
Optoelectronic Evolution in Halogen-Doped Organic–Inorganic Halide Perovskites: A First-Principles Analysis


Abstract
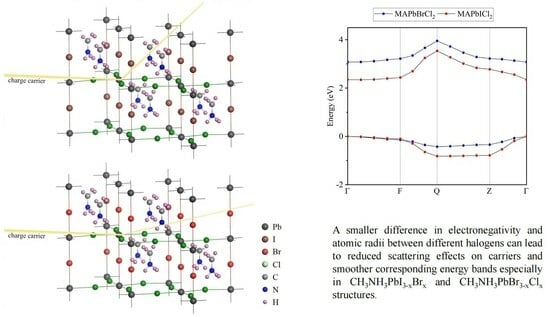
:
1. Introduction
2. Results and Discussion
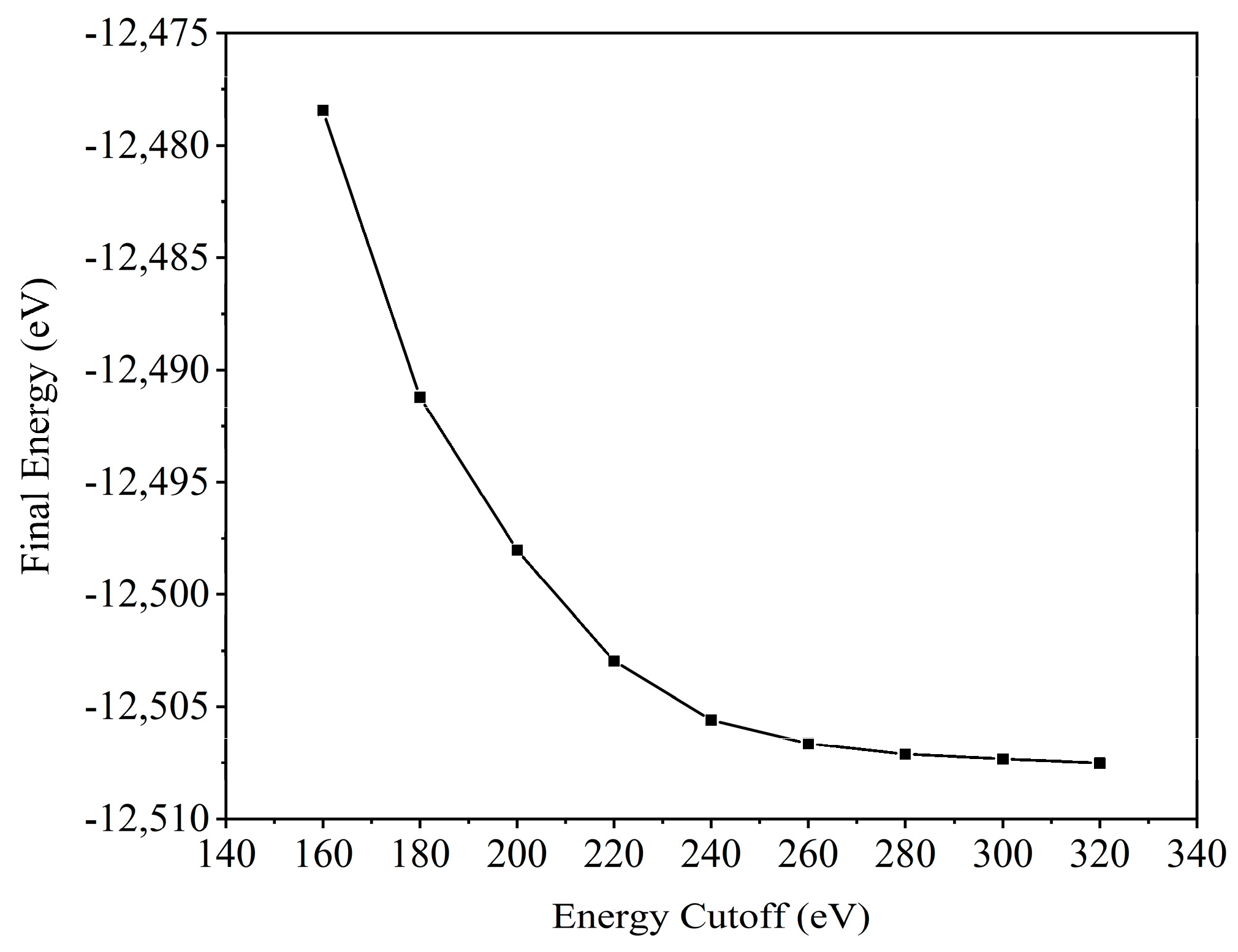
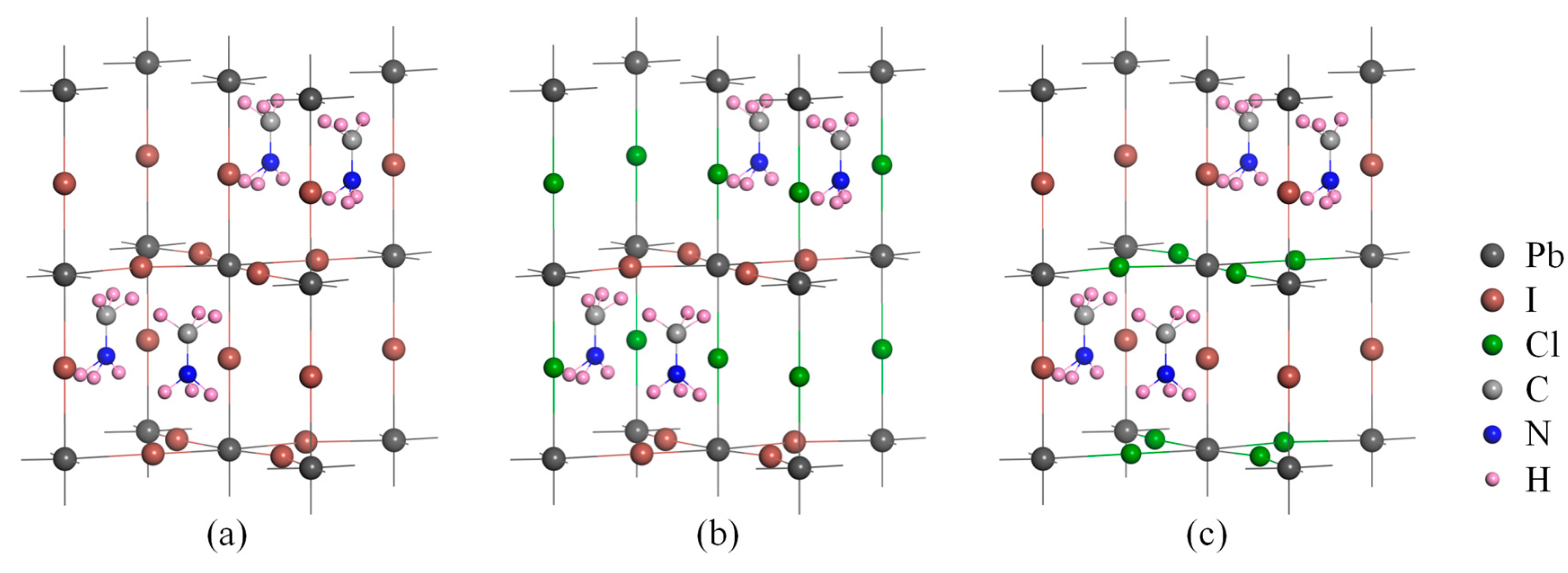
2.1. Geometric Properties
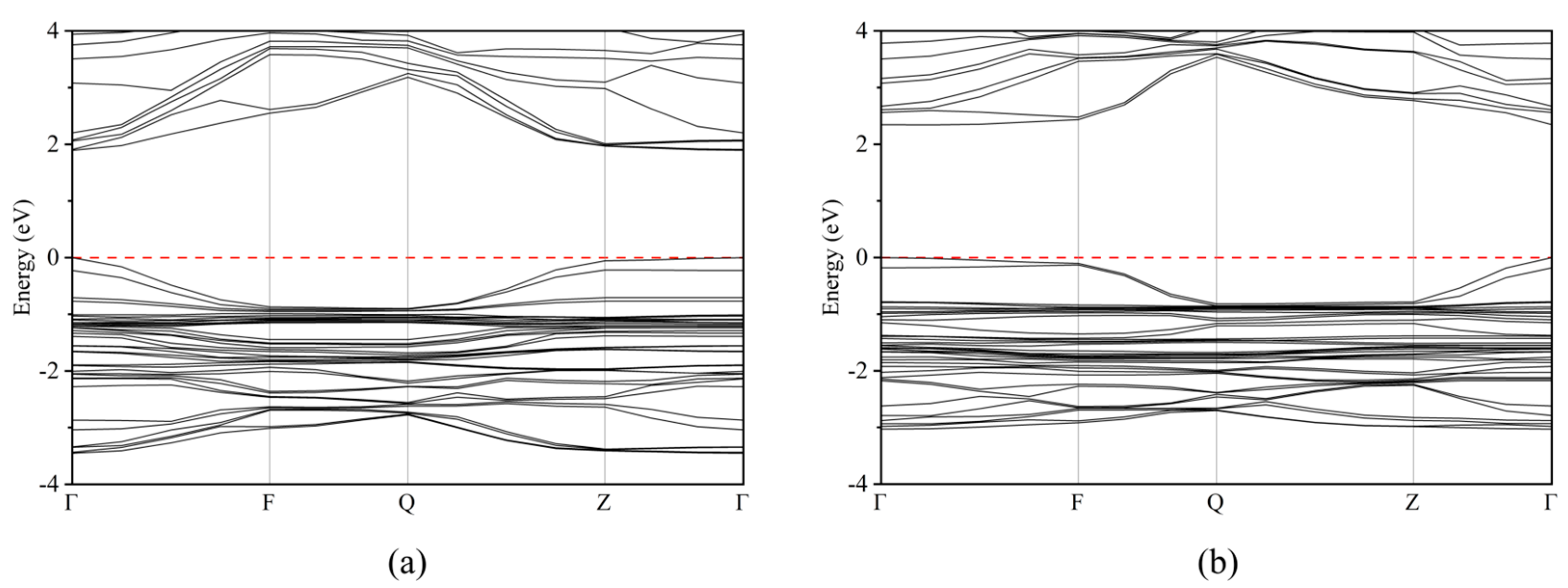
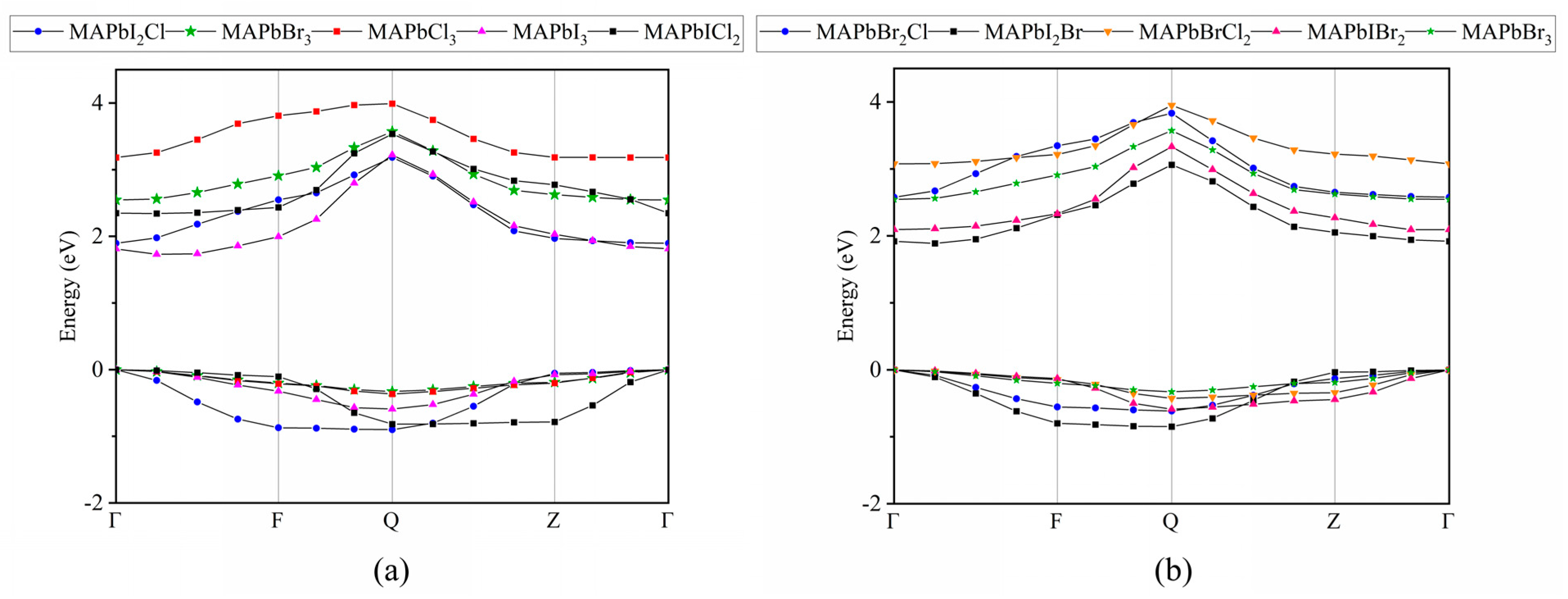
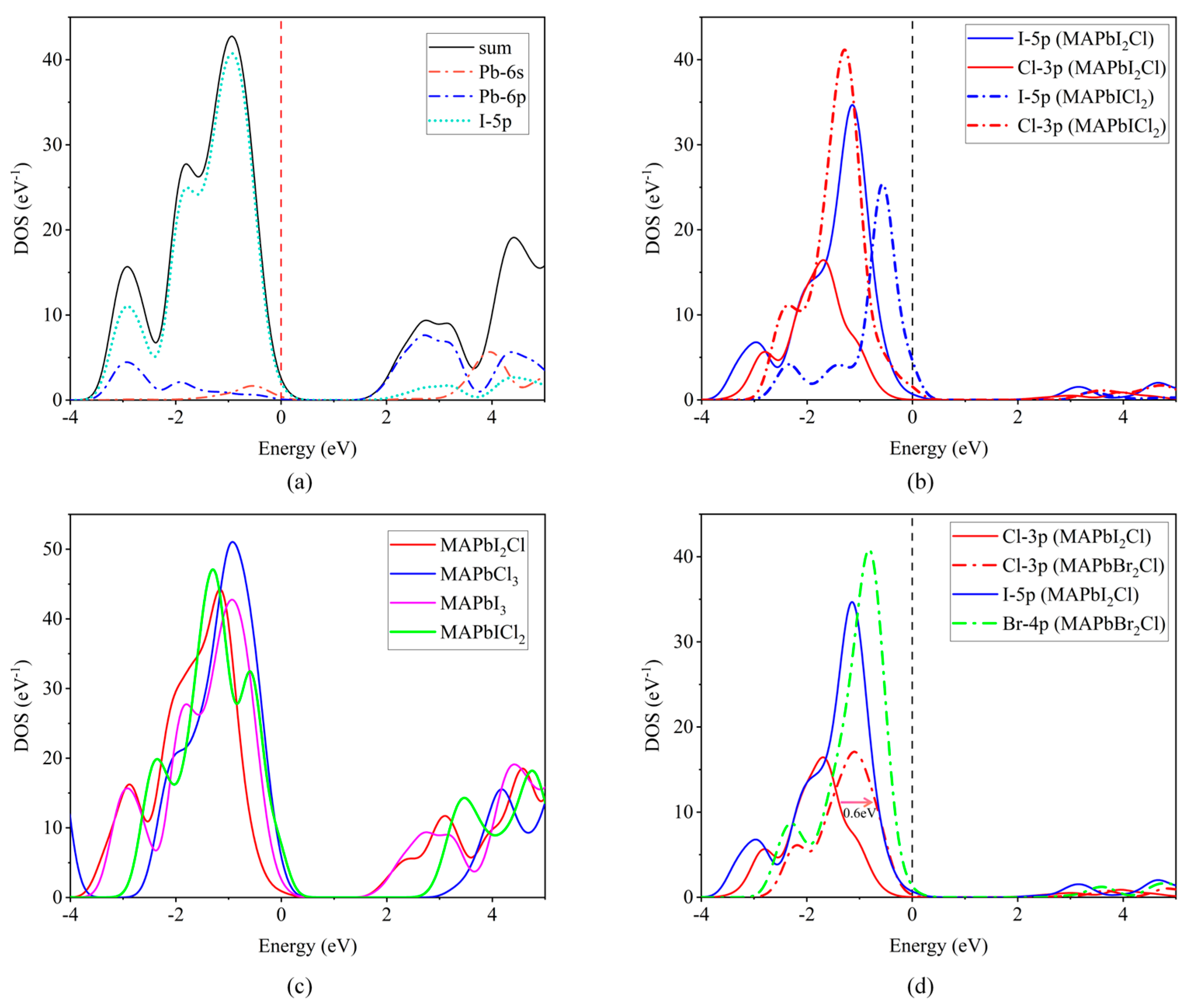
2.2. Electronic Properties
2.3. Effective Masses
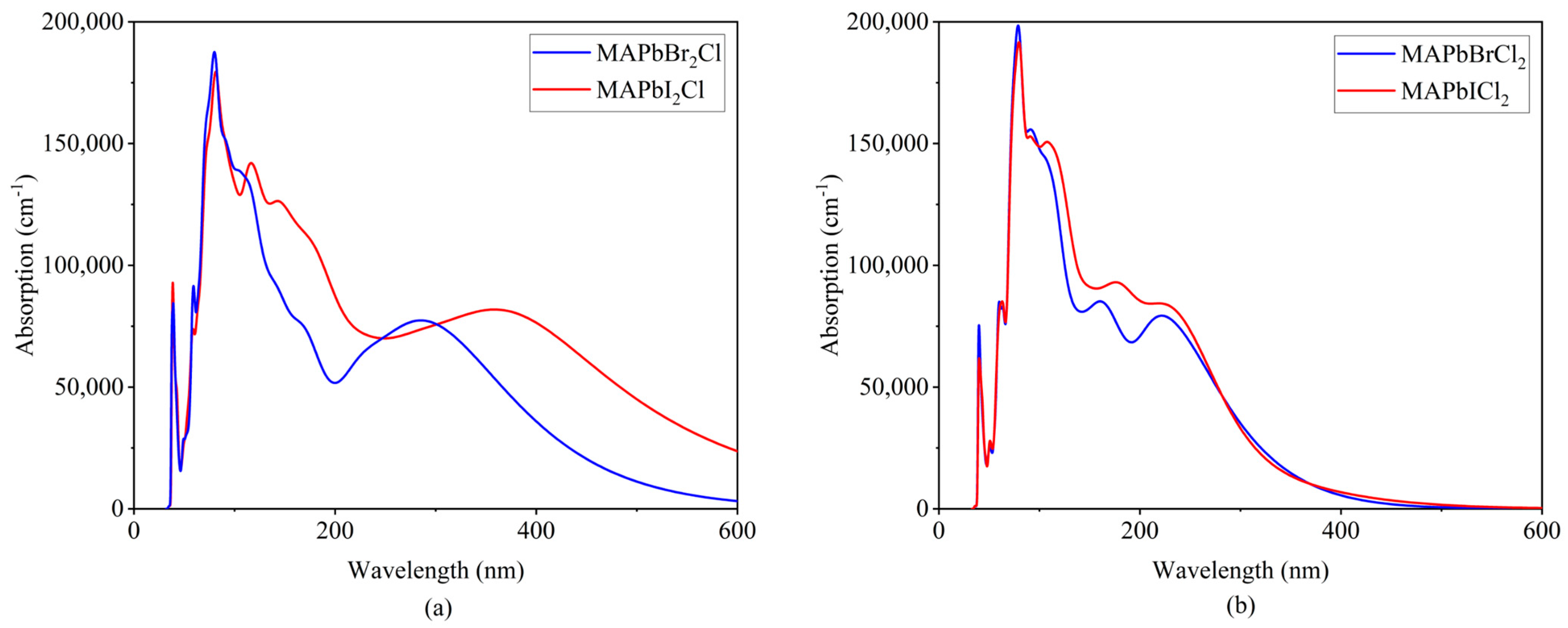
2.4. Absorption Spectrum
3. Simulation Strategy
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kojima, A.; Teshima, K.; Shirai, Y.; Miyasaka, T. Organometal Halide Perovskites as Visible-Light Sensitizers for Photovoltaic Cells. J. Am. Chem. Soc. 2009, 131, 6050–6051. [Google Scholar] [CrossRef]
- Wu, T.H.; Qin, Z.Z.; Wang, Y.B.; Wu, Y.Z.; Chen, W.; Zhang, S.F.; Cai, M.L.; Dai, S.Y.; Zhang, J.; Liu, J.; et al. The Main Progress of Perovskite Solar Cells in 2020–2021. Nano-Micro Lett. 2021, 13, 152. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.-L.; Xie, S.-Y.; Huang, R.-B.; Zheng, L.-S. Research Progress in Perovskite Solar Cell Materials and Devices. J. Xiamen Univ. Nat. Sci. 2015, 54, 619–629. [Google Scholar]
