
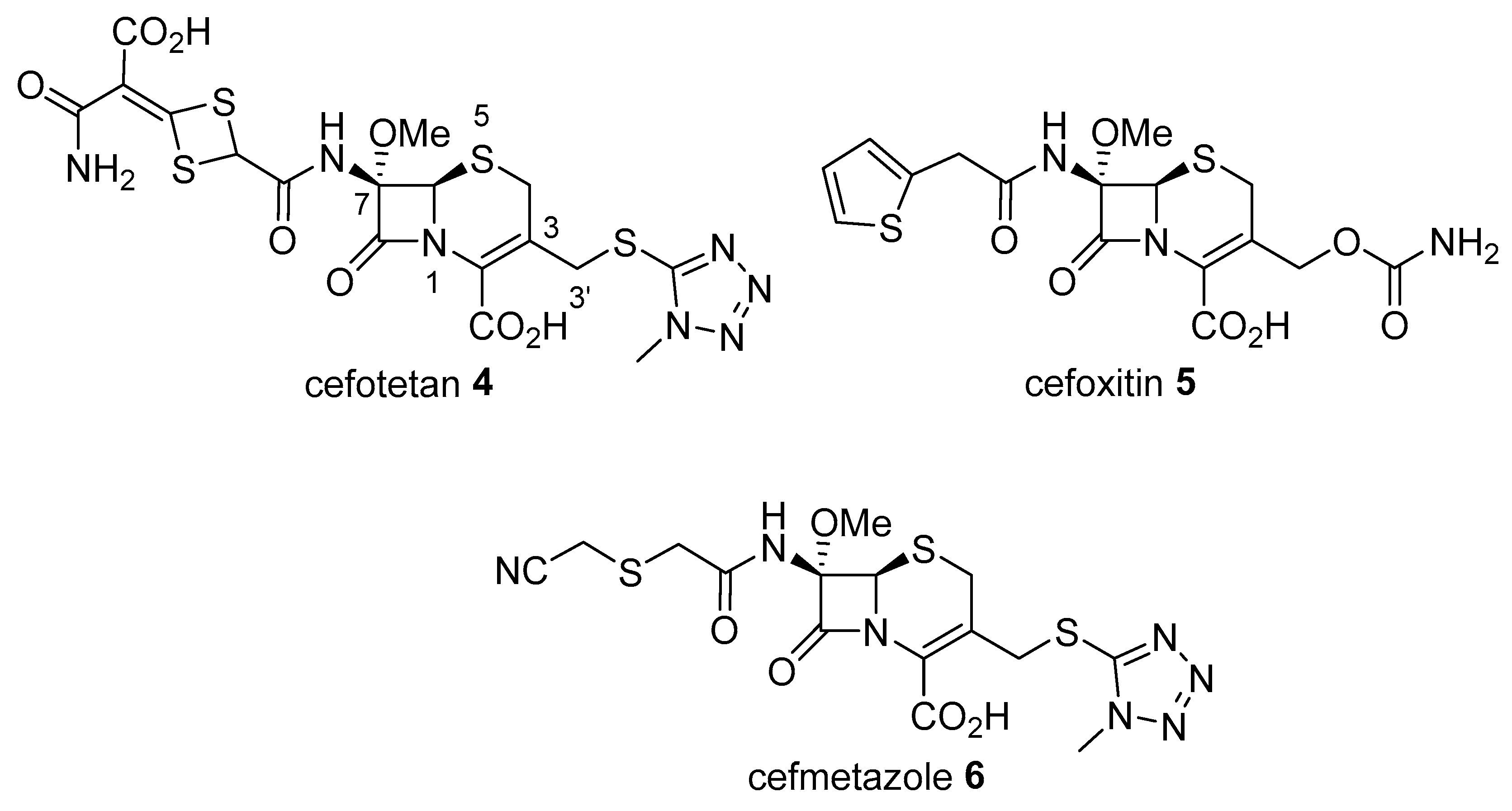
Synthesis of 7α-Methoxy-7-(4-phenyl-1H-1,2,3-triazol-1-yl)acetamino-3′-arylthio-cephalosporic Acid Derivatives from 7-Aminocephalosporic Acid


Abstract
:1. Introduction
2. Results and Discussion
3. Experimental
3.1. General Statement
3.2. Characterization and Analysis
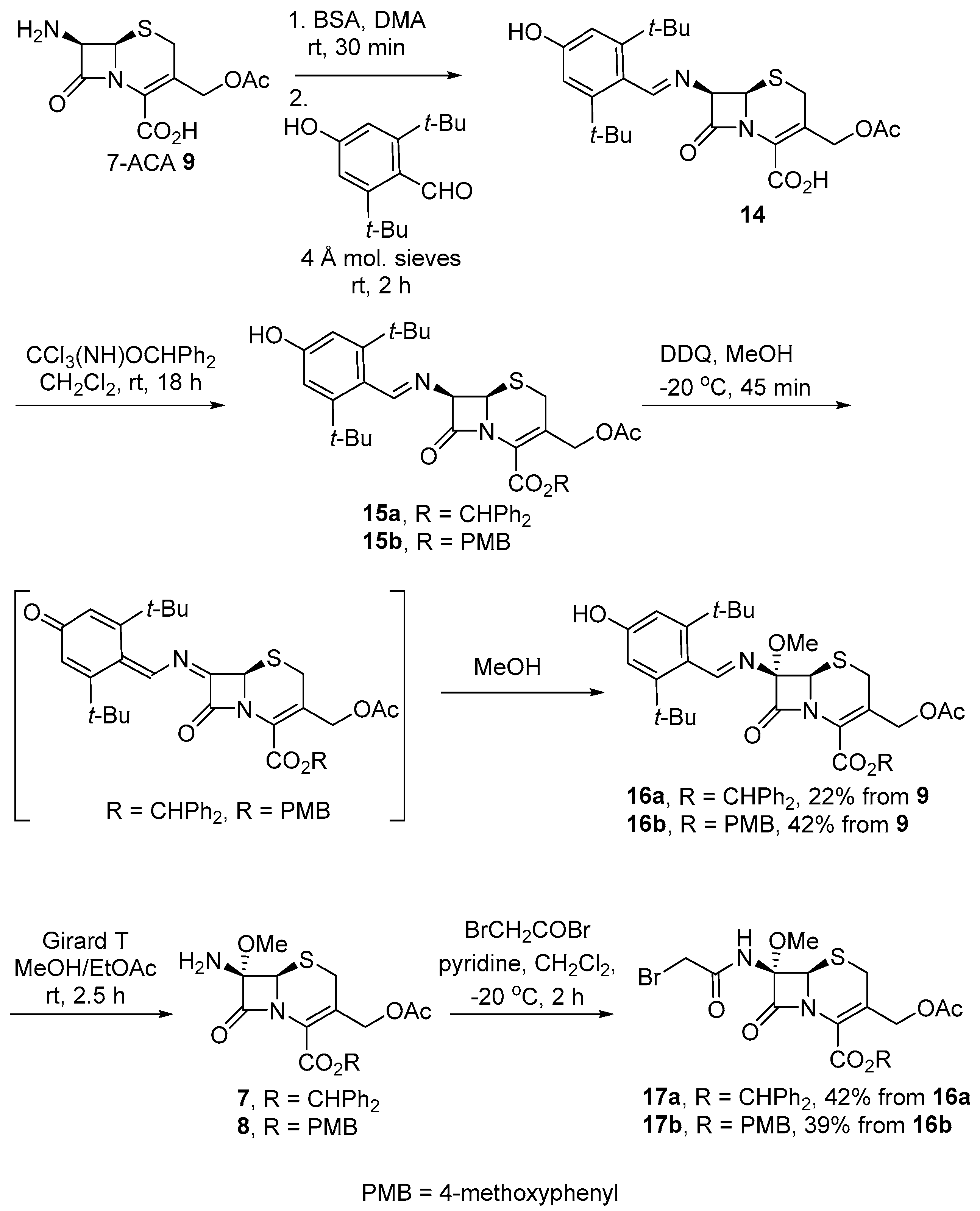
- (7S)-3-(Acetoxymethyl)-7-benzamido-7-methoxy-8-oxo-5-thia-1-azabicyclo[4.2.0]-oct-2-ene-2-carboxylic acid 12
- To a suspension of 7-ACA 9 (307.4, 1.13 mmol) in anhydrous DMA (5 mL) under N2 at rt, BSA (469.3 μL, 1.92 mmol, 1.7 equiv.) was added and stirred for 30 min until the mixture turned clear. The reaction mixture was then cooled to −20 °C (NaCl/ice), and phenylacetyl chloride (179.2 μL, 1.36 mmol, 1.2 equiv.) was added dropwise. The resulting solution was stirred at −20 °C for 2 h, at which point the reaction was shown to be complete via TLC analysis (TLC (MeOH/CH2Cl2—2:3): Rf = 0.63). The reaction mixture was poured into iced water (20 mL) and extracted using EtOAc (3 × 20 mL). The combined EtOAc layer was washed with water (3 × 20 mL), brine (20 mL), dried over anhydrous MgSO4, filtered, and concentrated in vacuo to give a pale-yellow residue. The obtained crude product was then dissolved in a minimum amount of EtOAc, and hexanes were added dropwise to this vigorously stirred solution until precipitation started to occur. The mixture was stirred at rt overnight. The solvent was removed using a syringe and the precipitate was washed with hexanes (5 × 10 mL), then dried in vacuo to afford the titled compound as an off-white powder (258.1 mg, 0.661 mmol, 58%). 1H NMR (500 MHz, CD3COCD3) δ 7.95 (d, J = 8.7 Hz, 1H, NH), 7.26–7.07 (m, 5H, ArCH), 5.71 (dd, J = 8.7, 4.8 Hz, 1H, H7), 5.00 (d, J = 4.9 Hz, 1H, H6), 4.83 (ABq, JA,B = 13.1 Hz, 2H, O-CH2, acetate), 3.58–3.47 (m, 3H, CH2Ph and H4A), 3.39 (d, J = 18.3 Hz, 1H, H4B), 1.90 (s, 3H, CH3); the 1H NMR spectroscopic data were in agreement with those previously reported [18]. MS (ESI +ve) m/z 413 ([M + Na]+, 100%), 429 ([M + K]+, 74%), 803 ([2M + Na]+, 21%); (ESI–ve) m/z 389 ([M − H]−, 65%), 779 ([2M − H]−, 100%).
