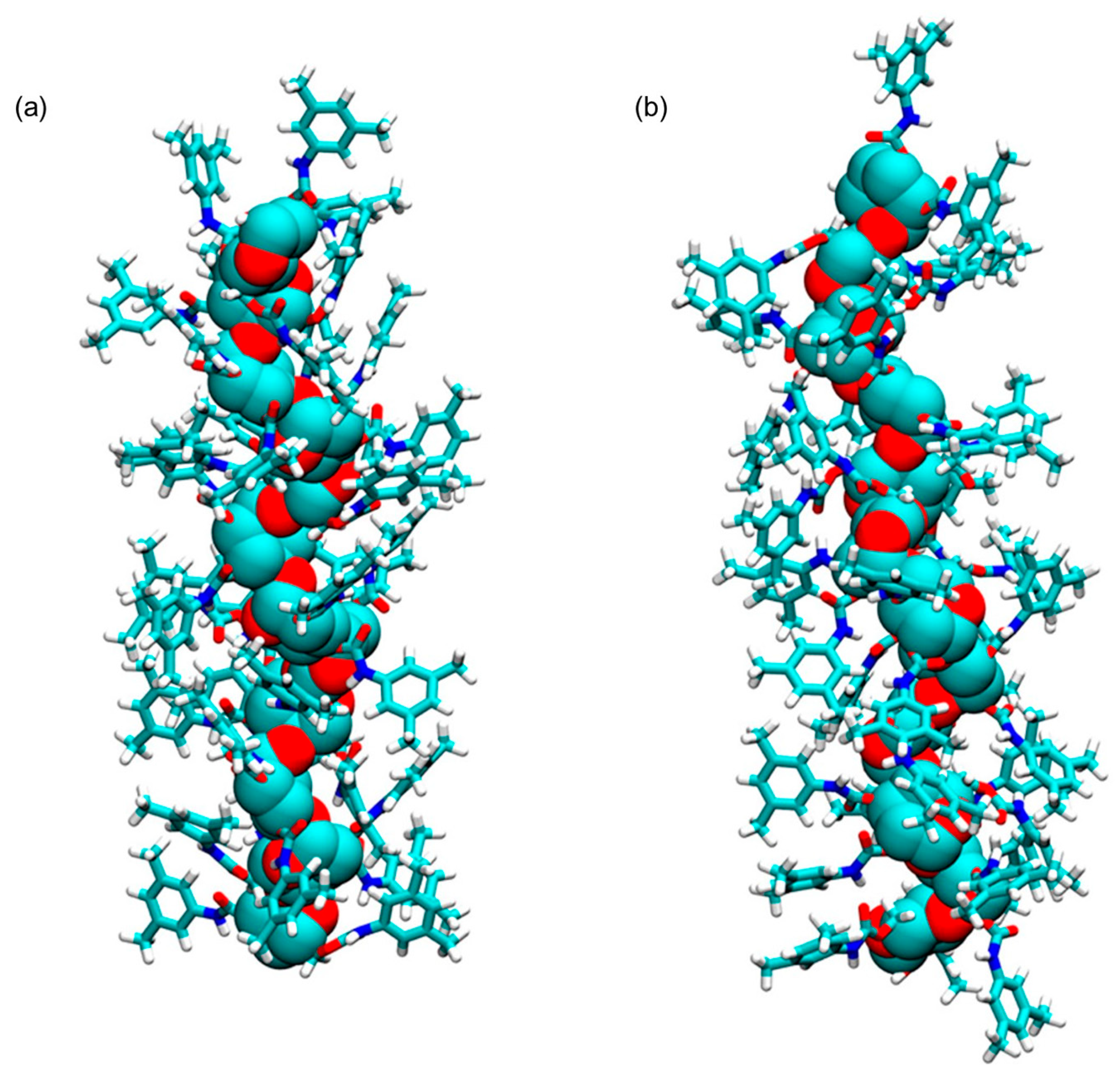
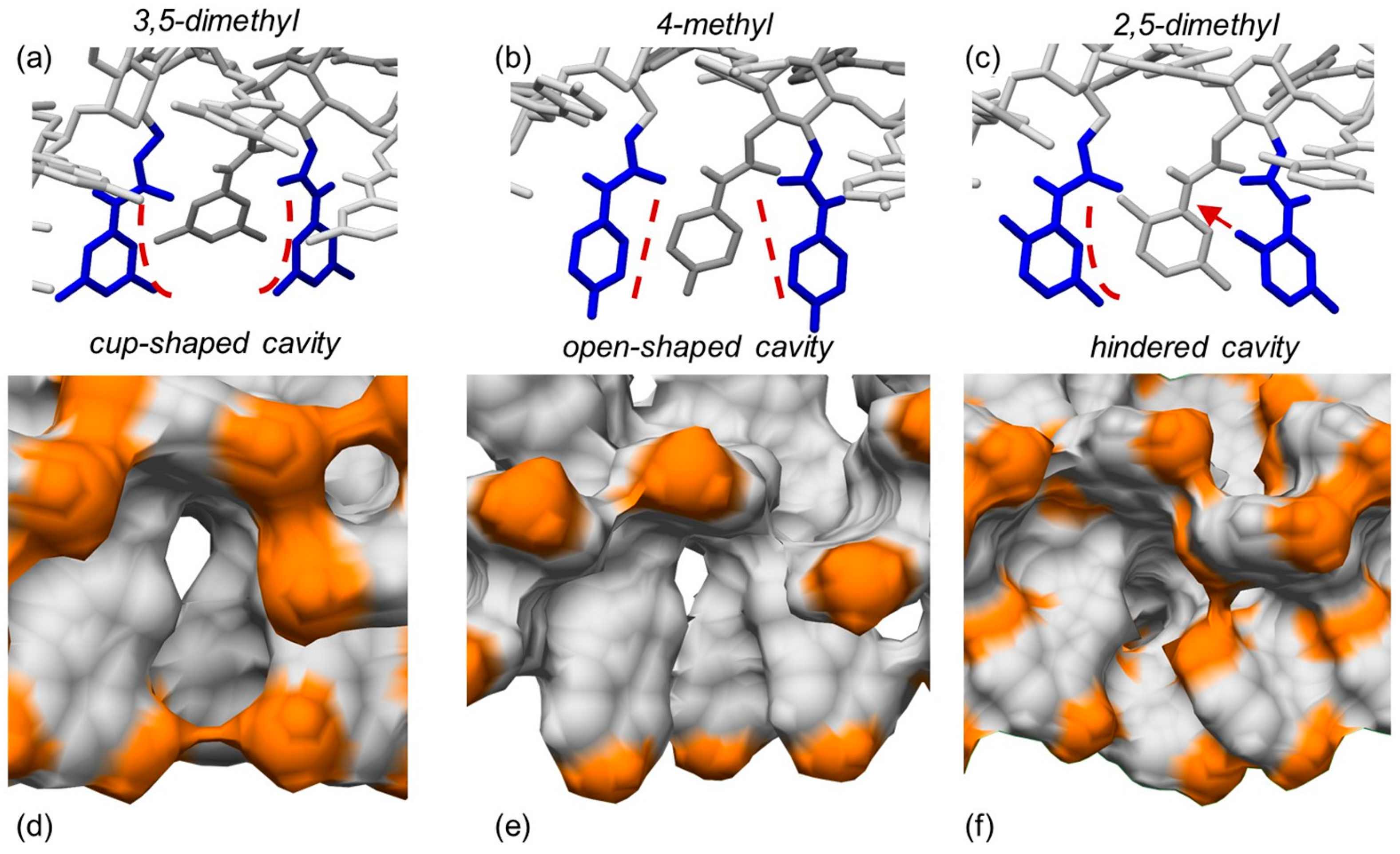
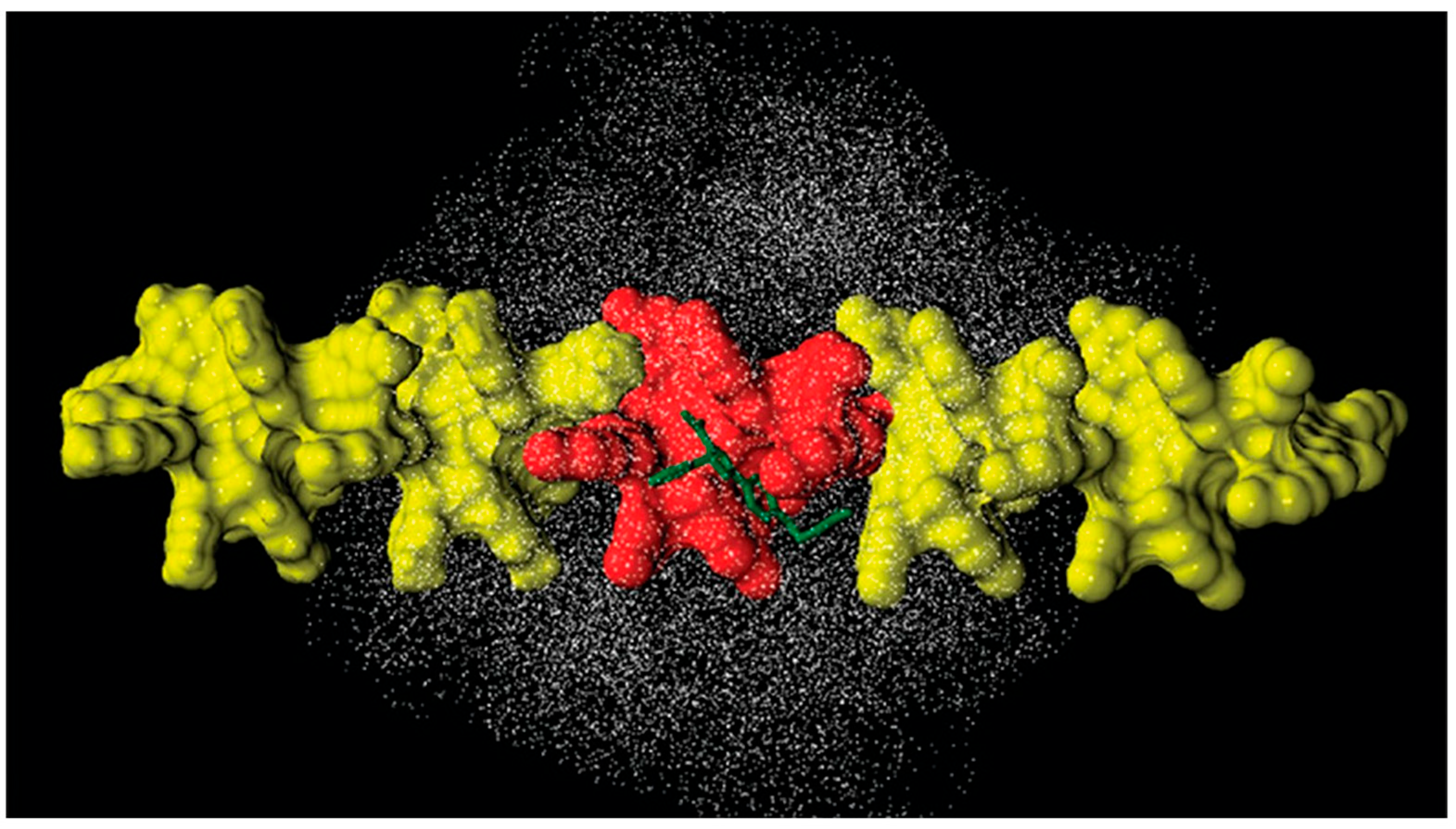
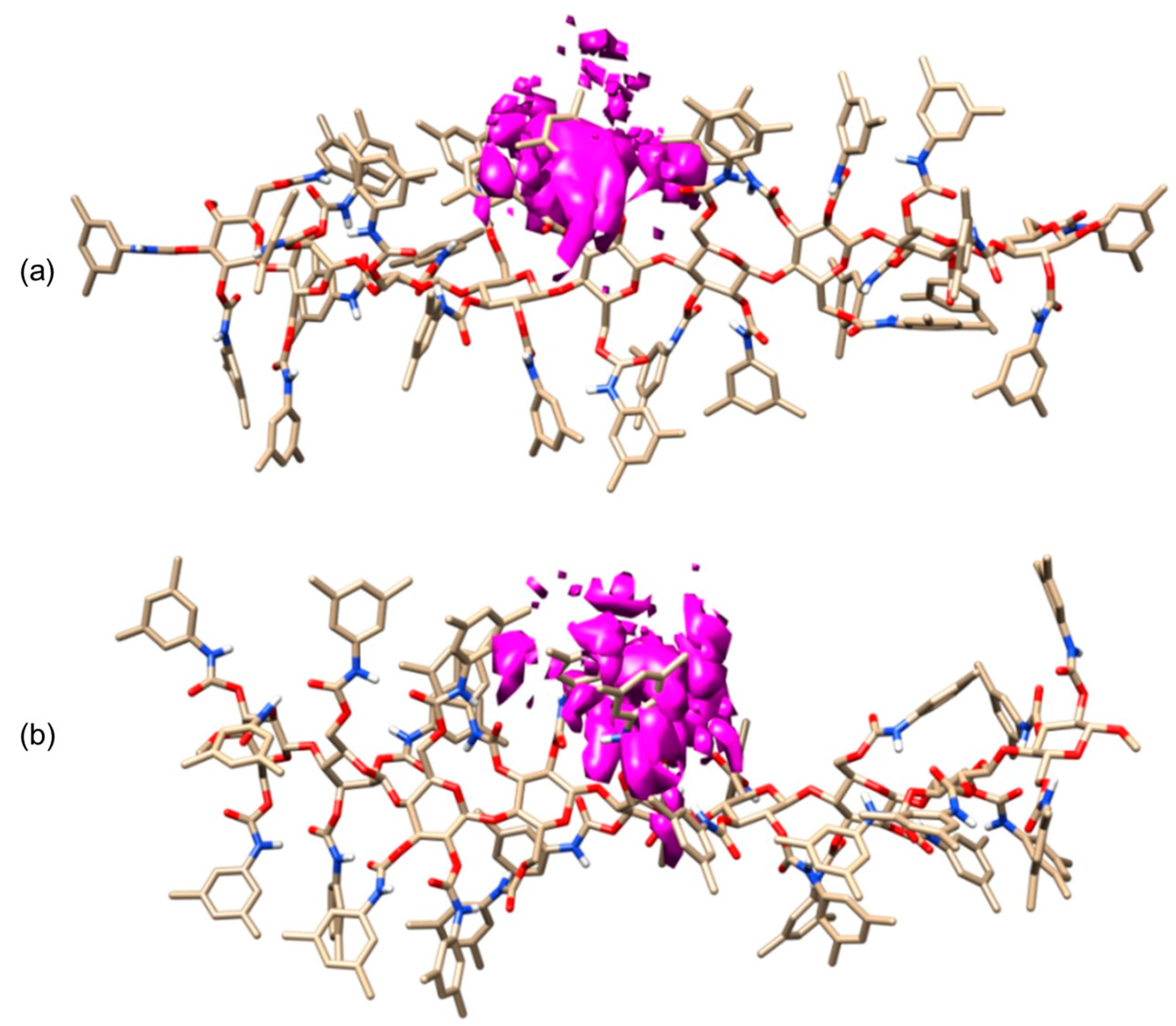
4. Application of MD to Model Polysaccharide-Based Selectors
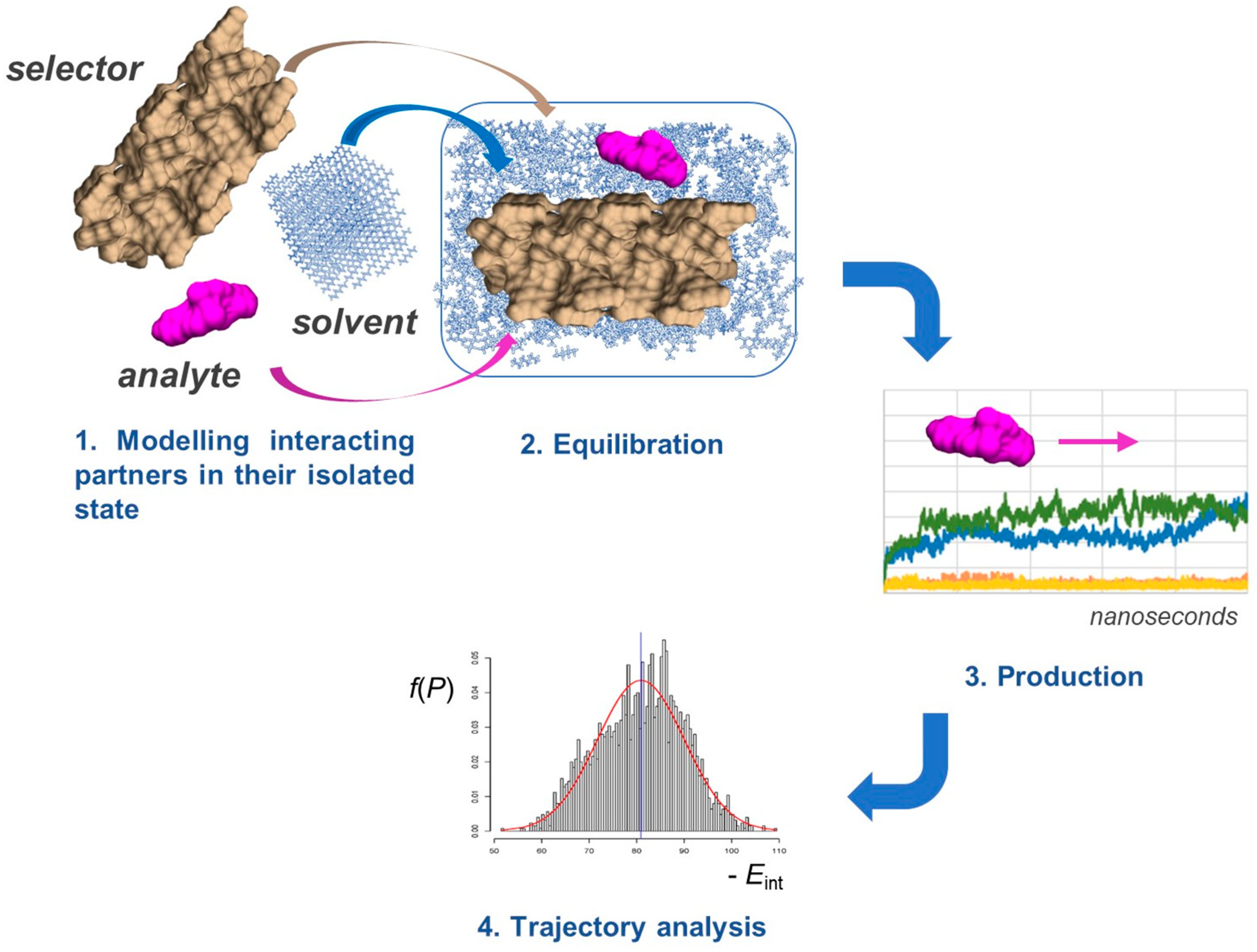
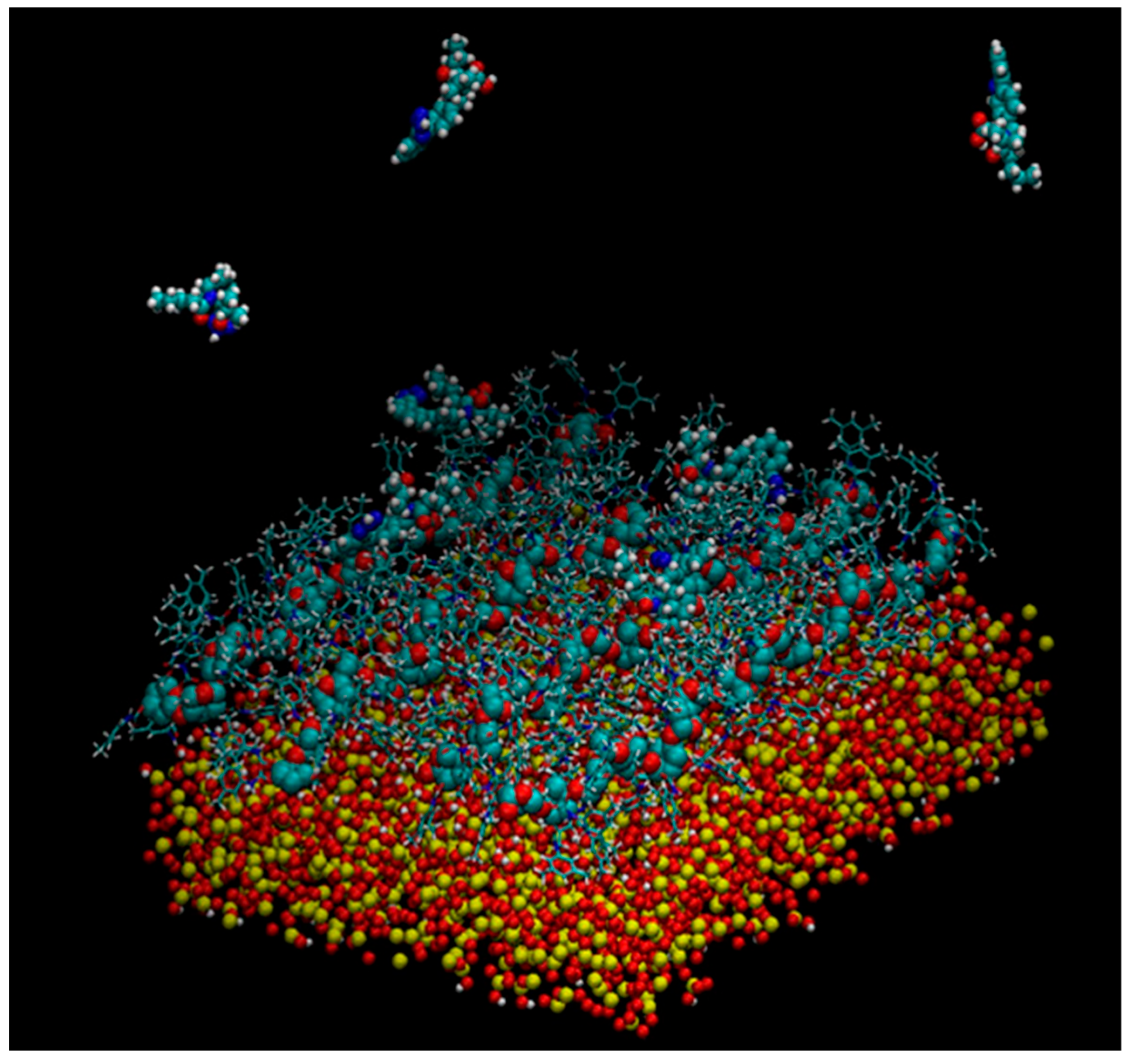
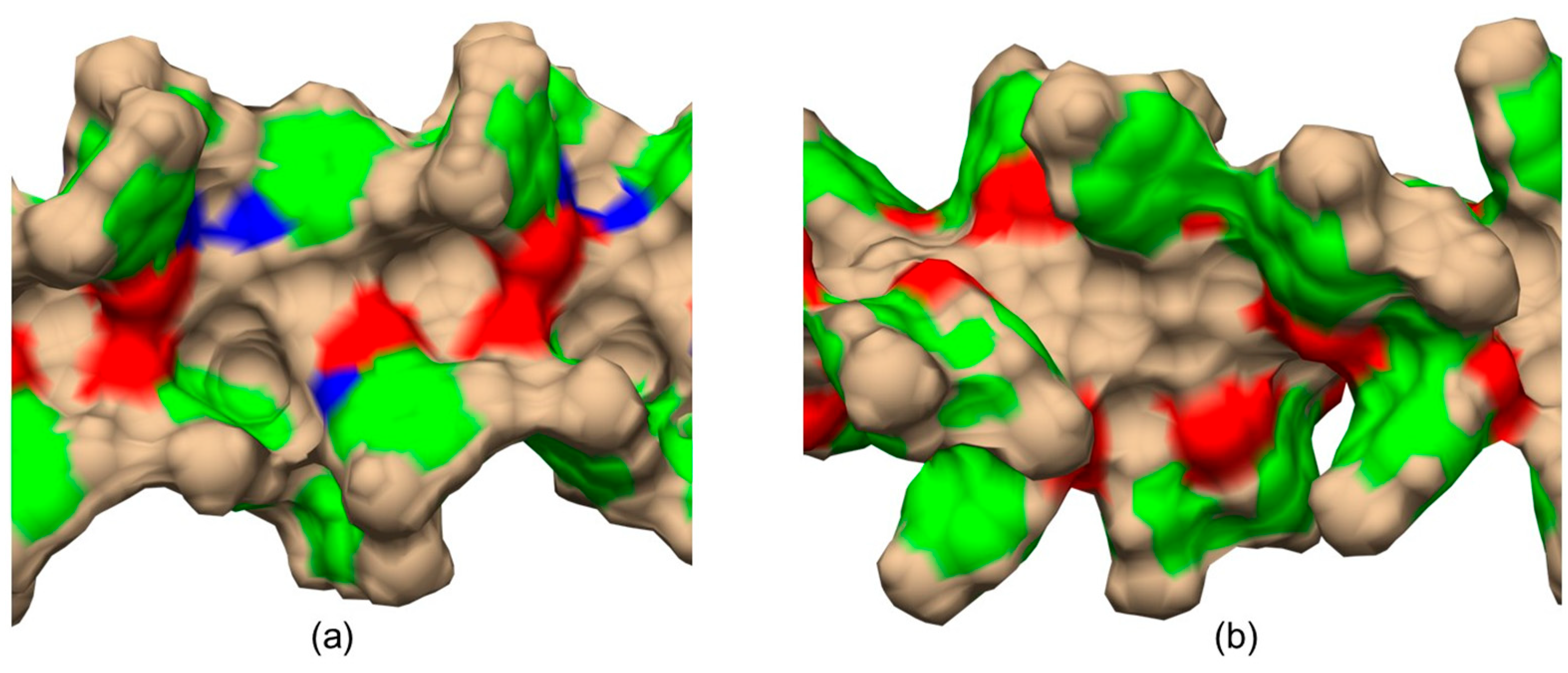
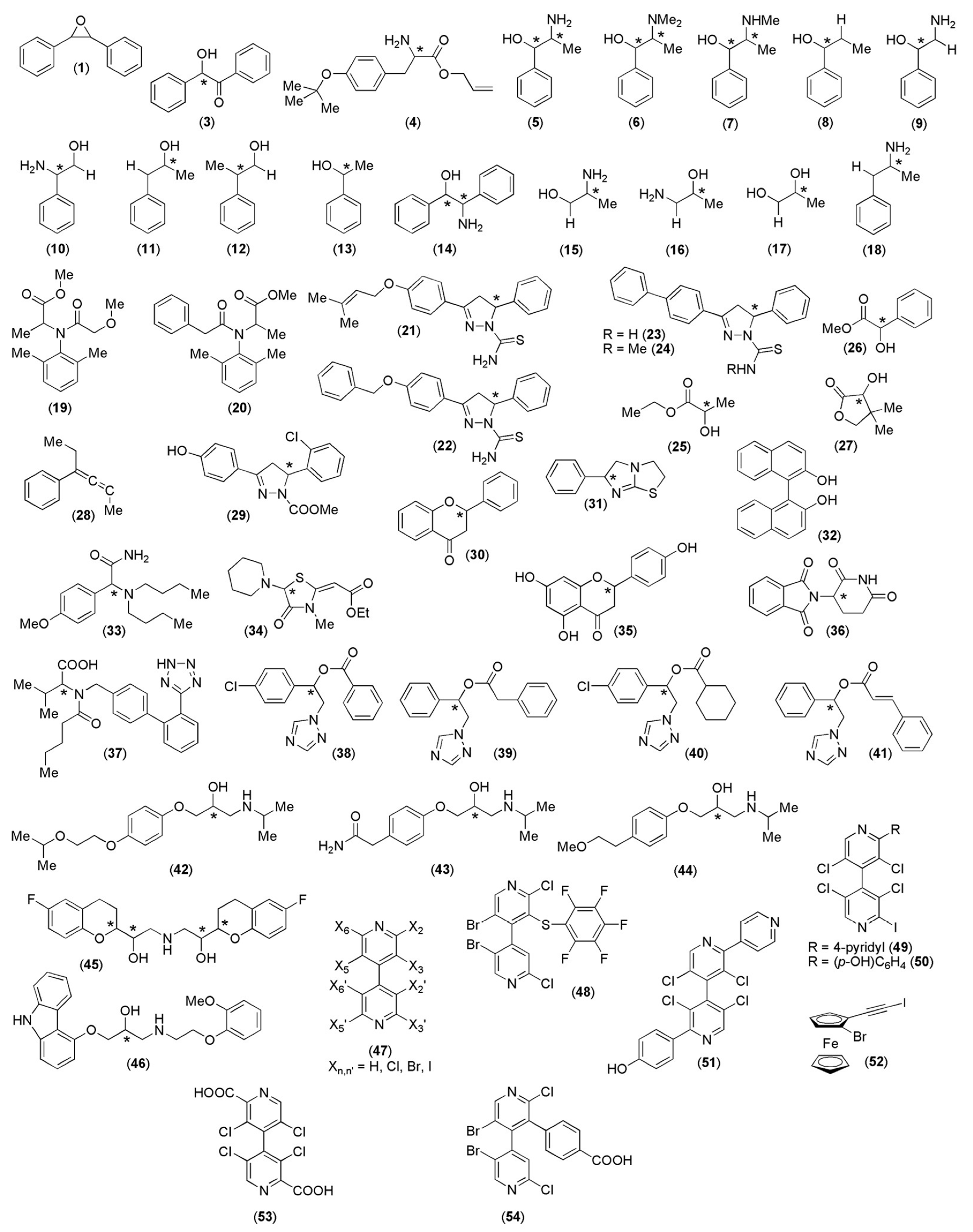
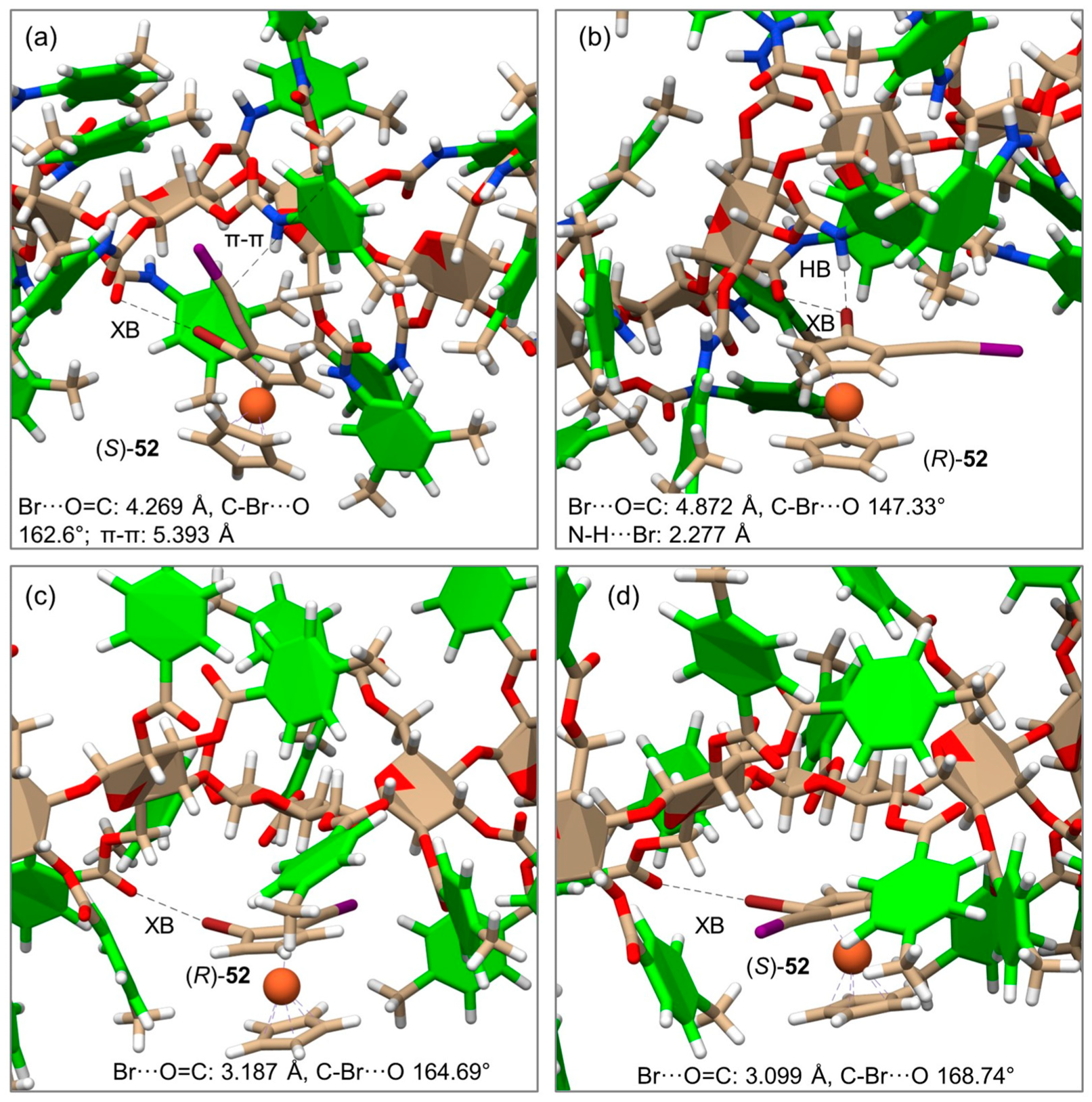
5. Application of MD to Model Enantioseparations Promoted by Polysaccharide-Based Selectors
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Peluso, P.; Dessì, A.; Dallocchio, R.; Mamane, V.; Cossu, S. Recent studies of docking and molecular dynamics simulation for liquid-phase enantioseparations. Electrophoresis 2019, 40, 1881–1896. [Google Scholar] [PubMed]
- Peluso, P.; Mamane, V.; Dallocchio, R.; Dessì, A.; Cossu, S. Noncovalent interactions in high-performance liquid chromatography enantioseparations on polysaccharide-based chiral selectors. J. Chromatogr. A 2020, 1623, 461202. [Google Scholar]
- Sardella, R.; Camaioni, E.; Macchiarulo, A.; Gioiello, A.; Marinozzi, M.; Carotti, A. Computational studies in enantioselectiveliquid chromatography: Forty years of evolution in docking- and molecular dynamics-based simulations. Trends Anal. Chem. 2020, 122, 115703. [Google Scholar]
- De Gauquier, P.; Vanommeslaeghe, K.; Vander Heyden, Y.; Mangelings, D. Modelling approaches for chiral chromatography on polysaccharide-based and macrocyclic antibiotic chiral selectors: A review. Anal. Chim. Acta 2022, 1198, 338861. [Google Scholar] [PubMed]
- Peluso, P.; Chankvetadze, B. Recognition in the domain of molecular chirality: From noncovalent interactions to separation of enantiomers. Chem. Rev. 2022, 122, 13235–13400. [Google Scholar]
- Peluso, P.; Chankvetadze, B. Fundamentals of Enantioselective Liquid Chromatography. In Liquid Chromatography: Fundamentals and Instrumentation, 3rd ed.; Fanali, S., Chankvetadze, B., Haddad, P.R., Poole, C.F., Riekkola, M.-L., Eds.; Elsevier: Amsterdam, The Netherlands, 2023; Volume 1, Chapter 16; pp. 383–439. [Google Scholar]
- Okamoto, Y.; Kawashima, M.; Hatada, K. Useful chiral packing materials for high-performance liquid chromatographic resolution of enantiomers: Phenyl-carbamates of polysaccharides coated on silica gel. J. Am. Chem. Soc. 1984, 106, 5357–5359. [Google Scholar]
- Okamoto, Y.; Kawashima, M.; Hatada, K. Chromatographic resolution: XI. Controlled chiral recognition of cellulose triphenylcarbamate derivatives supported on silica gel. J. Chromatogr. A 1986, 363, 173–186. [Google Scholar]
- Okamoto, Y.; Aburatani, R.; Hatada, K. Chromatographic chiral resolution: XIV. Cellulose tribenzoate derivatives as chiral stationary phases for high-performance liquid chromatography. J. Chromatogr. A 1987, 389, 95–102. [Google Scholar]
- Okamoto, Y.; Aburatani, R.; Fukumoto, T.; Hatada, K. Useful chiral stationary phases for HPLC. Amylose tris(3,5-dimethylphenylcarbamate) and tris(3,5-dichlorophenylcarbamate) supported on silica gel. Chem. Lett. 1987, 16, 1857–1860. [Google Scholar]
