

Raman Spectrum of the Li2SO4-MgSO4-H2O System: Excess Spectrum and Hydration Shell Spectrum

Abstract
:1. Introduction
2. Results and Discussion
2.1. Raman Spectroscopy of Li2SO4-H2O System
2.1.1. Raman Spectra and Excess Raman Spectra of SO42− Symmetric Stretching Vibration
2.1.2. Raman and Excess Raman Spectra of the O-H Stretching Vibration Range
2.1.3. Hydrate Shell Spectrum and Hydration Number
2.2. Raman Spectroscopic for Li2SO4-MgSO4-H2O System
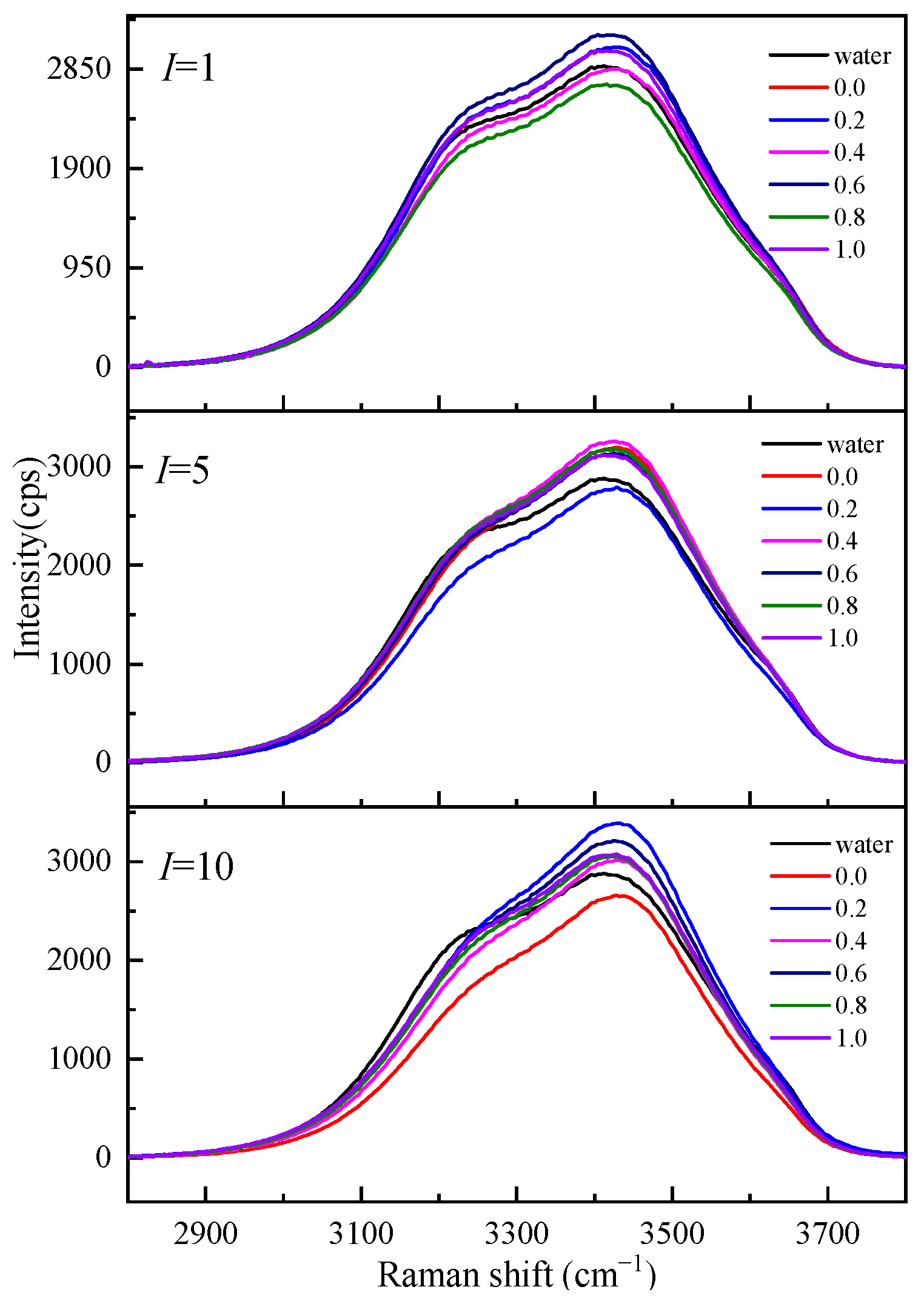
2.2.1. Raman Spectra of O-H Stretching Vibration Range
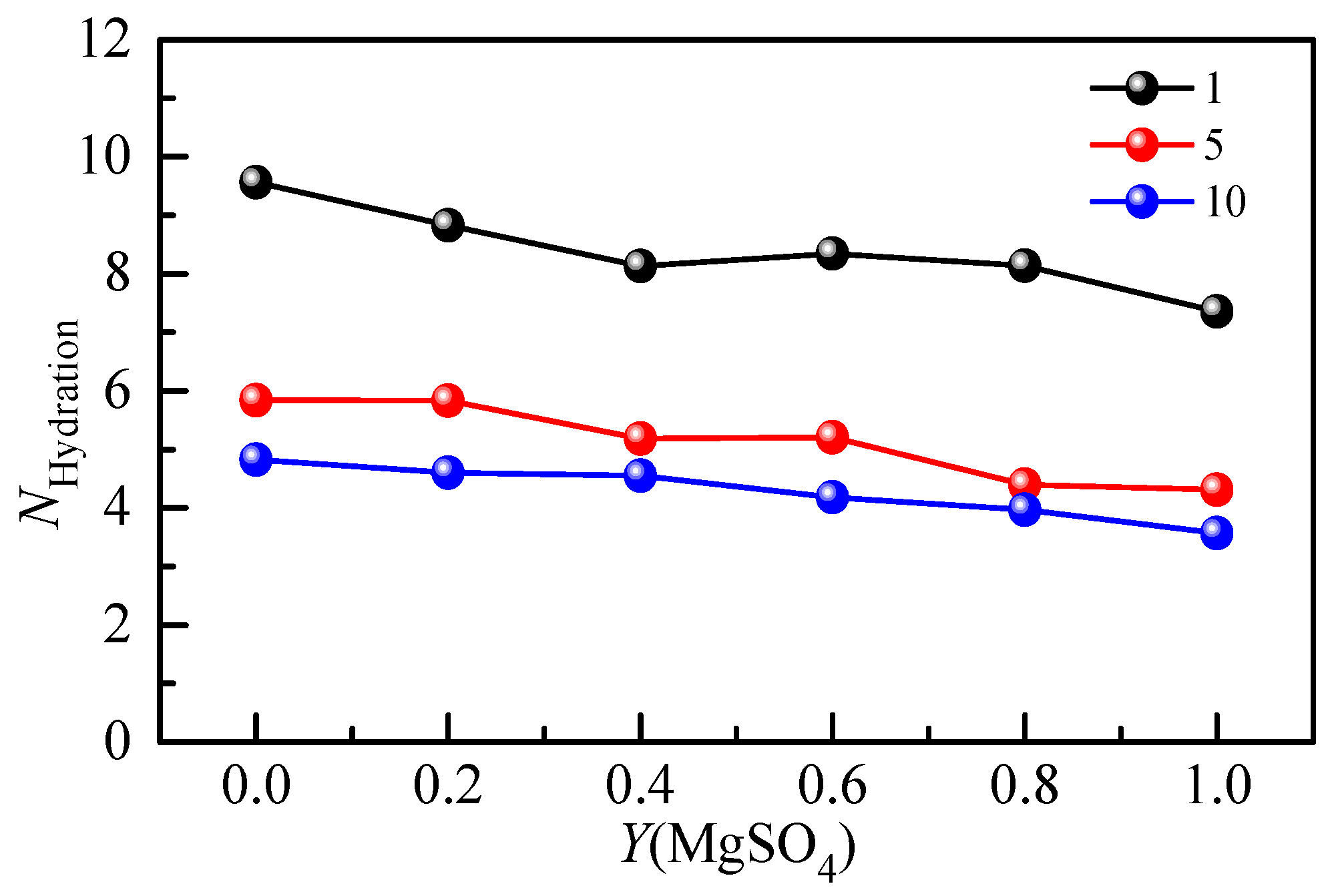
2.2.2. Hydration Shell Spectrum and Hydration Number
3. Experimental
3.1. Sample Preparation
3.2. Experimental Method
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, Q.B.; Ji, Z.Y.; Liu, J.; Zhao, Y.Y.; Wang, S.Z.; Yuan, J.S. Development of recovering lithium from brines by selective-electrodialysis: Effect of coexisting cations on the migration of lithium. J. Membr. Sci. 2018, 548, 408–420. [Google Scholar] [CrossRef]
- Gruber, P.W.; Medina, P.A.; Keoleian, G.A.; Kesler, S.E.; Everson, M.P.; Wallington, T.J. Global Lithium Availability. J. Ind. Ecol. 2011, 15, 760–775. [Google Scholar] [CrossRef]
- Torres, A.E.; Balbuena, P.B. Exploring the LiOH Formation Reaction Mechanism in Lithium–Air Batteries. Chem. Mater. 2018, 30, 708–717. [Google Scholar] [CrossRef]
- Lang, S.Y.; Xiao, R.J.; Gu, L.; Guo, Y.G.; Wen, R.; Wan, L.J. Interfacial Mechanism in Lithium–Sulfur Batteries: How Salts Mediate the Structure Evolution and Dynamics. J. Am. Chem. Soc. 2018, 140, 8147–8155. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Qu, W.; Guo, X.; Li, L.; Wu, F. The pursuit of solid-state electrolytes for lithium batteries: From comprehensive insight to emerging horizons. Mater. Horiz. 2016, 3, 487–516. [Google Scholar] [CrossRef]
- Jaskula, B.W.; Balbuena, P.B. Mineral Commodity Summaries; U.S. Geological Survey: Reston, VA, USA, 2018; pp. 98–99. [Google Scholar]