- To a solution of diphenylmethanol (317.3 mg, 1.72 mmol) in anhydrous CH2Cl2 (2 mL) DBU (25.8 μL, 0.172 mmol, 0.1 equiv.) and CCl3CN (1.73 mL, 17.2 mmol, 10 equiv.) were added at rt under an Ar atmosphere. The reaction mixture was stirred at 40 °C overnight, at which point the reaction was shown to be complete via TLC analysis (TLC (3% Et3N in toluene): Rf = 0.71). Reaction mixture was concentrated in vacuo and purified via flash chromatography over SiO2 (3% Et3N in hexane/EtOAc—80:1) to give the titled compound as a white solid (375.8 mg, 1.14 mmol, 66%). 1H NMR (500 MHz, CDCl3) δ 8.41 (s, 1H, NH), 7.50–7.23 (m, 10H, ArCH), 6.94 (s, 1H, CHPh2); the 1H NMR spectroscopic data agreed with those previously reported [19,20].
- Benzhydryl (6R,7R)-3-(acetoxymethyl)-8-oxo-7-(2-phenylacetamido)-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylate 13 [17]
- To a suspension of 12 (99.1 mg, 0.254 mmol) in anhydrous CH2Cl2 (2 mL) benzhydryl 2,2,2-trichloroacetimidate (108.4 mg, 0.330 mmol, 1.3 equiv.) was added at rt under an Ar atmosphere. The reaction mixture was stirred for 1 h until it turned into a clear pale-yellow solution. The completion of the reaction was indicated via TLC analysis (TLC (MeOH/CH2Cl2—1:9): Rf = 0.86). The obtained crude product was then dissolved in a minimum amount of EtOAc, and hexane was added dropwise to this vigorously stirred solution until precipitation started to occur. The mixture was stirred at rt overnight. The solvent was removed using a syringe and the precipitate was washed with hexanes (5 × 10 mL), then dried in vacuo to afford the titled compound as an off-white powder (123.9 mg, 0.222 mmol, 88%). 1H NMR (500 MHz, CDCl3) δ 7.44–7.25 (m, 15H, Ar-CH), 6.93 (s, 1H, CHPh2), 6.02 (d, J = 9.1 Hz, 1H, NH), 5.86 (dd, J = 9.0, 4.9 Hz, H7), 4.88 (ABq, JA,B = 13.6 Hz, 2H, O-CH2, acetate), 4.95 (d, J = 4.9 Hz, H6), 3.65 (ABq, JA,B = 16.2 Hz, 2H, CH2Ph), 3.43 (ABq, JA,B = 18.6 Hz, 2H, H4), 2.01 (s, 3H, CH3); the 1H NMR spectroscopic data agreed with those previously reported [17]; MS (ESI + ve) m/z 630 ([M + Na]+, 100%); (ESI –ve) m/z 686 ([M − H]−, 62%).

- (6R,7R)-3-(Acetoxymethyl)-7-(((E)-3,5-di-tert-butyl-4-hydroxybenzylidene)-amino)-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid 14
- To a suspension of 7-ACA 9 (134.0 mg, 0.492 mmol) in DMA (1 mL) BSA (204.6 µL, 1.29 mmol, 1.7 equiv.) was added, and the reaction mixture was stirred at rt for 30 min until the white suspension turned into an orange solution. Powdered molecular sieves (30 mg, 4 Å) and 3,5-di-tert-butyl-4-hydroxybenzaldehyde (121.1 mg, 0.517 mmol, 1.05 equiv.) were then added to the reaction mixture and stirred at rt for 2 h until the pale-orange color of the reaction turned into bright yellow. The completion of the reaction was indicated via TLC analysis (TLC (MeOH/CH2Cl2—2:3): Rf = 0.76). A portion of the reaction mixture was diluted with MeOH (10 mL) and filtered. The obtained filtrate was concentrated for NMR analysis, which indicated a mixture of the titled compound and 3,5-di-tert-butyl-4-hydroxybenzaldehyde. The reaction mixture was concentrated under a stream of N2 overnight to give crude product 14 as a yellow gum. 1H NMR (500 MHz, CD3SOCD3) δ 8.44 (s, 1H, H9), 7.57 (s, 2H, H10), 5.53 (d, J = 4.9, 1H, H7), 5.28 (d, J = 5.0 Hz, 1H, H6), 4.84 (ABq, JA,B = 12.7 Hz, 2H, H3′), 3.53 (ABq, JA,B = 17.3 Hz, 2H, H4), 2.04 (s, 3H, C4′-CH3), 1.39 (s, 18H, 2 × C-(CH3)3); (ESI +ve) m/z 489 ([M + H]+, 58%).
- Benzhydryl (6R,7R)-3-(acetoxymethyl)-7-(((E)-3,5-di-tert-butyl-4-hydroxy-benzylidene)amino)-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylate 15a
- To a suspension of crude product 14 (0.735 mmol, prepared from 200.1 mg of 7-ACA) in CH2Cl2 (2 mL) under an Ar atmosphere at rt benzhydryl 2,2,2-trichloroacetimidate (314.0 mg, 0.956 mmol, 1.3 equiv.) was added. The color of the reaction turned from bright yellow to brown, and the completion of the reaction was indicated using TLC analysis (TLC (MeOH/CH2Cl2—1:9): Rf = 0.74). The reaction mixture was concentrated in vacuo and the obtained brown residue was suspended in MeOH (30 mL) and filtered. The filtrate was concentrated for NMR analysis, which indicated a mixture of the titled compound, DMA and 3,5-di-tert-butyl-4-hydroxybenzaldehyde. Further purification using flash chromatography resulted in decomposition. 1H NMR (500 MHz, CD3OD) δ 8.45 (s, 1H, H9), 7.65 (s, 2H, H10), 7.46–7.22 (m, 10H, Ar-CH), 6.94 (s, 1H, CHPh2), 5.46 (d, J = 5.1 Hz, 1H, H7), 5.24 (d, J = 5.1 Hz, 1H, H6), 4.98 (d, J = 13.1 Hz, 1H, H3′A), 4.71 (d, J = 13.1 Hz, 1H, H3ʹB), 3.56 (ABq, JA,B = 18.5 Hz, 2H, H4), 1.98 (s, 3H, C4′-CH3), 1.46 (s, 18H, 2 × C-(CH3)3); the 1H NMR spectroscopic data agreed with those previously reported [22]; (ESI +ve) m/z 655 ([M + H]+, 51%), 677 ([M + Na]+, 100%); (ESI −ve) m/z 653 ([M−H]−, 100%).
- Benzhydryl (6R,7S)-3-(acetoxymethyl)-7-(((E)-3,5-di-tert-butyl-4-hydroxy-benzylidene)amino)-7-methoxy-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylate 16a
- To a suspension of crude product 15a from the previous steps (0.163 mmol, prepared from 44.5 mg of 7-ACA 9) in MeOH (1.5 mL) at −20 °C DDQ (37.0 mg, 0.163 mmol, 1.0 equiv.) was added, and the reaction mixture was stirred at −20 °C for 45 min. The completion of the reaction was indicated using MS analysis. EtOAc (1.5 mL) was then added to the reaction and the resulting mixture was stirred for another 30 min. The dark red reaction mixture was concentrated in vacuo and purified via flash chromatography over SiO2 (EtOAc/hexane—1:10–3:10) to give the title compound as a hydroscopic, yellow foam (24.6 mg, 0.0359 mmol, 22% from 7-ACA 9).