- Okamoto, Y.; Aburatani, R.; Kaida, Y.; Hatada, K. Direct optical resolution of carboxylic acids by chiral HPLC on tris(3,5-dimethylphenylcarbamate)s of cellulose and amylose. Chem. Lett. 1988, 17, 1125–1128. [Google Scholar]
- Chankvetadze, B.; Yashima, E.; Okamoto, Y. Chloromethylphenylcarbamate derivatives of cellulose as chiral stationary phases for high-performance liquid chromatography. J. Chromatogr. A 1994, 670, 39–49. [Google Scholar]
- Chankvetadze, B.; Yashima, E.; Okamoto, Y. Dimethyl-, dichloro- and chloromethylphenylcarbamates of amylose as chiral stationary phases for high-performance liquid chromatography. J. Chromatogr. A 1995, 694, 101–109. [Google Scholar]
- Chankvetadze, B.; Chankvetadze, L.; Sidamonidze, S.; Kasashima, E.; Yashima, E.; Okamoto, Y. 3-Fluoro-, 3-chloro- and 3-bromo-5-methylphenylcarbamates of cellulose and amylose as chiral stationary phases for high-performance liquid chromatographic enantioseparation. J. Chromatogr. A 1997, 787, 67–77. [Google Scholar]
- Okamoto, Y.; Yashima, E. Polysaccharide derivatives for chromatographic separation of enantiomers. Angew. Chem. Int. Ed. 1998, 37, 1020–1043. [Google Scholar]
- Chankvetadze, B. Recent developments on polysaccharide-based chiral stationary phases for liquid-phase separation of enantiomers. J. Chromatogr. A 2012, 1269, 26–51. [Google Scholar] [PubMed]
- Chankvetadze, B. Polysaccharide-based chiral stationary phases for enantioseparations by high-performance liquid chromatography: An overview. In Chiral Separations: Methods and Protocols, Methods in Molecular Biology; Scriba, G.K.E., Ed.; Springer Science + Business Media, LLC, part of Springer Nature: New York, NY, USA, 2019; Volume 1985, pp. 93–126. [Google Scholar]
- Chankvetadze, B. Recent trends in preparation, investigation and application of polysaccharide-based chiral stationary phases for separation of enantiomers in high-performance liquid chromatography. Trends Anal. Chem. 2020, 122, 115709. [Google Scholar]
- Wang, F.; O’Brien, T.; Dowling, T.; Bicker, G.; Wyvratt, J. Unusual effect of column temperature on chromatographic enantioseparation of dihydropyrimidinone acid and methyl ester on amylose chiral stationary phase. J. Chromatogr. A 2002, 958, 69–77. [Google Scholar]
- Wang, F.; Wenslow, R.M., Jr.; Dowling, T.M.; Mueller, K.T.; Santos, I.; Wyvratt, J.M. Characterization of a thermally induced irreversible conformational transition of amylose tris(3,5-dimethylphenylcarbamate) chiral stationary phase in enantioseparation of dihydropyrimidinone acid by quasi-equilibrated liquid chromatography and solid-state NMR. Anal. Chem. 2003, 75, 5877–5885. [Google Scholar]
- Matarashvili, I.; Kobidze, G.; Chelidze, A.; Dolidze, G.; Beridze, N.; Jibuti, G.; Farkas, T.; Chankvetadze, B. The effect of temperature on the separation of enantiomers with coated and covalently immobilized polysaccharide-based chiral stationary phases. J. Chromatogr. A 2019, 1599, 172–179. [Google Scholar]
- Kasat, R.B.; Wang, N.-H.L.; Franses, E.I. Experimental probing and modeling of key sorbent-solute interactions of norephedrine enantiomers with polysaccharide-based chiral stationary phases. J. Chromatogr. A 2008, 1190, 110–119. [Google Scholar]