- Ooi, K.; Sonoda, A.; Makita, Y.; Chitrakar, R.; Tasaki-Handa, Y.; Nakazato, T. Recovery of lithium from salt-brine eluates by direct crystallization as lithium sulfate. Hydrometallurgy 2017, 174, 123–130. [Google Scholar] [CrossRef]
- He, L.; Xu, W.; Song, Y.; Liu, X.; Zhao, Z. Selective removal of magnesium from a lithium-concentrated anolyte by magnesium ammonium phosphate precipitation. Sep. Purif. Technol. 2017, 187, 214–220. [Google Scholar] [CrossRef]
- Yu, J.; Gao, C.; Cheng, A.; Liu, Y.; Zhang, L.; He, X. Geomorphic, hydroclimatic and hydrothermal controls on the formation of lithium brine deposits in the Qaidam Basin, northern Tibetan Plateau, China. Ore Geol. Rev. 2013, 50, 171–183. [Google Scholar] [CrossRef]
- Zhao, Z.; Si, X.; Liu, X.; He, L.; Liang, X. Li extraction from high Mg/Li ratio brine with LiFePO4/FePO4 as electrode materials. Hydrometallurgy 2013, 133, 75–83. [Google Scholar] [CrossRef]
- An, J.W.; Kang, D.J.; Tran, K.T.; Kim, M.J.; Lim, T.; Tran, T. Recovery of lithium from Uyuni salar brine. Hydrometallurgy 2012, 117–118, 64–70. [Google Scholar] [CrossRef]
- Flexer, V.; Baspineiro, C.F.; Galli, C.I. Lithium recovery from brines: A vital raw material for green energies with a potential environmental impact in its mining and processing. Sci. Total Environ. 2018, 639, 1188–1204. [Google Scholar] [CrossRef]
- Swain, B. Recovery and recycling of lithium: A review. Sep. Purif. Technol. 2017, 172, 388–403. [Google Scholar] [CrossRef]
- Choubey, P.K.; Kim, M.S.; Srivastava, R.R.; Lee, J.C.; Lee, J.Y. Advance review on the exploitation of the prominent energy-storage element: Lithium. Part I: From mineral and brine resources. Miner. Eng. 2016, 89, 119–137. [Google Scholar] [CrossRef]
- Marcus, Y. Effect of Ions on the Structure of Water: Structure Making and Breaking. Chem. Rev. 2009, 109, 1346–1370. [Google Scholar] [CrossRef] [PubMed]