- Koscher, B.A.; Swabeck, J.K.; Bronstein, N.D.; Alivisatos, A.P. Essentially Trap-Free CsPbBr3 Colloidal Nanocrystals by Postsynthetic Thiocyanate Surface Treatment. J. Am. Chem. Soc. 2017, 139, 6566–6569. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, X.M.; Zhang, T.Y.; Wang, X.T.; Kan, M.; Shi, J.L.; Zhao, Y.X. The Role of Dimethylammonium Iodide in CsPbI3 Perovskite Fabrication: Additive or Dopant? Angew. Chem.-Int. Ed. 2019, 58, 16691–16696. [Google Scholar] [CrossRef]
- Wei, H.T.; DeSantis, D.; Wei, W.; Deng, Y.H.; Guo, D.Y.; Savenije, T.J.; Cao, L.; Huang, J.S. Dopant compensation in alloyed CH3NH3PbBr3−xClx perovskite single crystals for gamma-ray spectroscopy. Nat. Mater. 2017, 16, 826–833. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.L.; Xu, H.Y.; Ji, X.; Liang, J.K.; Yu, Q.F. Progress toward Applications of Perovskite Solar Cells. Energy Fuels 2020, 34, 6624–6633. [Google Scholar] [CrossRef]
- Zhang, W.; Peng, X.; Feng, X. Recent progress of perovskite solar cells. Electron. Compon. Mater. 2014, 33, 7–11. [Google Scholar] [CrossRef]
- Alla, M.; Manjunath, V.; Chawki, N.; Singh, D.; Yadav, S.C.; Rouchdi, M.; Boubker, F. Optimized CH3NH3PbI3−XClX based perovskite solar cell with theoretical efficiency exceeding 30%. Opt. Mater. 2022, 124, 112044. [Google Scholar] [CrossRef]
- Ayad, M.; Fathi, M.; Mellit, A. Study and performance analysis of Perovskite solar cell structure based on organic and inorganic thin films. Optik 2021, 233, 166619. [Google Scholar] [CrossRef]