- Peluso, P.; Chankvetadze, B. The molecular bases of chiral recognition in 2-(benzylsulfinyl)benzamide enantioseparation. Anal. Chim. Acta 2021, 1141, 194–205. [Google Scholar] [CrossRef]
- Shibata, T.; Shinkura, S.; Ohnishi, A.; Ueda, K. Achiral molecular recognition of aromatic position isomers by polysaccharide-based CSPs in relation to chiral recognition. Molecules 2017, 22, 38. [Google Scholar] [CrossRef]
- Ianni, F.; Cerra, B.; Moroni, G.; Varfaj, I.; Di Michele, A.; Gioiello, A.; Carotti, A.; Sardella, R. Combining molecular modeling approaches to establish the chromatographic enantiomer elution order in the absence of pure enantiomeric standards: A study case with two tetracyclic quinolines. Sep. Sci. Plus 2022, 5, 662–670. [Google Scholar] [CrossRef]
- Nguyen, B.T.; Choi, Y.J.; Kim, K.H.; Song, G.Y.; Kim, H.M.; Kang, J.S. Chiral separation and molecular modeling study of decursinol and its derivatives using polysaccharide-based chiral stationary phases. J. Chromatogr. A 2023, 1705, 464165. [Google Scholar] [CrossRef] [PubMed]
- Vogt, U.; Zugenmaier, P. Structural models for some liquid crystalline cellulose derivatives. Berichte Bunsenges. Phys. Chem. 1985, 89, 1217–1224. [Google Scholar] [CrossRef]
- Yashima, E.; Yamada, M.; Kaida, Y.; Okamoto, Y. Computational studies on chiral discrimination mechanism of cellulose trisphenylcarbamate. J. Chromatogr. A 1995, 694, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, C.; Yashima, E.; Okamoto, Y. Structural analysis of amylose tris(3,5-dimethylphenylcarbamate) by NMR relevant to its chiral recognition mechanism in HPLC. J. Am. Chem. Soc. 2002, 124, 12583–12589. [Google Scholar] [CrossRef]
- Peluso, P.; Mamane, V.; Dessì, A.; Dallocchio, R.; Aubert, E.; Gatti, C.; Mangelings, D.; Cossu, S. Halogen bond in separation science: A critical analysis across experimental and theoretical results. J. Chromatogr. A 2020, 1616, 460788. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF chimera-A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Sechi, B.; Dessì, A.; Dallocchio, R.; Tsetskhladze, N.; Chankvetadze, B.; Pérez-Baeza, M.; Cossu, S.; Jibuti, G.; Mamane, V.; Peluso, P. Unravelling dispersion forces in liquid-phase enantioseparation. Part I: Impact of ferrocenyl versus phenyl groups. Anal. Chim. Acta 2023, 1278, 341725. [Google Scholar] [CrossRef]
- Ribeiro, J.; Tiritan, M.E.; Pinto, M.M.M.; Fernandes, C. Chiral stationary phases for liquid chromatography based on chitin- and chitosan-derived marine polysaccharides. Symmetry 2017, 9, 190. [Google Scholar] [CrossRef]
- Tang, S.; Liu, J.D.; Chen, W.; Huang, S.H.; Zhang, J.; Bai, Z.W. Performance comparison of chiral separation materials derived from N-cyclohexylcarbonyl and N-hexanoyl chitosans. J. Chromatogr. A 2018, 1532, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Lindsey, R.K.; Rafferty, J.L.; Eggimann, B.L.; Siepmann, J.I.; Schure, M.R. Molecular simulation studies of reversed-phase liquid chromatography. J. Chromatogr. A 2013, 1287, 60–82. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E., III; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The AMBER biomolecular simulation programs. J. Computat. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef]