- Hribar, B.; Southall, N.T.; Vlachy, V.; Dill, K.A. How ions affect the structure of water. J. Am. Chem. Soc. 2002, 124, 12302–12311. [Google Scholar] [CrossRef]
- Tielrooij, K.J.; Van Der Post, S.T.; Hunger, J.; Bonn, M.; Bakker, H.J. Anisotropic water reorientation around ions. J. Phys. Chem. B 2011, 115, 12638–12647. [Google Scholar] [CrossRef]
- Smith, J.D.; Saykally, R.J.; Geissler, P.L. The effects of dissolved halide anions on hydrogen bonding in liquid water. J. Am. Chem. Soc. 2007, 129, 13847–13856. [Google Scholar] [CrossRef]
- Mahanta, D.D.; Samanta, N.; Mitra, R.K. The effect of monovalent cations on the collective dynamics of water and on a model protein. J. Mol. Liq. 2016, 215, 197–203. [Google Scholar] [CrossRef]
- Funkner, S.; Niehues, G.; Schmidt, D.A.; Heyden, M.; Schwaab, G.; Callahan, K.M.; Havenith, M. Watching the low-frequency motions in aqueous salt solutions: The terahertz vibrational signatures of hydrated ions. J. Am. Chem. Soc. 2012, 134, 1030–1035. [Google Scholar] [CrossRef]
- Omta, A.W.; Kropman, M.F.; Woutersen, S.; Bakker, H.J. Negligible Effect of Ions on the Hydrogen-Bond Structure in Liquid Water. Science 2003, 301, 347–349. [Google Scholar] [CrossRef]
- Kropman, M.F.; Bakker, H.J. Femtosecond mid-infrared spectroscopy of aqueous solvation shells. J. Chem. Phys. 2001, 115, 8942–8948. [Google Scholar] [CrossRef]
- Ben-Amotz, D. Hydration-Shell Vibrational Spectroscopy. J. Am. Chem. Soc. 2019, 141, 10569–10580. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.; Singh, A.K.; Mondal, J.A.; Sarkar, S.K. Water in the Hydration Shell of Halide Ions Has Significantly Reduced Fermi Resonance and Moderately Enhanced Raman Cross Section in the OH Stretch Regions. J. Phys. Chem. B 2013, 117, 9728–9733. [Google Scholar] [CrossRef]
- Dillon, S.R.; Dougherty, R.C. Raman Studies of the Solution Structure of Univalent Electrolytes in Water. J. Phys. Chem. A 2002, 106, 7647–7650. [Google Scholar] [CrossRef]