- Mosconi, E.; Amat, A.; Nazeeruddin, M.K.; Grätzel, M.; De Angelis, F. First-Principles Modeling of Mixed Halide Organometal Perovskites for Photovoltaic Applications. J. Phys. Chem. C 2013, 117, 13902–13913. [Google Scholar] [CrossRef]
- Giorgi, G.; Fujisawa, J.I.; Segawa, H.; Yamashita, K. Cation Role in Structural and Electronic Properties of 3D Organic-Inorganic Halide Perovskites: A DFT Analysis. J. Phys. Chem. C 2014, 118, 12176–12183. [Google Scholar] [CrossRef]
- Pan, Y.Y.; Su, Y.H.; Hsu, C.H.; Huang, L.W.; Kaun, C.C. The electronic structure of organic-inorganic hybrid perovskite solar cell: A first-principles analysis. Comput. Mater. Sci. 2016, 117, 573–578. [Google Scholar] [CrossRef]
- Zheng, F.; Takenaka, H.; Wang, F.; Koocher, N.Z.; Rappe, A.M. First-Principles Calculation of the Bulk Photovoltaic Effect in CH3NH3PbI3 and CH3NH3PbI3–xClx. J. Phys. Chem. Lett. 2015, 6, 31–37. [Google Scholar] [CrossRef]
- Wang, S.; Xiao, W.-b.; Wang, F. Structural, electronic, and optical properties of cubic formamidinium lead iodide perovskite: A first-principles investigation. RSC Adv. 2020, 10, 32364–32369. [Google Scholar] [CrossRef]
- Liu, S.; Zheng, F.; Koocher, N.Z.; Takenaka, H.; Wang, F.; Rappe, A.M. Ferroelectric Domain Wall Induced Band Gap Reduction and Charge Separation in Organometal Halide Perovskites. J. Phys. Chem. Lett. 2015, 6, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Filippetti, A.; Delugas, P.; Mattoni, A. Radiative