- Brooks, B.R.; Brooks, C.L., III; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef] [PubMed]
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.E.; Klepeis, I.L.; Kolossváry, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. In Proceedings of the ACM/IEEE Conference on Supercomputing (SC06), Tampa, Florida, 11–17 November 2006.
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef]
- Thompson, A.P.; Aktulga, H.M.; Berger, R.; Bolintineanu, D.S.; Brown, W.M.; Crozier, P.S.; in ’t Veld, P.J.; Kohlmeyer, A.; Moore, S.G.; Nguyen, T.D.; et al. LAMMPS-A flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales. Comp. Phys. Comm. 2022, 271, 108171. [Google Scholar] [CrossRef]
- Lipkowitz, K.B. Theoretical studies of type II-V chiral stationary phases. J. Chromatogr. A 1995, 694, 15–37. [Google Scholar] [CrossRef]
- Peluso, P.; Landy, D.; Nakhhle, L.; Dallocchio, R.; Dessì, A.; Krait, S.; Salgado, A.; Chankvetadze, B.; Scriba, G.K.E. Isothermal titration calorimetry and molecular modeling study of the complex formation of daclatasvir by γ-cyclodextrin and trimethyl-β-cyclodextrin. Carbohydr. Polym. 2023, 313, 120870. [Google Scholar] [CrossRef]
- Dallocchio, R.; Dessì, A.; Solinas, M.; Arras, A.; Cossu, S.; Aubert, E.; Mamane, V.; Peluso, P. Halogen bond in high-performance liquid chromatography enantioseparations: Description, features and modelling. J. Chromatogr. A 2018, 1563, 71–81. [Google Scholar] [CrossRef]
- Anandakrishnan, R.; Drozdetski, A.; Walker, R.C.; Onufriev, A.V. Speed of conformational change: Comparing explicit and implicit solvent molecular dynamics simulations. Biophys. J. 2015, 108, 1153–1164. [Google Scholar] [CrossRef] [PubMed]
- Camilleri, P.; Murphy, J.A.; Saunders, M.R.; Thorpe, C.J. Molecular modelling studies and the chromatographic behaviour of oxiracetam and some closely related molecules. J. Comput. Aided Mol. Des. 1991, 5, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Peterson, M.A.; Lipkowitz, K.B. Structure and dynamics of cellulose triacetate. J. Mol. Struct. (Theochem) 1997, 395–396, 411–423. [Google Scholar] [CrossRef]
- Yamamoto, C.; Yashima, E.; Okamoto, Y. Computational studies on chiral discrimination mechanism of phenylcarbamate derivatives of cellulose. Bull. Chem. Soc. Jpn. 1999, 72, 1815–1825. [Google Scholar] [CrossRef]
- Ye, Y.K.; Bai, S.; Vyas, S.; Wirth, M.J. NMR and computational studies of chiral discrimination by amylose tris(3,5-dimethylphenylcarbamate). J. Phys. Chem. B 2007, 111, 1189–1198. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, D.; Wang, P.; Zhou, Z. Computational study of enantioseparation by amylose tris(3,5-dimethylphenylcarbamate)-based chiral stationary phase. J. Sep. Sci. 2010, 33, 3245–3255. [Google Scholar] [CrossRef]
- Tsui, H.-W.; Wang, N.-H.L.; Franses, E.I. Chiral recognition mechanism of acyloin-containing chiral solutes by amylose tris[(S)-α-methylbenzylcarbamate]. J. Phys. Chem. B 2013, 117, 9203–9216. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Huang, M.; Luo, C.; Wang, Q.; Zou, J.-W. Interactions between pyrazole derived enantiomers and Chiralcel OJ: Prediction of enantiomer absolute configurations and elution order by molecular dynamics simulations. J. Mol. Graph. Model. 2016, 66, 123–132. [Google Scholar] [CrossRef]