- A series of solvents in a EtOAc/hexane system were prepared (using 3.6 mmol crude product as an example: 100 mL—1:19, 200 mL—1:9, 100 mL—3:17, 100 mL—1:4, 100 mL—11:39, 100 mL—6:19, 100 mL—13:37, 100 mL—7:18, and 200 mL—3:7) and chilled at −20 °C overnight. On the following day, an appropriately sized column was packed with a SiO2 slurry in hexane and chilled at −20 °C until ready to use. Once the crude product was loaded onto the column, the pre-chilled solvents was kept cool on an ice bath and the flow rate was accelerated by using a stream of compressed air to ensure the entire purification process was kept within 30 min. 1H NMR (500 MHz, CDCl3) δ 8.54 (s, 1H, H9), 7.69 (s, 2H, H10), 7.51–7.29 (m, 10H, Ar-CH), 6.97 (s, 1H, CHPh2), 5.64 (s, 1H, OH), 5.08 (s, 1H, H6), 4.85 (ABq, JA,B = 13.4 Hz, 2H, H3′), 3.57 (s, 3H, O-CH3), 3.39 (ABq, JA,B = 18.2 Hz, 2H, H4), 2.00 (s, 3H, C4′-CH3), 1.46 (s, 18H, 2 × C-(CH3)3); the 1H NMR spectroscopic data agreed with those previously reported [13]; (ESI +ve) m/z 685 ([M + H]+, 100%), 707 ([M + Na]+, 79%); (ESI−ve) m/z 683 ([M − H]−, 81%).
- Benzhydryl (6R,7S)-3-(acetoxymethyl)-7-amino-7-methoxy-8-oxo-5-thia-azabicyclo[4.2.0]oct-2-ene- 2-carboxylate 7
- To a solution of 16a (56.4 mg, 0.0824 mmol) in EtOAc (0.5 mL) a solution of Girard-T reagent (27.6 mg, 0.165 mmol, 2.0 equiv.) was added in MeOH (0.6 mL) at rt, and the resulting solution was stirred for 2.5 h until the completion of the reaction was indicated by MS analysis. Upon completion, the reaction mixture was diluted with EtOAc (15 mL) and poured into water (20 mL), which was extracted with additional EtOAc (15 mL × 2). The combined EtOAc layer was washed with brine (20 mL), dried over anhydrous Na2SO4, filtered, and concentrated in vacuo to give the crude product as a dark green gum. Further purification of the crude product resulted in decomposition and it was, therefore, used directly for the subsequent reaction. 1H NMR (500 MHz, CDCl3) δ 7.41–7.27 (m, 10H, ArCH), 6.94 (s, 1H, CHPh2), 5.03 (d, J = 13.7 Hz, 1H, H3′A), 4.85 (s, 1H, H6), 4.83 (d, J = 13.7 Hz, 1H, H3′B), 3.52 (s, 3H, O-CH3), 3.44 (d, J = 17.2 Hz, 1H, H4A), 3.31 (d, J = 17.2 Hz, 1H, H4B), 2.02 (s, 3H, C4′-CH3); (ESI +ve) m/z 469 ([M + H]+, 11%), 491 ([M + Na]+, 100%), and 501 ([M + MeOH + H]+, 9%). The sample of 7 also contained (E)-2-(2-(3,5-di-tert-butyl-4-hydroxybenzylidene)hydrazineyl)-N,N,N-trimethyl-2-oxoethan-1-aminium chloride in a 1:1 ratio from 1H NMR analysis: 1H NMR (500 MHz, CDCl3) δ 8.38 (s, 1H, H3), 7.57 (s, 2H, H4), 4.65 (s, 2H, H2), 3.42 (s, 9H, H1), 1.43 (s, 18H).
- Benzhydryl (6R,7S)-3-(acetoxymethyl)-7-(2-bromoacetamido)-7-methoxy-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylate 17a
- To a solution of crude product 7 (0.0813 mmol, prepared from 55.6 mg of 16a from the previous reaction in anhydrous CH2Cl2 (1 mL) under N2 at −20 °C (NaCl/ice) pyridine (20 μL, 0.248 mmol, 3.1 equiv.) was added and the resulting solution was stirred for 2 min. Bromoacetyl bromide (19 μL, 0.216 mmol, 2.7 equiv.) was then added dropwise to the solution. The reaction mixture was stirred at −20 °C for 2 h, at which point the reaction was shown to be complete via TLC analysis (TLC (EtOAc/hexane—2:3): Rf = 0.39). The reaction mixture was poured into EtOAc (5 mL); washed sequentially with HCl (1.0 M—5 × 5 mL), saturated NaHCO3 (5 × 5 mL), and brine (10 mL); dried over anhydrous Na2SO4; filtered; and concentrated in vacuo to give a brown residue. The crude product was purified via flash chromatography over SiO2 (EtOAc/hexane—1:19–9:11) to give the titled compound as a pale-yellow foam (20.3 mg, 0.0344 mmol, 42% from 16a). +109.45 (c 0.22, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 7.51–7.23 (m, 10H, ArCH), 6.95 (s, 1H, CHPh2), 5.09 (s, 1H, H6), 4.95 (ABq, JA,B = 13.8 Hz, 2H, H3ʹ), 3.92 (ABq, JA,B = 13.6 Hz, 2H, Br-CH2), 3.58 (s, 3H, O-CH3), 3.39 (ABq, JA,B = 18.2 Hz, 2H, H4), 2.02 (s, 3H, C4′-CH3); NH signal was not observed; 13C NMR (101 MHz, CDCl3) δ 170.4 (C4′), 166.6 (C=O, amide), 160.2 (C2ʹ), 160.1 (C8), 139.3 (Cq, benzhydryl), 139.1 (Cq, benzhydryl), 132.0 (C3), 128.57 (ArCH), 128.55 (ArCH), 128.5 (ArCH), 128.2 (ArCH), 128.2 (ArCH), 127.5 (ArCH), 127.04 (ArCH), 127.01(ArCH), 125.9 (C2), 95.8 (C7), 79.7(CH-Ph2), 64.3 (C6), 62.4 (C3′), 54.0 (O-CH3), 27.9 (Br-CH2), 27.0 (C4), 20.64 (CH3); IR (cm−1) max 3278 (w), 2960 (w), 1775 (s, β-lactam C=O), 1735 (s, C=O, acetate), 1608 (m), 1517 (s), 1380 (m), 1237 (s, C-O stretching, acetate), 1129 (m), 1090 (m), 1032 (m), 749 (w), 700 (m); MS (ESI + ve) m/z 611 ([79BrM + Na]+, 90%), 613 ([81BrM + Na]+, 100%); HRMS (ESI +ve TOF) calcd for 79BrC26H25N2O7SNa 611.0464, found 611.0435 ([M + Na]+).
- 4-Methoxybenzyl (6R,7R)-3-(acetoxymethyl)-7-(((E)-3,5-di-tert-butyl-4-hydroxy-benzylidene)amino)-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylate 15b
- To a suspension of crude product 14 (0.757 mmol, prepared from 206.1 mg of 7-ACA 9) in CH2Cl2 (2 mL) under an Ar atmosphere at rt 4-methoxybenzyl 2,2,2-trichloroacetimidate (204.3 µL, 0.984 mmol, 1.3 equiv.) was added, and the resulting mixture was stirred at rt for 48 h. The color of the reaction turned from bright yellow to brown, and the completion of the reaction was indicated using MS analysis. The reaction mixture was filtered and concentrated in vacuo to give the crude product 15 as a sticky brown gum. A portion of this filtrate was used for NMR analysis, which indicated a mixture of the titled compound, DMA, and 3,5-di-tert-butyl-4-hydroxybenzaldehyde. Further purification using flash chromatography resulted in decomposition. 1H NMR (500 MHz, CD3OD) δ 8.42 (s, 1H, H9), 7.63 (s, 2H, H10), 7.35 (d, J = 8.7 Hz, 2H, ArCH, PMB), 6.90 (d, J = 8.8 Hz, 2H, ArCH, PMB), 5.40 (d, J = 5.2 Hz, 1H, H7), 5.25–5.15 (m, 3H, H6 and O-CH2), 4.99 (d, J = 13.0 Hz, 1H, H3′A), 4.76 (d, J = 13.0 Hz, 1H, H3′B), 3.78 (s, 3H, OCH3), 3.64 (d, J = 18.4 Hz, 1H, H4A), 3.46 (d, J = 18.4 Hz, 1H, H4B), 2.02 (s, 3H, C4′-CH3), 1.44 (s, 18H, 2 × C-(CH3)3); (ESI + ve) m/z 609 ([M + H]+, 81%) and 631([M + H]+, 76%); (ESI − ve) m/z 607 ([M – H]–, 100%).].