Recombination and Photoconversion of Methylammonium Lead Iodide Perovskite by First Principles: Properties of an Inorganic Semiconductor within a Hybrid Body. J. Phys. Chem. C 2014, 118, 24843–24853. [Google Scholar] [CrossRef]
- Quarti, C.; Mosconi, E.; Umari, P.; De Angelis, F. Chlorine Incorporation in the CH3NH3PbI3 Perovskite: Small Concentration, Big Effect. Inorg. Chem. 2017, 56, 74–83. [Google Scholar] [CrossRef]
- Yan, W.-L.; Lu, G.-H.; Liu, F. Effect of Chlorine Substitution on Lattice Distortion and Ferroelectricity of CH3NH3PbI3. J. Phys. Chem. C 2016, 120, 17972–17977. [Google Scholar] [CrossRef]
- Long, R.; Prezhdo, O.V. Dopants Control Electron–Hole Recombination at Perovskite–TiO2 Interfaces: Ab Initio Time-Domain Study. ACS Nano 2015, 9, 11143–11155. [Google Scholar] [CrossRef]
- Xu, F.; Zhang, T.; Li, G.; Zhao, Y. Synergetic Effect of Chloride Doping and CH3NH3PbCl3 on CH3NH3PbI3−xClx Perovskite-Based Solar Cells. ChemSusChem 2017, 10, 2365–2369. [Google Scholar] [CrossRef] [PubMed]
- Caputo, M.; Cefarin, N.; Radivo, A.; Demitri, N.; Gigli, L.; Plaisier, J.R.; Panighel, M.; Di Santo, G.; Moretti, S.; Giglia, A.; et al. Electronic structure of MAPbI3 and MAPbCl3: Importance of band alignment. Sci. Rep. 2019, 9, 15159. [Google Scholar] [CrossRef] [PubMed]
- Petrovai, I.; Todor-Boer, O.; David, L.; Botiz, I. Growth of Hybrid Perovskite Crystals from CH3NH3PbI3−xClx Solutions Subjected to Constant Solvent Evaporation Rates. Materials 2023, 16, 2625. [Google Scholar] [CrossRef] [PubMed]