- Pisani, L.; Rullo, M.; Catto, M.; de Candia, M.; Carrieri, A.; Cellamare, S.; Altomare, C.D. Structure–property relationship study of the HPLC enantioselective retention of neuroprotective 7-[(1-alkylpiperidin-3-yl)methoxy]coumarin derivatives on an amylose-based chiral stationary phase. J. Sep. Sci. 2018, 41, 1376–1384. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Oroskar, P.A.; Wang, X.; House, D.; Oroskar, A.; Oroskar, A.; Jameson, C.J.; Murad, S. The composition of the mobile phase affects the dynamic chiral recognition of drug molecules by the chiral stationary phase. Langmuir 2017, 33, 11246–11256. [Google Scholar] [CrossRef]
- Wang, X.; Jameson, C.J.; Murad, S. Modeling enantiomeric separations as an interfacial process using amylose tris(3,5-dimethylphenyl carbamate) (ADMPC) polymers coated on amorphous silica. Langmuir 2020, 36, 1113–1124. [Google Scholar] [CrossRef]
- Peluso, P.; Mamane, V.; Dallocchio, R.; Dessì, A.; Villano, R.; Sanna, D.; Aubert, E.; Pale, P.; Cossu, S. Polysaccharide-based chiral stationary phases as halogen bond acceptors: A novel strategy for detection of stereoselective σ-hole bonds in solution. J. Sep. Sci. 2018, 41, 1247–1256. [Google Scholar] [CrossRef]
- Dallocchio, R.; Dessì, A.; Sechi, B.; Chankvetadze, B.; Jibuti, G.; Cossu, S.; Mamane, V.; Peluso, P. Enantioseparation of planar chiral ferrocenes on cellulose-based chiral stationary phases: Benzoate versus carbamate pendant groups. Electrophoresis 2023, 44, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Wolf, R.M.; Francotte, E.; Glasser, L.; Simon, I.; Scherage, H.A. Computation of low-energy crystalline arrangements of cellulose triacetate. Macromolecules 1992, 25, 709–720. [Google Scholar] [CrossRef]
- Kasat, R.B.; Wee, S.Y.; Loh, J.X.; Wang, N.-H.L.; Franses, E.I. Effect of solute molecular structure on its enantioresolution on cellulose tris(3,5-dimethylphenylcarbamate). J. Chromatogr. B 2008, 875, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Kasat, R.B.; Wang, N.-H.L.; Franses, E.I. Effects of backbone and side chain on the molecular environments of chiral cavities in polysaccharide-based biopolymers. Biomacromolecules 2007, 8, 1676–1685. [Google Scholar] [CrossRef] [PubMed]
- Kasat, R.B.; Franses, E.I.; Wang, N.-H.L. Experimental and computational studies of enantioseparation of structurally similar chiral compounds on amylose tris(3,5-dimethylphenylcarbamate). Chirality 2010, 22, 565–579. [Google Scholar] [CrossRef] [PubMed]
- Tsui, H.-W.; Willing, J.N.; Kasat, R.B.; Wang, N.-H.L.; Franses, E.I. Infrared spectroscopy and molecular simulations of a polymeric sorbent and its enantioselective interactions with benzoin enantiomers. J. Phys. Chem. B 2011, 115, 12785–12800. [Google Scholar] [CrossRef]