- Benzhydryl (6R,7S)-3-(acetoxymethyl)-7-(2-bromoacetamido)-7-methoxy-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylate 16b
- The crude compound 15b was suspended in MeOH (5 mL) at −20 °C under an Ar atmosphere. To the suspension DDQ (171.8 mg, 0.757 mmol, 1.0 equiv.) was added, and the reaction mixture was stirred at −20 °C for 45 min. The completion of the reaction was indicated via TLC analysis (TLC (EtOAc/hexane—2:3): Rf = 0.39). The reaction mixture was concentrated in vacuo and purified via column chromatography over SiO2. Prior to the purification, a series of solvents in a EtOAc/hexane system were prepared (using 3.6 mmol crude product as an example: 100 mL—1:19, 200 mL—1:9, 100 mL—3:17, 100 mL—1:4, 100 mL—11:39, 100 mL—6:19, 100 mL—13:37, 100 mL—7:18, 200 mL—3:7, and 200 mL—7:20) and chilled at −20 °C overnight. On the following day, an appropriately sized column was packed with a SiO2 slurry in hexane and chilled at −20 °C until ready to use. Once the crude product was loaded onto the column, the pre-chilled solvents was kept cool on an ice bath and the flow rate was accelerated by using a stream of compressed air to ensure the entire purification process was kept within 30 min. Fractions with Rf value of 0.39 (TLC (EtOAc/hexane—2:3)) were combined and concentrated in vacuo to give the title compound as a hydroscopic, pale-yellow foam (204.1 mg, 0.320 mmol, 42% from 7-ACA). +89.2 (c 0.27, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 8.51 (s, 1H, H9), 7.69 (s, 2H, H10), 7.39 (d, J = 8.8 Hz, 2H, ArCH, PMB), 6.90 (d, J = 8.8 Hz, 2H, ArCH, PMB), 5.63 (s, 1H, OH), 5.27 (ABq, JA,B = 11.6 Hz, 2H, O-CH2), 5.05 (s, 1H, H6), 4.87 (ABq, JA,B = 13.2 Hz, 2H, H), 3.81 (s, 3H, OCH3, PMB), 3.55 (s, 3H,OCH3, β-lactam), 3.38 (ABq, JA,B = 18.3 Hz, 2H, H4), 2.03 (s, 3H, C4′-CH3), 1.46 (s, 18H, C-(CH3)3); 13C NMR (101 MHz, CDCl3) δ 170.6 (C4′), 165.8 (C9), 163.4 (C8), 161.5 (C2′), 160.0 (Cq, PMB), 157.9 (C-OH), 136.3 (C-C(CH3)3), 130.7 (ArCH, PMB), 126.9 (C10), 126.5 (C-C9), 126.2 (Cq, PMB), 124.2 (C2), 114.0 (C12), 104.6 (C7), 68.1 (CH2, PMB ester), 64.5 (C6), 63.1 (C3′), 55.3 (OCH3, PMB), 53.5 (OCH3, β-lactam), 34.4 (C(CH3)3), 30.2 (C(CH3)3), 26.6 (C4), 20.7 (CH3, acetate); C3 resonance was not observed in the 13C NMR or HMBC spectra; IR (cm−1) max 3613 (w), 2958 (m), 1772 (s, β-lactam C=O), 1737 (s, C=O, acetate), 1631 (m), 1516 (m), 1429 (m), 1389 (m), 1302 (w), 1235 (s, C-O stretching, acetate), 1176 (m), 1129 (m), 1093 (m), 1034 (m), 888 (w), 827 (m), 775 (w); MS (ESI +ve) m/z 639 ([M + H]+, 23%), 661 ([M + Na]+, 100%), 1300 ([2M + Na]+, 77%); (ESI − ve) m/z 637 ([M − H]−, 100%); HRMS (ESI + ve TOF) calcd for C34H43N2O8S 639.2740, found 639.2752 ([M + H]+).
- 4-Methoxybenzyl (6R,7S)-3-(acetoxymethyl)-7-amino-7-methoxy-8-oxo-5-thia-1 azabicyclo[4.2.0]oct-2-ene-2-carboxylate 8
- To a solution of 16b (333.5 mg, 0.487 mmol) in EtOAc (2.4 mL) a solution of Girard-T reagent (163.3 mg, 0.974 mmol, 2.0 equiv.) in MeOH (2.9 mL) at rt was added, and the resulting solution was stirred for 3.5 h until the completion of the reaction was indicated via TLC analysis (TLC (EtOAc/hexane—1:1): Rf = 0.24). Upon completion, the reaction mixture was diluted with EtOAc (50 mL) and poured into water (50 mL), which was extracted using additional EtOAc (50 mL × 2). The combined EtOAc layer was washed with brine (70 mL), dried over anhydrous Na2SO4, filtered, and concentrated in vacuo to give the crude product as a dark green gum. A portion of this crude product (0.216 mmol) was purified via flash chromatography over SiO2 (EtOAc/hexane—1:10–2:3) to give the titled compound as a green gum (15.4 mg, 0.0365 mmol, 17%). + 48.74 (c 0.77, CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 7.35 (d, J = 8.4 Hz, 3H, ArCH, PMB), 6.88 (d, J = 8.5 Hz, 2H, ArCH, PMB), 5.23 (ABq, JA,B = 12.3 Hz, 2H, O-CH2), 5.02 (d, J = 13.4 Hz, 1H, H3′A), 4.85–4.76 (m, 2H, H6 and H3′B), 3.80 (s, 3H, OCH3, PMB), 3.53–3.25 (m, 5H, H4 and OCH3, β-lactam), 2.04 (s, 3H, C4′-CH3); 13C NMR (126 MHz, CDCl3) δ 170.8 (C4′), 164.1 (C8), 161.5 (C2′), 160.1 (Cq, PMB), 130.8 (ArCH, PMB), 127.1 (Cq, PMB), 126.7 (C2), 114.1 (ArCH, PMB), 98.6 (C7), 68.2 (CH2, PMB ester), 63.9 (C6), 63.0 (C3′), 55.5 (OCH3, PMB), 52.6 (OCH3, β-lactam), 26.8 (C4), 20.9 (CH3, acetate); C3 resonance was not observed in the 13C NMR or HMBC spectra; IR (cm−1) max 3307 (w), 2956 (w), 2930 (w), 1777 (s, β-lactam C=O), 1725 (s, C=O, acetate), 1612 (m), 1514 (s), 1455 (w), 1379 (m), 1354 (m), 1219 (s, C-O stretching, acetate), 1174 (s), 1112 (m), 1026 (s), 821 (s), 728 (m); MS (ESI + ve) m/z; 445 ([M + Na]+, 100%), 468 ([M + 2Na − H]+, 83%), 867 (2M + Na]+, 35%); HRMS (ESI + ve TOF) calcd for C19H22N2O7SNa 455.1045, found 455.1063 ([M + Na]+).
- 4-Methoxybenzyl (6R,7S)-3-(acetoxymethyl)-7-(2-bromoacetamido)-7-methoxy-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylate 17b
- To a solution of the crude compound 7b (0.320 mmol, prepared from 204.1 mg of 16b) in anhydrous CH2Cl2 (3 mL) under N2 at –20 °C (NaCl/ice) pyridine (80.2 μL, 0.992 mmol, 3.1 equiv.) was added and the resulting solution was stirred for 2 min. Bromoacetyl bromide (75.3 μL, 0.864 mmol, 2.7 equiv.) was then added dropwise to the solution. The reaction mixture was stirred at –20 °C for 2 h, at which point the reaction was shown to be complete via TLC analysis (TLC (EtOAc/hexane—2:3): Rf = 0.32). The reaction mixture was poured into EtOAc (5 mL); washed sequentially with HCl (1.0 M—5 × 50 mL), saturated NaHCO3 (5 × 50 mL), and brine (100 mL); dried over anhydrous Na2SO4, filtered, and concentrated in vacuo to give a brown residue. The crude product was purified via flash chromatography over SiO2 (EtOAc/hexane—1:19–9:11) to give the titled compound as an orange gum (67.5 mg, 0.126 mmol, 39% over from 16b). +122.96 (c 1.08, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 7.36 (d, J = 8.7 Hz, 2H, ArCH, PMB), 6.89 (d, J = 8.7 Hz, 2H, ArCH, PMB), 5.25 (ABq, JA,B = 11.9 Hz, 2H, H9), 5.07 (s, 1H, H6), 4.94 (ABq, JA,B = 13.7 Hz, 2H, H3′), 3.93 (ABq, JA,B = 13.7 Hz, 2H, Br-CH2), 3.81 (s, 3H, OCH3, PMB), 3.56 (s, 3H, OCH3, β-lactam), 3.40 (ABq, JA,B = 18.0 Hz, 2H, H4), 2.05 (s, 3H, C4′-CH3), NH signal was not observed; 13C NMR (101 MHz, CDCl3) δ 170.5 (C4′), 166.7 (C=O, amide), 160.9 (C2′), 160.1 (C8), 159.9 (Cq, PMB), 130.6 (ArCH, PMB), 126.8 (ArCH, PMB), 126.0 (C2), 114.0 (C12), 95.6 (C7), 68.1 (CH2, PMB ester), 62.6 (C3′), 55.3 (OCH3, PMB), 53.9 (OCH3, β-lactam), 27.9 (Br-CH2), 26.9 (C4), 20.7 (CH3, acetate); C3 resonance was not observed in the 13C NMR or HMBC spectra; IR (cm−1) max 3279 (w), 2962 (w), 2838 (w), 1777 (s, β-lactam C=O), 1735 (s, C=O, acetate), 1613 (m), 1516 (s), 1392 (m), 1240 (s, C-O stretching, acetate), 1178 (m), 1130 (m), 1088 (m), 10320 (m), 853 (w); MS (ESI + ve) m/z 565 ([79BrM + Na]+, 86%), 567 ([81BrM + Na]+, 100%), 588 ([79BrM + 2Na − H]+, 21%), 590 ([81BrM + 2Na − H]+, 24%); HRMS (ESI + ve TOF) calcd for 79BrC21H23N2O8SNa 565.0256, found 565.0266 ([M + Na]+).