- Yerezhep, D.; Omarova, Z.; Aldiyarov, A.; Shinbayeva, A.; Tokmoldin, N. IR Spectroscopic Degradation Study of Thin Organometal Halide Perovskite Films. Molecules 2023, 28, 1288. [Google Scholar] [CrossRef]
- Siddique, Z.; Payne, J.L.; Irvine, J.T.S.; Jagadamma, L.K.; Akhter, Z.; Samuel, I.D.W.; Iqbal, A. Effect of halide-mixing on tolerance factor and charge-carrier dynamics in (CH3NH3PbBr3−xClx) perovskites powders. J. Mater. Sci.-Mater. Electron. 2020, 31, 19415–19428. [Google Scholar] [CrossRef]
- Pramchu, S.; Cheiwchanchamnangij, T.; Laosiritaworn, Y.; Jaroenjittichai, A.P. Enhancing surface stabilization of CH3NH3PbI3 perovskite by Cl and Br doping: First-principles study. J. Appl. Phys. 2019, 125, 115302. [Google Scholar] [CrossRef]
- Keremane, K.S.; Rao, R.; Adhikari, A.V. Simple 3,6-disubstituted Carbazoles as Potential Hole Transport Materials: Photophysical, Electrochemical and Theoretical Studies. Photochem. Photobiol. 2021, 97, 289–300. [Google Scholar] [CrossRef]
- Somsongkul, V.; Lang, F.; Jeong, A.R.; Rusu, M.; Arunchaiya, M.; Dittrich, T. Hole blocking PbI2/CH3NH3PbI3 interface. KU Knowl. Repos. 2014, 8, 763–766. [Google Scholar]