- Perera, P.N.; Browder, B.; Ben-Amotz, D. Perturbations of Water by Alkali Halide Ions Measured using Multivariate Raman Curve Resolution. J. Phys. Chem. B 2009, 113, 1805–1809. [Google Scholar] [CrossRef]
- Max, J.J.; Gessinger, V.; Driessche, C.V.; Larouche, P.; Chapados, C. Infrared spectroscopy of aqueous ionic salt solutions at low concentrations. J. Chem. Phys. 2007, 126, 184507. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhu, W.; Lin, K.; Yuan, L.; Zhou, X.; Liu, S. Ratiometric detection of Raman hydration shell spectra. J. Raman Spectrosc. 2016, 47, 1231–1238. [Google Scholar] [CrossRef]
- Okazaki, Y.; Taniuchi, T.; Mogami, G.; Matubayasi, N.; Suzuki, M. Comparative Study on the Properties of Hydration Water of Na- and K-Halide Ions by Raman OH/OD-stretching Spectroscopy and Dielectric Relaxation Data. J. Phys. Chem. A 2014, 118, 2922–2930. [Google Scholar] [CrossRef]
- Lin, K.; Zhou, X.; Luo, Y.; Liu, S. The Microscopic Structure of Liquid Methanol from Raman Spectroscopy. J. Phys. Chem. B 2010, 114, 3567–3573. [Google Scholar] [CrossRef]
- Liu, J.H.; Zhang, Y.H.; Wang, L.Y.; Wei, Z.F. Drawing out the structural information of the first layer of hydrated ions: ATR-FTIR spectroscopic studies on aqueous NH4NO3, NaNO3, and Mg(NO3)2 solutions. Spectrochim. Acta A 2005, 61, 893–899. [Google Scholar] [CrossRef]
- Näslund, L.Å.; Edwards, D.C.; Wernet, P.; Bergmann, U.; Ogasawara, H.; Pettersson, L.G.M.; Myneni, S.; Nilsson, A. X-ray Absorption Spectroscopy Study of the Hydrogen Bond Network in the Bulk Water of Aqueous Solutions. J. Phys. Chem. A 2005, 109, 5995–6002. [Google Scholar] [CrossRef]
- Heisler, I.A.; Mazur, K.; Meech, S.R. Low-Frequency Modes of Aqueous Alkali Halide Solutions: An Ultrafast Optical Kerr Effect Study. J. Phys. Chem. B 2011, 115, 1863–1873. [Google Scholar] [CrossRef] [PubMed]