- Ma, S.; Tsui, H.-W.; Spinelli, E.; Busacca, C.A.; Franses, E.I.; Wang, N.-H.L.; Wu, L.; Lee, H.; Senanayake, C.; Yee, N.; et al. Insights into chromatographic enantiomeric separation of allenes on cellulose carbamate stationary phase. J. Chromatogr. A 2014, 1362, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Zhuravlev, N.D.; Siepmann, J.I.; Schure, M.R. Surface coverages of bonded-phase ligands on silica: A computational study. Anal. Chem. 2001, 73, 4006–4011. [Google Scholar] [CrossRef]
- Imberty, A.; Chanzy, H.; Pérez, S.; Buléon, A.; Tran, V. The double helical nature of the crystalline part of A-starch. J. Mol. Biol. 1988, 201, 365–378. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision B. 01; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Wang, X.; House, D.H.; Oroskar, P.A.; Oroskar, A.; Oroskar, A.; Jameson, C.J.; Murad, S. Molecular dynamics simulations of the chiral recognition mechanism for a polysaccharide chiral stationary phase in enantiomeric chromatographic separations. Mol. Phys. 2019, 117, 3569–3588. [Google Scholar] [CrossRef]
- Gambacorta, N.; Özdemir, Z.; Doğan, İ.S.; Ciriaco, F.; Zenni, Y.N.; Karakurt, A.; Saraç, S.; Nicolotti, O. Integrated experimental and theoretical approaches to investigate the molecular mechanisms of the enantioseparation of chiral anticonvulsant and antifungal compounds. J. Mol. Struct. 2022, 1270, 133905. [Google Scholar] [CrossRef]
- Okamoto, Y.; Kawashima, M.; Yamamoto, K.; Hatada, K. Useful chiral packing materials for high-performance liquid chromatographic resolution. Cellulose triacetate and tribenzoate coated on macroporous silica gel. Chem. Lett. 1984, 13, 739–742. [Google Scholar] [CrossRef]
- Yamamoto, C.; Yamada, K.; Motoya, K.; Kamiya, Y.; Kamigaito, M.; Okamoto, Y.; Aratani, T. Preparation of HPLC chiral packing materials using cellulose tris(4-methylbenzoate) for the separation of chrysanthemate isomers. J. Polym. Sci. A Polym. Chem. 2006, 44, 5087–5897. [Google Scholar] [CrossRef]
- Francotte, E.; Wolf, R.M.; Lohmann, D.; Mueller, R. Chromatographic resolution of racemates on chiral stationary phases: I. Influence of the supramolecular structure of cellulose triacetate. J. Chromatogr. A 1985, 347, 25–37. [Google Scholar] [CrossRef]
- Alcaro, S.; Bolasco, A.; Cirilli, R.; Ferretti, R.; Fioravanti, R.; Ortuso, F. Computer-aided molecular design of asymmetric pyrazole derivatives with exceptional enantioselective recognition toward the Chiralcel OJ-H stationary phase. J. Chem. Inf. Model. 2012, 52, 649–654. [Google Scholar] [CrossRef]
- Ortuso, F.; Alcaro, S.; Menta, S.; Fioravanti, R.; Cirilli, R. A Chromatographic and computational study on the driving force operating in the exceptionally large enantioseparation of N-thiocarbamoyl-3-(4’-biphenyl)-5-phenyl-4,5-dihydro-(1H) pyrazole on a 4-methylbenzoate cellulose-based chiral stationary phase. J. Chromatogr. A 2014, 1324, 71–77. [Google Scholar] [CrossRef]
- Wang, X.; Jameson, C.J.; Murad, S. Molecular dynamics simulations of chiral recognition of drugs by amylose polymers coated on amorphous silica. Mol. Phys. 2021, 119, 19–20. [Google Scholar] [CrossRef]
- Saleh, O.A.; Badawey, A.M.; Aboul-Enein, H.Y.; Fouad, M.A. Enantioseparation, quantification, molecular docking and molecular dynamics study of five β-adrenergic blockers on Lux-Cellulose-2 column. BMC Chem. 2023, 17, 22. [Google Scholar] [CrossRef]