- Cao, D.Y.H.; Guo, P.J.; Mannodi-Kanakkithodi, A.; Wiederrecht, G.P.; Gosztola, D.J.; Jeon, N.; Schaller, R.D.; Chan, M.K.Y.; Martinson, A.B.F. Charge Transfer Dynamics of Phase-Segregated Halide Perovskites: CH3NH3PbCl3 and CH3NH3PbI3 or (C4H9NH3)2(CH3NH3)n−1PbnI3n+1 Mixtures. ACS Appl. Mater. Interfaces 2019, 11, 9583–9593. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Laasonen, K.; Pasquarello, A.; Car, R.; Lee, C.; Vanderblit, D. Car-parrinello molecular-dynamics with vanderbilt ultrasoft pseudopotentials. Phys. Rev. B 1993, 47, 10142–10153. [Google Scholar] [CrossRef] [PubMed]
- Garrity, K.F.; Bennett, J.W.; Rabe, K.M.; Vanderbilt, D. Pseudopotentials for high-throughput DFT calculations. Comput. Mater. Sci. 2014, 81, 446–452. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.J.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Z. Fur Krist. 2005, 220, 567–570. [Google Scholar] [CrossRef]
- Feng, J.; Xiao, B. Effective Masses and Electronic and Optical Properties of Nontoxic MASnX3 (X = Cl, Br, and I) Perovskite Structures as Solar Cell Absorber: A Theoretical Study Using HSE06. J. Phys. Chem. C 2014, 118, 19655–19660. [Google Scholar] [CrossRef]
- Han, X.; Amrane, N.; Qamhieh, N.; Zhang, Z.; Benkraouda, M. Optoelectronic functionality and photovoltaic performance of Sr-doped tetragonal CH3NH3PbI3: A first-principles study. Phys. B Condens. Matter 2023, 649, 414453. [Google Scholar] [CrossRef]
- Shi, J.; Yun, S. First-Principles DFT Calculations for Perovskite Solar Cells. In Counter Electrodes for Dye-Sensitized and Perovskite Solar Cells; John Wiley & Sons: Hoboken, NJ, USA, 2018; pp. 487–509. [Google Scholar]