- (6R,7S)-3-(Acetoxymethyl)-7-(2-bromoacetamido)-7-methoxy-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid 18
- Method 1—prepared from compound 17a. To a solution of compound 17a (76.5 mg, 0.130 mmol) in anisole (451 μL, 4.15 mmol, 32 equiv.) TFA was added (745 μL, 9.73 mmol, 75 equiv.), and the solution was stirred at rt for 10 min. The reaction mixture was poured into EtOAc (50 mL) and extracted with saturated NaHCO3 (3 × 50 mL). The combined aqueous layer was washed with EtOAc (2 × 50 mL). Additional EtOAc (200 mL) was added to a stirred solution of the aqueous layer, and the aqueous layer was acidified with conc. HCl to pH < 1. The two layers were then separated. The aqueous layer was extracted with EtOAc (2 × 50 mL), and the combined organic layer was washed sequentially with HCl (1.0 M—3 × 50 mL) and brine (50 mL), dried over anhydrous Na2SO4, filtered, and concentrated in vacuo to give a yellow gum. This crude product was redissolved in a minimum amount of EtOAc, and hexane was added dropwise to the vigorously stirred solution of EtOAc until precipitation started to occur. The mixture was stirred at rt overnight. The solvent was removed using a syringe and the precipitate was washed with hexanes (5 × 10 mL) then dried in vacuo to afford the titled compound as a yellow gum (41.0 mg, 0.0969 mmol, 75%).
- Method 2—prepared from compound 17b. To a solution of compound 17b (67.5 mg, 0.124 mmol) in anhydrous CH2Cl2 (1 mL) at 0 °C TFA was slowly added (247 μL, 3.23 mmol, 26 equiv.). The resulting dark-pink solution was stirred at 0 °C for 30 min. Work-up and precipitation as in Method 1 gave the titled compound as a yellow gum (47.1 mg, 0.111 mmol, 90%). +143.50 (c 1.41, MeOH); 1H NMR (400 MHz, CD3CN) δ 7.79 (s, 1H, NH), 5.06 (s, 1H, H6), 4.87 (ABq, JA,B = 13.4 Hz, 2H, H3′), 3.89 (s, 2H, Br-CH2), 3.56 (d, J = 18.0 Hz, 1H, H4A), 3.50 (s, 3H, OCH3), 3.33 (d, J = 18.0 Hz, 1H, H4B), 2.02 (s, 3H, CH3, acetate); 13C NMR (101 MHz, CD3CN) δ 171.1 (C4′), 167.9 (C=O, amide), 162.4 (C2′), 160.9 (C8), 128.7 (C3), 126.1 (C2), 95.6 (C7), 64.2 (C6), 63.0 (C3′), 53.6 (O-CH3), 28.5 (Br-CH2), 26.6 (C4), 20.5 (CH3, acetate); IR (cm−1) max 3538 (w), 3271 (w), 3022 (w), 1774 (s, β-lactam C=O),1728 (s, C=O, acetate), 1693 (s), 1539 (m), 1386 (m), 1235 (s, C-O stretching, acetate), 1134 (m), 1088 (m), 1024 (m); MS (ESI + ve) m/z 445 ([79BrM + Na]+, 93%), 447 ([81BrM + Na]+, 100%); HRMS (ESI + ve TOF) calcd for 79BrC13H15N2O7S2Na 444.9681, found 444.9699 ([M + Na]+).
- (6R,7S)-3-(Acetoxymethyl)-7-(2-azidoacetamido)-7-methoxy-8-oxo-5-thia-1-azabicyclo [4.2.0]oct-2-ene-2-carboxylic acid 19
- To a solution of compound 18 (36.7 mg, 0.0867 mmol) in DMF (0.3 mL) at −15 °C sodium azide (28.2 mg, 0.434 mmol, 5.0 equiv.) was added, and the solution was stirred for 24 h, at which point the reaction was shown to be complete via MS analysis. The reaction mixture was diluted with distilled H2O (20 mL), to which EtOAc (20 mL) was added. The resulting mixture was stirred vigorously, and the aqueous layer was acidified with conc. HCl to pH < 1. The two layers were separated, and the aqueous layer was extracted with EtOAc (3 × 20 mL). The combined organic layer was washed sequentially with distilled H2O (3 × 20 mL) and brine (40 mL), dried over anhydrous Na2SO4, filtered, and concentrated in vacuo to give a sticky gum. This crude product was dissolved in a minimum amount of EtOAc, and hexane was added dropwise to the vigorously stirred solution until the precipitation started to occur. The mixture was stirred at rt overnight. The solvent was removed using a syringe and the precipitate was washed with hexanes (5 × 10 mL), then dried in vacuo to afford the titled compound as a pale-yellow gum (27.1 mg, 0.0703 mmol, 81%). +158.3 (c 0.77, MeOH); 1H NMR (400 MHz, CD3CN) δ 7.58 (s, 1H, NH), 5.06 (s, 1H, H6), 4.88 (ABq, JA,B = 13.3 Hz, 2H, H3′), 3.95 (s, 2H, Br-CH2), 3.57 (d, J = 18.0 Hz, 1H, H4A), 3.50 (s, 3H, OCH3), 3.34 (d, J = 18.0 Hz, 1H, H4B), 2.02 (s, 3H, CH3, acetate); 13C NMR (101 MHz, CD3CN) δ 171.1 (C4′), 169.2 (C=O, amide), 162.5 (C2′), 161.0 (C8), 128.6 (C3), 126.3 (C2),96.3 (C7), 64.1 (C6), 63.0 (C3′), 53.6 (O-CH3), 51.6 (N3-CH2), 26.7 (C4), 20.5 (CH3, acetate); IR (cm−1) max 3385 (m), 3223 (m), 3026 (w), 2111 (N=N=N stretching), 1772 (s, β-lactam C=O), 1705 (s) 1514 (m), 1424 (m), 1385 (m), 1230 (s, C-O stretching, acetate), 1134 (m), 1087 (m), 1026 (m), 551 (w); MS (ESI +ve) m/z 403 ([M+NH4]+, 100%), 408 ([M + Na]+, 79%), 431 ([M + 2Na − H]+, 74%), 449 ([M + MeCN + Na]+, 20%); (ESI − ve) m/z 384 ([M − H]−, 37%), 420 ([M + Cl]−, 65%), 422 ([M + K − 2H]−, 28%), 498 ([M + TFA − H]−, 64%); HRMS (ESI + ve TOF) calcd for C13H15N5O7S2Na 408.0590, found 408.0581 ([M + Na]+).