- Dallocchio, R.; Sechi, B.; Dessì, A.; Chankvetadze, B.; Cossu, S.; Mamane, V.; Pale, P.; Peluso, P. Enantioseparations of polyhalogenated 4,4’-bipyridines on polysaccharide-based chiral stationary phases and molecular dynamics simulations of selector-selectand interactions. Electrophoresis 2021, 42, 1853–1863. [Google Scholar] [CrossRef]
- Dallocchio, R.; Dessì, A.; Sechi, B.; Chankvetadze, B.; Cossu, S.; Mamane, V.; Aubert, E.; Rozzo, C.; Palmieri, G.; Spissu, Y.; et al. Exploring interaction modes between polysaccharide-based selectors and biologically active 4,4’-bipyridines by experimental and computational analysis. J. Chromatogr. Open 2022, 2, 100030. [Google Scholar] [CrossRef]
- Sechi, B.; Mamane, V.; Dallocchio, R.; Dessì, A.; Cossu, S.; Jibuti, G.; Peluso, P. Enantioseparation of new axially chiral carboxylic acids on polysaccharide-based chiral stationary phases under normal phase elution conditions. J. Pharm. Biomed. Anal. Open 2023, 1, 100011. [Google Scholar] [CrossRef]
- Peluso, P.; Gatti, C.; Dessì, A.; Dallocchio, R.; Weiss, R.; Aubert, E.; Pale, P.; Cossu, S.; Mamane, V. Enantioseparation of fluorinated 3-arylthio-4,4’-bipyridines: Insights into chalcogen and π-hole bonds in high-performance liquid chromatography. J. Chromatogr. A 2018, 1567, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Baeza, M.; Martίn-Biosca, Y.; Escuder-Gilabert, L.; Medina-Hernández, M.J.; Sagrado, S. Artificial neural networks to model the enantioresolution of structurally unrelated neutral and basic compounds with cellulose tris(3,5-dimethylphenylcarbamate) chiral stationary phase and aqueous-acetonitrile mobile phases. J. Chromatogr. A 2022, 1672, 463048. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Lin, J.; Zhang, D.; Mo, F. Retention time prediction for chromatographic enantioseparation by quantile geometry-enhanced graph neural network. Nat. Commun. 2023, 14, 3095. [Google Scholar] [CrossRef] [PubMed]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer | Model (mod. n) | Force Field/Software | Production Time | Solvent | Year [Ref.] |
|---|---|---|---|---|---|
| CTPC | 8-mer (mod. 1) | CHARMM/CHARMM | 10 ps | Not considered | 1995 [28] |
| CTA | multistrand 8-mers (mod. 2) | AMBER/Macromodel | 300 ps | Not considered | 1997 [46] |
| CTPC, CDMPC | 9-mer (mod. 3 and 4) | CHARMM/CHARMM | 10 ps | Not considered | 1999 [47] |
| ADMPC | 12-mer (mod. 5) | COMPASS/MS-Modeling | 200 ps | Implicit CHCl3 | 2007 [48] |
| ADMPC, ASMBC | 12-mer (mod. 6 and 7) | CVFF/MS-Modeling | 1 ns | Not considered | 2008 [22] |
| CDMPC | 9-mer (mod. 8) | CVFF/MS-Modeling | 1 ns | Not considered | 2008 [22] |
| ADMPC | 36-mer/silica surface (mod. 9) | COMPASS/MS-Modeling | 3 ns | Not considered | 2010 [49] |
| ASMBC | 12-mer (mod. 10) | CVFF/MS-Modeling | 3 ns | Explicit Hex | 2013 [50] |
| CMB | 12-mer (mod. 11) | PCFF/Material Studio | 100 ps | Not considered | 2016 [51] |
| ADMPC | 12-mer (mod. 12) | Not specified/Maestro | 180 ns | Explicit MeOH | 2018 [52] |