- Peng, C.; Chen, J.; Wang, H.; Hu, P. First-Principles Insight into the Degradation Mechanism of CH3NH3PbI3 Perovskite: Light-Induced Defect Formation and Water Dissociation. J. Phys. Chem. C 2018, 122, 27340–27349. [Google Scholar] [CrossRef]
- Lin, Q.Q.; Armin, A.; Nagiri, R.C.R.; Burn, P.L.; Meredith, P. Electro-optics of perovskite solar cells. Nat. Photonics 2015, 9, 106–112. [Google Scholar] [CrossRef]
- Arakcheeva, A.; Chernyshov, D.; Spina, M.; Forró, L.; Horváth, E. CH3NH3PbI3: Precise structural consequences of water absorption at ambient conditions. Acta Crystallogr. Sect. B-Struct. Sci. Cryst. Eng. Mater. 2016, 72, 716–722. [Google Scholar] [CrossRef]
- Stoumpos, C.C.; Malliakas, C.D.; Kanatzidis, M.G. Semiconducting Tin and Lead Iodide Perovskites with Organic Cations: Phase Transitions, High Mobilities, and Near-Infrared Photoluminescent Properties. Inorg. Chem. 2013, 52, 9019–9038. [Google Scholar] [CrossRef]
- Jaffe, A.; Lin, Y.; Beavers, C.M.; Voss, J.; Mao, W.L.; Karunadasa, H.I. High-Pressure Single-Crystal Structures of 3D Lead-Halide Hybrid Perovskites and Pressure Effects on their Electronic and Optical Properties. ACS Cent. Sci. 2016, 2, 201–209. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | Bandgap (eV) | Structure | Bandgap (eV) | Structure | Bandgap (eV) |