- (6R,7S)-3-(Acetoxymethyl)-7-methoxy-8-oxo-7-(2-(4-phenyl-1H-1,2,3-triazol-1-yl)acetamido)-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid 20
- To a reaction vessel charged consecutively with azide 19 (27.1 mg, 0.0703 mmol), CuSO4·5H2O (3.50 mg, 0.0141 mmol, 0.2 equiv.) and sodium ascorbate (5.55 mg, 0.0281 mmol, 0.4 equiv.) a mixture of t-BuOH and H2O (t-BuOH/H2O—1:1, 0.8 mL) was added. To this stirred mixture, phenylacetylene (23.3 μL, 0.211 mmol, 3.0 eq.) was then added, and the reaction mixture was stirred at 30 ℃ for 24 h. The reaction mixture was diluted with EtOAc (20 mL), washed with saturated aqueous NH4Cl solution (20 mL), dried over anhydrous MgSO4, filtered, and concentrated in vacuo. The obtained residue was then dissolved in a minimum amount of EtOAc, and to this vigorously stirred solution hexanes were added dropwise until precipitation started to occur. The mixture was stirred at rt overnight. The solvent was removed using a syringe and the precipitate was washed with hexanes (5 × 10 mL), then dried in vacuo to afford the titled compound as a thin, yellow film (22.5 mg, 0.0461 mmol, 66%). +103.1 (c 0.54, MeOH); 1H NMR (400 MHz, CD3CN) δ 8.16 (s, 1H, CH, triazole), 7.93 (s, 1H, NH), 7.89–7.84 (m, 2H, H2″), 7.46 (t, J = 7.5 Hz, 2H, H3″), 7.39–7.33 (m, 1H, H4″), 5.28 (s, 2H, CH2-triazole), 5.06 (s, 1H, H6), 4.88 (ABq, JA,B = 13.3 Hz, 2H, H3′), 3.59–3.50 (m, 4H, H4A and O-CH3), 3.32 (d, J = 17.9 Hz, 1H, H4B), 2.02 (s, 3H, CH3, acetate); 13C NMR (101 MHz, CD3CN) δ 171.1 (C4′), 167.2 (C=O, amide), 162.6 (C2′), 160.9 (C8), 147.7 (Cq, triazole), 131.3 (C1″), 129.5 (C3″), 128.71 (C3) 128.68 (C4″), 126.3 (C2), 126.1 (C2″), 122.9 (CH, triazole), 96.4 (C7), 64.1 (C6), 63.0 (C3′), 53.8 (O-CH3), 52.4 (CH2-triazole), 26.7 (C4), 20.5 (CH3, acetate); IR (cm−1) max 3288 (w), 2946 (w), 1779 (s, β-lactam C=O), 1708 (s), 1533 (m), 1442 (w), 1382 (m), 1231 (s, C-O stretching, acetate), 1136 (m), 1087 (m) 1028 (m), 768 (s), 696 (m); MS (ESI + ve) m/z 488 ([M + H]+, 100%), 510 ([M + Na]+, 12%), 975 ([2M + H]+, 7%); (ESI − ve) m/z 486 ([2M − H]−, 65%); HRMS (ESI + ve TOF) calcd for C17H17N5O7SNa 510.1059, found 510.1050 ([M + Na]+).
- (6R,7S)-7-Methoxy-3-(((4-methoxyphenyl)thio)methyl)-8-oxo-7-(2-(4-phenyl-1H-1,2,3-triazol-1-yl)acetamido)-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid 21a
- This compound was prepared following a procedure reported in the literature with some modifications [27]. To a flame-dried flask Pd2(dba)3·CHCl3 (10.7 mg, 0.0103 mmol, 10 mol%), BIPHEPHOS (17.0 mg, 0.0216 mmol, 21 mol%), and anhydrous MeCN (2 mL) were added under Ar at 50 °C. The resulting suspension was stirred at 50 °C for 30 min until it turned into a bright-yellow solution. The reaction flask was allowed to cool to rt and was then placed in a sonicator with the water temperature at 30–35 °C. Compound 20 (50.4 mg, 0.103 mmol) and 4-methoxybenzenethiol (25.4 μL, 0.207 mmol, 2.0 equiv.) were added to the reaction flask. The resulting mixture was sonicated at 30–35 °C until the completion of the reaction (10 h) was indicated using MS analysis. The reaction mixture was poured into EtOAc (10 mL) and extracted with NaHCO3 (3 × 10 mL). The combined aqueous layer was acidified with conc. HCl to pH = 3 and extracted with EtOAc (3 × 10 mL). The combined EtOAc fraction was then washed with HCl (1.0 M—3 × 10 mL) and brine (20 mL), dried over anhydrous Na2SO4, filtered, and concentrated in vacuo to give a yellow gum. This crude product was purified via semi-preparative RP-HPLC using a MeCN/H2O gradient mobile phase containing 0.01% TFA (MeCN/H2O—3:5–7:10, 15 min, injection volume = 200 μL) at a flow rate of 3.8 mL/min to give the titled compound as a pale-yellow oil (13.6 mg, 0.0240 mmol, 23%). Compound purity by HPLC (see Supplementary Materials): 99.8%, 279 nm; +22.6 (c 0.46, MeCN); 1H NMR (400 MHz, CD3CN) δ 8.16 (s, 1H, CH, triazole), 7.87 (dd, J = 8.4, 1.3 Hz, 2H, H2″), 7.45 (t, J = 7.5 Hz, 2H, H3″), 7.42–7.33 (m, 3H, H4″ and H6″), 6.85 (d, J = 8.8 Hz, 2H, H7″), 5.28 (s, 2H, CH2-triazole), 4.97 (s, 1H, H6), 4.21 (d, J = 13.3 Hz, 1H, H3′A), 3.77 (s, 3H, OCH3, β-lactam), 3.71 (d, J = 13.3 Hz, 1H, H3′B), 3.55 (d, J = 13.3 Hz, 1H, H4A), 3.53 (s, 3H, Ar-OCH3), 3.35 (d, J = 16.5 Hz, 1H, H4B); NH resonance was not observed; 13C NMR (101 MHz, CD3CN) δ 167.2 (C=O, amide), 162.5 (C2′), 161.2 (C8), 160.9 (C8″), 148.0 (Cq, tetrazole), 137.3 (C3), 136.7 (C6″), 131.6 (C1″), 129.7 (C3″), 128.9 (C4″), 126.3 (C2″), 125.3 (C2), 124.4 (C5″), 123.1 (CH, triazole), 115.4 (C7″), 97.0 (C7), 65.9 (C6), 55.9 (OCH3, β-lactam), 54.0 (Ar-OMe), 52.8 (CH2-triazole), 38.8 (C3′), 29.48 (C4); IR (cm−1) max 3267 (w), 2993 (w), 2839 (w), 1771 (s, β-lactam C=O), 1703 (s), 1591 (m), 1494 (s), 1465 (w), 1442 (w), 1286 (w), 1247 (s), 1181 (w), 1148 (w), 1106 (m), 1088 (m), 1026 (m), 830 (m), 766 (s), 696 (m); MS (ESI + ve) m/z 568 ([M + H]+, 40%), 590 ([M + Na]+, 53%); (ESI − ve) m/z 566 ([M − H]−, 63%), 680 ([M + TFA − H]−, 63%); HRMS (ESI − ve TOF) calcd for C26H24N5O6S2 566.1168, found 566.1160 ([M − H]−).
- (6R,7S)-3-(((4-Carboxyphenyl)thio)methyl)-7-methoxy-8-oxo-7-(2-(4-phenyl-1H-1,2,3-triazol-1-yl)acetamido)-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid 21b
- This compound was prepared according to the General Procedure using 20 (50.0 mg, 0.103 mmol), Pd2(dba)3·CHCl3 (10.6 mg, 0.0103 mmol, 10 mol%), BIPHEPHOS (17.0 mg, 0.0216 mmol, 21 mol%), and 4-mercaptobenzoic acid (31.6 mg, 0.205 mmol, 2.0 equiv.) in MeCN (2 mL) with 19 h of reaction time. Work-up and purification via RP-HPLC (MeCN/H2O—1:1–9:11, 15 min, injection volume = 160 μL) as described above gave the titled compound as a pale-yellow oil (18.5 mg, 0.0318 mmol, 31%). Compound purity by HPLC (see Supplementary Materials): 98.7%, 254 nm; +39.3 (c 0.67, MeCN); 1H NMR (400 MHz, CD3CN) δ 8.14 (s, 1H, CH, triazole), 7.99–7.82 (m, 4H, H2″ and H7″), 7.80 (s, 1H, NH), 7.54–7.40 (m, 4H, H3″ and H6″), 7.40–7.31 (m, 1H, H4″), 5.26 (s, 2H, CH2-triazole), 4.98 (s, 1H, H6), 4.31 (d, J = 13.4 Hz, 1H, H3′A), 4.01 (d, J = 13.4 Hz, 1H, H3′B), 3.54 (d, 1H, J = 16.9 Hz, H4A), 3.53 (s, 3H, O-CH3), 3.36 (d, J = 16.9 Hz, 1H, H4B); 13C NMR (101 MHz, CD3CN) δ 167.1 (C=O, amide or C9″), 167.0 (C9″ or C=O, amide), 162.5 (C2′), 161.0 (C8), 147.8 (Cq, tetrazole), 142.0 (C5″), 135.1 (C3), 131.3 (C1″), 130.7 (C3″), 130.5 (C7″), 129.5 (C6″), 128.9 (C4″), 128.7 (C8″), 126.1 (C2″), 125.5 (C2), 122.9 (CH, triazole), 96.7 (C7), 65.4 (C6), 53.8 (O-CH3), 52.5 (CH2-triazole), 35.9 (C3′), 29.0 (C4); IR (cm−1) max 3527 (w), 3032 (w), 2940 (w), 1772 (s, β-lactam C=O), 1701 (s), 1592 (m), 1560 (w), 1402 (w), 1364 (w), 1235 (s), 1182 (w), 1110 (m), 1187 (m), 1015 (w), 851 (w), 796 (w), 765 (s), 695 (m); MS (ESI + ve) m/z 582 ([M + H]+, 19%), 604 ([M + Na]+, 42%); (ESI − ve) m/z 580 ([M − H]−, 72%), 680 ([M + TFA − H]−, 19%); HRMS (ESI − ve TOF) calcd for C26H22N5O6S2 580.0961, found 580.0952 ([M − H]−).