| ADMPC | 12-mer (mod. 13) | GAFF/AMBER | 100 ns | Explicit Hept/2-PrOH, | 2017 [53] |
| MeOH | |||||
| ADMPC | multistrand 18-mers/silica | GAFF/AMBER | 40 ns | Explicit Hept/2-PrOH | 2020 [54] |
| surface (mod. 14) | |||||
| ADMPC, CDMPC | 9-mer (mod. 15 and 16) | GAFF/AMBER | 10 ns | Explicit Hex or MeOH | 2018 [43,55] |
| CMB | 9-mer (mod. 17) | GAFF/AMBER | 100 ns | Explicit Hex/2-PrOH | 2023 [56] |
| Polymer Model (mod. n) 1 | Chiral Analyte | Force Field/Software | Time | Solvent | NCI | [Ref.] |
|---|---|---|---|---|---|---|
| ADMPC (mod. 5) | 4 | COMPASS/MS-Modeling | 2 ns | Implicit CHCl3 | HB, vdW | [48] |
| ADMPC, ASMBC, CDMPC | 5 | CVFF/MS-Modeling | 1 ns | Not considered | HB, π–π | [22] |
| (mod. 6–8) | ||||||
| CDMPC (mod. 8) | 5–17 | CVFF/MS-Modeling | 1 ns | Not considered | Steric, HB, π–π | [58] |
| ADMPC (mod. 6) | 5–18 | CVFF/MS-Modeling | 1 ns | Not considered | Steric, HB, π–π | [60] |
| ADMPC (mod. 9) | 19, 20 | COMPASS/MS-Modeling | 3 ns | Not considered | HB, π–π, NH–π | [49] |
| ASMBC (mod. 7) | 3 | CVFF/MS-Modeling | 3 ns | Not considered | HB, π–π | [61] |
| CMB 2 | 21, 22 | OLPS_2005/Desmond | 60 ns | Explicit EtOH | Hph, π–π | [71] |
| CMB 2 | 23, 24 | OLPS_2005/Desmond | 60 ns | Explicit EtOH | HB, π–π | [72] |
| ASMBC (mod. 10) | 3, 25–27 | CVFF/MS-Modeling | 300 ps | Not considered | HB, π–π | [50] |
| CDMPC (mod. 8) | 28 | CVFF/MS-Modeling | 500 ps | Not considered | El, Rep | [62] |
| CMB (mod. 11) | 29 | PCFF/Material Studio | 100 ps | Implicit Hex/ROH, | HB, π–π | [51] |
| ROH, water | ||||||
| ADMPC (mod. 13) | 1, 3, 30–37 | GAFF/AMBER | 100 ns | Explicit Hept/2-PrOH, | Steric, π–π, HB | [53,66] |
| MeOH, ACN | ||||||
| ADMPC (mod. 14) | 1, 3, 30, 31, 35–37 | GAFF/AMBER | 200 ns | Explicit Hept/2-PrOH | HB, π–π | [54,73] |
| ADMPC (modified mod.14) | 38–41 | GROMOS54A7/LAMMPS | 100 ns | Explicit solvents | Steric, HB, π–π | [67] |
| CCMPC 3 | 42–46 | EHT/AMBER | 500 ps | Explicit EtOH | Hph, HB, π–π | [74] |
| ADMPC (mod. 15) | 47, 49–51, 53, 54 | GAFF/AMBER | 10–100 ns | Explicit solvents | Hph, HaB, HB, π–π | [43,55,75,76,77] |
| CDMPC (mod. 16) | 47–52 | GAFF/AMBER | 10–100 ns | Explicit solvents | HaB, ChB, HB, π–π | [43,55,56,75,76,78] |
| CMB (mod. 17) | 52 | GAFF/AMBER | 100 ns | Explicit solvents | HaB, π–π | [56] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dallocchio, R.; Dessì, A.; Sechi, B.; Peluso, P. Molecular Dynamics Simulations of Amylose- and Cellulose-Based Selectors and Related Enantioseparations in Liquid Phase Chromatography. Molecules 2023, 28, 7419. https://doi.org/10.3390/molecules28217419
Dallocchio R, Dessì A, Sechi B, Peluso P. Molecular Dynamics Simulations of Amylose- and Cellulose-Based Selectors and Related Enantioseparations in Liquid Phase Chromatography. Molecules. 2023; 28(21):7419. https://doi.org/10.3390/molecules28217419
Chicago/Turabian StyleDallocchio, Roberto, Alessandro Dessì, Barbara Sechi, and Paola Peluso. 2023. "Molecular Dynamics Simulations of Amylose- and Cellulose-Based Selectors and Related Enantioseparations in Liquid Phase Chromatography" Molecules 28, no. 21: 7419. https://doi.org/10.3390/molecules28217419