- Petit, L.; Vuilleumier, R.; Maldivi, P.; Adamo, C. Ab Initio Molecular Dynamics Study of a Highly Concentrated LiCl Aqueous Solution. J. Chem. Theory Comput. 2008, 4, 1040–1048. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, W.; Brooker, M.H.; Pye, C.C. Hydration of Lithium Ion in Aqueous Solutions. J. Phys. Chem. 1995, 99, 3793–3797. [Google Scholar] [CrossRef]
- Wang, C.; Lin, K.; Hu, N.; Zhou, X.; Liu, S. Ion Pairs in Aqueous Magnesium Nitrate Solution by Excess Raman Spectroscopy. Acta Phys.-Chim. Sin. 2012, 28, 1823–1829. [Google Scholar]
- Li, D.; Zhu, Z.; Sun, D.W. Visualization of the in situ distribution of contents and hydrogen bonding states of cellular level water in apple tissues by confocal Raman microscopy. Analyst 2020, 145, 897–907. [Google Scholar] [CrossRef]
- Sun, Q. Local statistical interpretation for water structure. Chem. Phys. Lett. 2013, 568–569, 90–94. [Google Scholar] [CrossRef]
- Kitadai, N.; Sawai, T.; Tonoue, R.; Nakashima, S.; Katsura, M.; Fukushi, K. Effects of Ions on the OH Stretching Band of Water as Revealed by ATR-IR Spectroscopy. J. Solut. Chem. 2014, 43, 1055–1077. [Google Scholar] [CrossRef]
- Vchirawongkwin, V.; Rode, B.; Persson, I. Structure and Dynamics of Sulfate Ion in Aqueous SolutionsAn ab initio QMCF MD Simulation and Large Angle X-ray Scattering Study. J. Phys. Chem. B 2007, 111, 4150–4155. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Source | Final Purity | Impurity Analysis Method |
|---|---|---|---|
| Li2SO4·H2O | McLean Biochemical Technology Co., Ltd. | 0.995 | ICP |
| MgSO4·7H2O | McLean Biochemical Technology Co., Ltd. | 0.995 | ICP |
| BaCl2 | Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China) | A.R. | -- |
| Water | Double distillation | 1.5 × 10−4 S·m−1 | Conductivity meter |
| No. a | 100w(Li2SO4) | ρ/(g·cm−3) | m(mol·kg−1) | n(Li2SO4):n(H2O) |
|---|---|---|---|---|
| 1 | 0.5 | 1.00257 | 0.04584 | 0.0008258 |