- (6R,7S)-3-(((4-Cyanophenyl)thio)methyl)-7-methoxy-8-oxo-7-(2-(4-phenyl-1H-1,2,3-triazol-1-yl)acetamido)-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid 21c
- This compound was prepared according to the General Procedure using 20 (50.6 mg, 0.104 mmol), Pd2(dba)3·CHCl3 (10.8 mg, 0.0104 mmol, 10 mol%), BIPHEPHOS (17.2 mg, 0.0218 mmol, 21 mol%) and 4-mercaptobenzonitrile (28.1 mg, 0.208 mmol, 2.0 equiv.) in MeCN (2 mL) with 13 h of reaction time. Work-up and purification via RP-HPLC (MeCN/H2O —2:3–7:10, 15 min, injection volume = 200 μL) as described above gave the titled compound as a thin, transparent film (14.8 mg, 0.0263 mmol, 25%). Compound purity by HPLC (see Supplementary Materials): 99.9%, 254 nm; +35.5 (c 0.42, MeCN); 1H NMR (400 MHz, CD3CN) δ 8.14 (s, 1H, CH, triazole), 7.86 (d, J = 7.0 Hz, 2H, H2″), 7.81 (s, 1H, NH), 7.61 (d, J = 8.1 Hz, 2H, H7″), 7.46 (dd, J = 7.9, 8.4 Hz, 4H, H3″ and H6″), 7.40–7.31 (m, 1H, H4″), 5.26 (s, 2H, CH2-triazole), 4.98 (s, 1H, H6), 4.29 (d, J = 13.4 Hz, 1H, H3′A), 4.05 (d, J = 13.4 Hz, 1H, H3′B), 3.53 (d, 1H, J = 16.9 Hz, H4A), 3.52 (s, 3H, O-CH3), 3.35 (d, J = 16.9 Hz, 1H, H4B); 13C NMR (101 MHz, CD3CN) δ 167.0 (C=O, amide), 161.0 (C8), 147.8 (Cq, tetrazole), 142.8 (C5″), 134.0 (C3), 133.1 (C7″), 131.4 (C1″), 130.6 (C3″), 129.5 (C6″), 128.7 (C4″), 126.1 (C2″), 125.4 (C2), 122.9 (CH, triazole), 119.1 (C8″), 110.2 (C≡N), 96.6 (C7), 65.4 (C6), 53.8 (O-CH3), 52.5 (CH2-triazole), 35.6 (C3′), 28.9 (C4); C2′ resonance was not observed in the 13C NMR or HMBC spectra; IR (cm−1) max 3500 (w), 3279 (w), 2228 (s, C≡N stretching), 1771 (s, β-lactam C=O), 1705, 1592 (m), 1537 (w), 1468 (w), 1372 (m), 1154 (w), 1017 (s), 827 (m), 767 (s), 696 (m), 549 (m); MS (ESI + ve) m/z 585 ([M + Na]+, 53%), 607 ([M + 2Na − H]+, 19%), 639 ([M + 2K − H]+, 37%); (ESI − ve) m/z 561 ([M − H]−, 53%), 675 ([M + TFA − H]−, 21%); HRMS (ESI +ve TOF) calcd for C26H23N6O5S2 563.1171, found 563.1166 ([M + H]+).
- (6R,7S)-3-(((4-Fluorophenyl)thio)methyl)-7-methoxy-8-oxo-7-(2-(4-phenyl-1H-1,2,3-triazol-1-yl)acetamido)-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid 21d
- This compound was prepared according to the General Procedure using 20 (51.0 mg, 0.105 mmol), Pd2(dba)3·CHCl3 (10.9 mg, 0.0105 mmol, 10 mol%), BIPHEPHOS (17.3 mg, 0.0220 mmol, 21 mol%), and 4-fluorobenzenethiol (22.4 μL, 0.209 mmol, 2.0 equiv.) in MeCN (2 mL) with 23 h of reaction time. Work-up and purification via RP-HPLC (MeCN/H2O—13:7–7:10, 15 min, injection volume = 120 μL) as described above gave the titled compound as a pale-yellow oil (7.3 mg, 0.0131 mmol, 13%). Compound purity by HPLC (see Supplementary Materials): 99.6%, 279 nm; +41.3 (c 0.20, MeCN); 1H NMR (400 MHz, CD3CN) δ 8.15 (s, 1H, CH, triazole), 7.86 (d, J = 7.0 Hz, 2H, H2″), 7.49–7.42 (m, 4H, H3″ and H6″), 7.36 (t, J = 7.3 Hz, 1H, H4″), 7.05 (t, J = 8.8 Hz, 2H, H7″), 5.27 (s, 2H, CH2-triazole), 4.97 (s, 1H, H6), 4.24 (d, J = 13.4 Hz, 1H, H3′A), 3.79 (d, J = 13.4 Hz, 1H, H3′B), 3.53 (d, 1H, J = 16.6 Hz, H4A), 3.52 (s, 3H, O-CH3), 3.35 (d, J = 16.6 Hz, 1H, H4B), NH resonance was not observed; 13C NMR (101 MHz, CD3CN) δ 167.0 (C=O, amide), 163.3 (d, JC,F = 246.0 Hz, C8″), 162.5 (C2′), 161.0 (C8), 147.8 (Cq, tetrazole), 136.5 (d, JC,F = 8.5 Hz, C6″), 136.1 (C3), 131.4 (C1″), 129.5 (C3″), 128.7 (C4″), 126.1 (C2″), 125.5 (C2), 122.9 (CH, triazole), 116.55 (d, JC,F = 22.1 Hz, C7″), 96.7 (C7), 65.7 (C6), 53.8 (O-CH3), 52.5 (CH2-triazole), 38.1 (C3′), 29.1 (C4), C5″ resonance was not observed in the 13C NMR or HMBC spectra; IR (cm−1) max 3283 (w), 3144 (w), 2942 (w), 1768 (s, β-lactam C=O), 1699 (s), 1589 (m), 1534 (m), 1490 (s), 1370 (m), 1220 (s, C-F stretching), 1156 (m), 1109 (m), 1088 (m), 1016 (w), 832 (m, C-F), 766 (s), 695 (m), 629 (w), 517 (w); MS (ESI +ve) m/z 556 ([M + H]+, 28%), 578 ([M + Na]+, 58%); (ESI − ve) m/z 554 ([M − H]−, 67%), 668 ([M + TFA − H]−, 100%); HRMS (ESI − ve TOF) calcd for C25H21N5O5S2F 554.0968, found 554.0976 ([M − H]−).