|---|---|---|---|---|---|
| MAPbI3 | 1.730 | MAPbBr3 | 2.544 | MAPbCl3 | 3.183 |
| MAPbI2Cl | 1.895 | MAPbBr2Cl | 2.578 | MAPbI2Br | 1.888 |
| MAPbICl2 | 2.343 | MAPbBrCl2 | 3.076 | MAPbIBr2 | 2.093 |
| Structure Type | me | mh | Structure Type | me | mh |
|---|---|---|---|---|---|
| MAPbI3 | 0.643 m0 | −0.514 m0 | MAPbI2Cl | 0.265 m0 | −0.207 m0 |
| MAPbBr3 | 0.445 m0 | −0.866 m0 | MAPbICl2 | 1.692 m0 | −1.642 m0 |
| MAPbCl3 | 0.269 m0 | −0.816 m0 | MAPbBr2Cl | 0.223 m0 | −0.316 m0 |
| MAPbI2Br | 0.372 m0 | −0.216 m0 | MAPbBrCl2 | 1.125 m0 | −1.181 m0 |
| MAPbIBr2 | 0.688 m0 | −1.308 m0 | Path: G-F; m0 is the mass of a free electron | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, C.-L.; Liu, S.; Liu, X.-Y.; Li, Y.-N.; Zhang, P. Optoelectronic Evolution in Halogen-Doped Organic–Inorganic Halide Perovskites: A First-Principles Analysis. Molecules 2023, 28, 7341. https://doi.org/10.3390/molecules28217341
Xiao C-L, Liu S, Liu X-Y, Li Y-N, Zhang P. Optoelectronic Evolution in Halogen-Doped Organic–Inorganic Halide Perovskites: A First-Principles Analysis. Molecules. 2023; 28(21):7341. https://doi.org/10.3390/molecules28217341
Chicago/Turabian StyleXiao, Cheng-Liang, Sicheng Liu, Xiao-Yan Liu, Yi-Ning Li, and Peng Zhang. 2023. "Optoelectronic Evolution in Halogen-Doped Organic–Inorganic Halide Perovskites: A First-Principles Analysis" Molecules 28, no. 21: 7341. https://doi.org/10.3390/molecules28217341