| 2 | 1.0 | 1.00702 | 0.09343 | 0.0016831 |
| 3 | 6.0 | 1.05047 | 0.5814 | 0.0104739 |
| 4 | 8.0 | 1.06820 | 0.7962 | 0.0142517 |
| 5 | 10.0 | 1.08593 | 1.0110 | 0.0182143 |
| 6 | 14.0 | 1.12227 | 1.4806 | 0.0266741 |
| 7 | 18.0 | 1.15968 | 1.9962 | 0.0359628 |
| 8 | 24.0 | 1.21748 | 2.8738 | 0.0517729 |
| No. a | I(mol·kg−1) | Y(MgSO4) | mLi2SO4(mol·kg−1) | mMgSO4(mol·kg−1) | n(solute):n(H2O) |
|---|---|---|---|---|---|
| 1 | 1 | 0.0 | 0.3335 | 0.0000 | 0.00600796 |
| 2 | 1 | 0.2 | 0.2666 | 0.0548 | 0.00579009 |
| 3 | 1 | 0.4 | 0.2012 | 0.0984 | 0.00539825 |
| 4 | 1 | 0.6 | 0.1328 | 0.1504 | 0.00510299 |
| 5 | 1 | 0.8 | 0.0672 | 0.2006 | 0.00482338 |
| 6 | 1 | 1.0 | 0.0000 | 0.2489 | 0.00448382 |
| 7 | 5 | 0.0 | 1.3318 | 0.0000 | 0.02399285 |
| 8 | 5 | 0.2 | 1.0656 | 0.2027 | 0.02284913 |
| 9 | 5 | 0.4 | 0.8005 | 0.3990 | 0.02160967 |
| 10 | 5 | 0.6 | 0.5330 | 0.6016 | 0.02044111 |
| 11 | 5 | 0.8 | 0.2665 | 0.8003 | 0.01921944 |
| 12 | 5 | 1.0 | 0.0000 | 1.0015 | 0.01804287 |
| 13 | 10 | 0.0 | 2.3342 | 0.0000 | 0.04205187 |
| 14 | 10 | 0.2 | 1.8659 | 0.3500 | 0.03992034 |
| 15 | 10 | 0.4 | 1.3986 | 0.6979 | 0.03776931 |
| 16 | 10 | 0.6 | 0.9333 | 1.0488 | 0.03570847 |
| 17 | 10 | 0.8 | 0.4687 | 1.3977 | 0.03362393 |
| 18 | 10 | 1.0 | 0.0000 | 1.7526 | 0.03157351 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ge, H.; Wang, M. Raman Spectrum of the Li2SO4-MgSO4-H2O System: Excess Spectrum and Hydration Shell Spectrum. Molecules 2023, 28, 7356. https://doi.org/10.3390/molecules28217356
Ge H, Wang M. Raman Spectrum of the Li2SO4-MgSO4-H2O System: Excess Spectrum and Hydration Shell Spectrum. Molecules. 2023; 28(21):7356. https://doi.org/10.3390/molecules28217356
Chicago/Turabian StyleGe, Haiwen, and Min Wang. 2023. "Raman Spectrum of the Li2SO4-MgSO4-H2O System: Excess Spectrum and Hydration Shell Spectrum" Molecules 28, no. 21: 7356. https://doi.org/10.3390/molecules28217356