- (6R,7S)-7-Methoxy-8-oxo-7-(2-(4-phenyl-1H-1,2,3-triazol-1-yl)acetamido)-3-((p-tolylthio)methyl)-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid 21e
- This compound was prepared according to the General Procedure using 20 (51.3 mg, 0.105 mmol), Pd2(dba)3·CHCl3 (10.9 mg, 0.0105 mmol, 10 mol%), BIPHEPHOS (17.4 mg, 0.0221 mmol, 21 mol%), and 4-methylbenzenethiol (26.1 mg, 0.210 mmol, 2.0 equiv.) in MeCN (2 mL) with 13 h of reaction time. Work-up and purification via RP-HPLC (MeCN/H2O—13:7–7:10, 15 min, injection volume = 120 μL) as described above gave the titled compound as a pale-yellow oil (16.0 mg, 0.0290 mmol, 28%). Compound purity by HPLC (see Supplementary Materials): 99.9%, 254 nm; +33.7 (c 0.33, MeCN); 1H NMR (400 MHz, CD3CN) δ 8.15 (s, 1H, CH, triazole), 7.89–7.84 (m, 2H, H2″), 7.81 (s, 1H, NH), 7.45 (t, J = 7.5 Hz, 2H, H3″), 7.36 (t, J = 7.5 Hz, 1H, H4″), 7.31 (d, J = 8.0 Hz, 2H, H6″), 7.12 (d, J = 7.9 Hz, 2H, H7″), 5.27 (s, 2H, CH2-triazole), 4.96 (s, 1H, H6), 4.22 (d, J = 13.3 Hz, 1H, H3′A), 3.81 (d, J = 13.3 Hz, 1H, H3′B), 3.52 (d, 1H, J = 16.7 Hz, H4A), 3.51 (s, 3H, O-CH3), 3.33 (d, J = 16.7 Hz, 1H, H4B), 2.30 (s, 3H, CH3); 13C NMR (101 MHz, CD3CN) δ 167.0 (C=O, amide), 162.4 (C2′), 161.0 (C8), 147.8 (Cq, tetrazole), 138.7 (C8″), 136.5 (C3), 133.5 (C6″), 131.4 (C1″), 130.8 (C5″), 130.3 (C7″), 129.5 (C3″), 128.7 (C4″), 126.1 (C2″), 125.1 (C2), 122.9 (CH, triazole), 96.7 (C7), 65.6 (C6), 53.8 (O-CH3), 52.5 (CH2-triazole), 37.7 (C3′), 29.1 (C4), 20.8 (CH3); IR (cm−1) max 3519 (w), 3282 (w), 3024 (w), 1770 (s, β-lactam C=O), 1703 (s), 1536 (m), 1492 (w), 1442 (w), 1373 (m), 1233 (m), 1153 (w), 1088 (m), 1018 (m), 810 (m), 766 (s), 695 (m), 503 (w); MS (ESI + ve) m/z 552 ([M + H]+, 42%), 574 ([M + Na]+, 56%); (ESI − ve) m/z 550 ([M − H]−, 93%), 664 ([M + TFA − H]−, 40%); HRMS (ESI − ve TOF) calcd for C26H24N5O5S2 550.1219, found 550.1227 ([M − H]−).
- (6R,7S)-7-Methoxy-3-(((4-(methylthio)phenyl)thio)methyl)-8-oxo-7-(2-(4-phenyl-1H-1,2,3-triazol-1-yl)acetamido)-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid 21f
- This compound was prepared according to the General Procedure using 20 (51.1 mg, 0.105 mmol), Pd2(dba)3·CHCl3 (10.9 mg, 0.0105 mmol, 10 mol%), BIPHEPHOS (17.3 mg, 0.0220 mmol, 21 mol%), and 4-(methylsulfanyl)thiophenol (20.2 μL, 0.210 mmol, 2.0 equiv.) in MeCN (2 mL) with 13 h of reaction time. Work-up and purification via RP-HPLC (MeCN/H2O—17:8–7:10, 15 min, injection volume = 120 μL) as described above gave the titled compound as a pale-yellow oil (9.3 mg, 0.0159 mmol, 15%). Compound purity by HPLC (see Supplementary Materials): 99.8%, 279 nm; +22.2 (c 0.28, MeCN); 1H NMR (400 MHz, CD3CN) δ 8.15 (s, 1H, CH, triazole), 7.89–7.83 (m, 2H, H2″), 7.78 (s, 1H, NH), 7.45 (t, J = 7.6 Hz, 2H, H3″), 7.39–7.31 (m, 3H, H4″ and H6″), 7.18 (d, J = 8.5 Hz, 2H, H7″), 5.27 (s, 2H, CH2-triazole), 4.97 (s, 1H, H6), 4.23 (d, J = 13.4 Hz, 1H, H3′A), 3.81 (d, J = 13.4 Hz, 1H, H3′B), 3.53 (d, 1H, J = 16.7 Hz, H4A), 3.52 (s, 3H, O-CH3) 3.35 (d, J = 16.7 Hz, 1H, H4B), 2.45 (s, 3H, S-CH3); 13C NMR (101 MHz, CD3CN) δ 167.0 (C=O, amide), 162.4 (C2′), 161.0 (C8), 147.8 (Cq, tetrazole), 139.7 (C8″), 136.4 (C3), 134.2 (H6″), 131.4 (C1″), 130.2 (C5″), 129.5 (C3″), 128.7 (C4″), 127.0 (C7″), 126.1 (C2″), 125.2 (C2), 122.9 (CH, triazole), 96.7 (C7), 65.6 (C6), 53.8 (O-CH3), 52.5 (CH2-triazole), 37.8 (C3′), 29.2 (C4), 15.1 (CH3); IR (cm−1) max 3507 (w), 3278 (w), 3139 (w), 1770 (s, β-lactam C=O), 1704 (s), 1625 (w), 1532 (m), 1478 (w), 1440 (w), 1370 (m), 1230 (s), 1152 (s), 1106 (w), 1087 (s), 1013 (w), 1000 (w), 812 (m), 766 (s), 695 (m), 504 (w); MS (ESI + ve) m/z 584 ([M + H]+, 30%), 606 ([M + Na]+, 51%), 622 ([M + K]+, 53%); (ESI −ve) m/z 582 ([M − H]−, 47%), 696 ([M + TFA − H]−, 93%); HRMS (ESI − ve TOF) calcd for C26H24N5O5S3 582.0940, found 582.0931 ([M − H]−).
- (6R,7S)-3-(((4-Ethylphenyl)thio)methyl)-7-methoxy-8-oxo-7-(2-(4-phenyl-1H-1,2,3-triazol-1-yl)acetamido)-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid 21g
- This compound was prepared according to the General Procedure using 20 (58.7 mg, 0.120 mmol), Pd2(dba)3·CHCl3 (12.5 mg, 0.0120 mmol, 10 mol%), BIPHEPHOS (19.9 mg, 0.0253 mmol, 21 mol%), and 4-ethylbenzenethiol (33.3 μL, 0.241 mmol, 2.0 equiv.) in MeCN (2 mL) with 10 h of reaction time. Work-up and purification via RP-HPLC (MeCN/H2O—17:8–7:10, 15 min, injection volume = 120 μL) as described above gave the titled compound as a pale-yellow oil (8.4 mg, 0.0149 mmol, 12%). Compound purity by HPLC (see Supplementary Materials): 99.7%, 279 nm; +65.5 (c 0.15, MeCN); 1H NMR (400 MHz, CD3CN) δ 8.15 (s, 1H, CH, triazole), 7.86 (d, J = 7.0 Hz, 1H, H2″), 7.78 (s, 1H, NH), 7.45 (t, J = 7.5 Hz, 2H, H3″), 7.40–7.30 (m, 3H, H4″ and H6″), 7.28–7.18 (m, *H6″ and *H7″), 7.15 (d, J = 8.2 Hz, 2H, H7″), 5.26 (s, 2H, CH2-triazole), 4.96 (s, 1H, H6), 4.93 (s, *H6), 4.22 (d, J = 13.2 Hz, 1H, H3′A), 4.16 (d, J = 13.1 Hz, *H3′A), 3.88 (d, J = 13.1 Hz, *H3′B), 3.82 (d, J = 13.3 Hz, 1H, H3′B), 3.52 (d, 1H, J = 16.8 Hz, H4A), 3.51 (s, 3H, O-CH3), 3.35 (d, J = 16.8 Hz, *H4B), 3.34 (d, J = 16.7 Hz, 1H, H4B), 2.79 (qd, J = 7.4, 2.1 Hz, *H9″), 2.61 (q, J = 7.6 Hz, 2H, H9″), 1.18 (t, J = 7.6 Hz, 3H, CH3), 1.18 (td, J = 7.5, 5.4 Hz, *CH3); 13C NMR (101 MHz, CD3CN) δ 167.0 (C=O, amide), 162.4 (C2′), 161.0 (C8), 147.8 (Cq, tetrazole), 145.0 (C8″), 136.5 (C3), 133.7 (*C6″), 133.5 (C6″), 131.4 (C5″), 131.1 (*C5″), 129.6 (*C7″), 129.5 (C3″), 129.2 (C7″), 128.7 (C4″), 126.1(C2″), 125.1 (C2), 122.9 (CH, triazole), 96.7 (C7), 65.6 (C6), 65.4 (*C6), 53.8 (O-CH3), 52.5 (CH2-triazole), 37.6 (C3′), 37.0 (*C3′), 29.2 (C4), 28.8 (*C4), 28.7 (C9″), 27.4 (*C9″), 15.5 (CH3), 15.2 (*CH3); C1″ resonance was not observed in the 13C NMR or HMBC spectra (*resonance of minor rotamer observed in the 1H NMR and/or 13C NMR spectra); IR (cm−1) max 3280 (w), 2965 (w), 1772 (s, β-lactam C=O), 1704 (s), 1632 (w), 1532 (m), 1466 (w), 1441 (w), 1371 (m), 1232 (m), 1153 (w), 1109 (m), 1088 (m) 1000 (w), 829 (m), 765 (s), 627 (m), 520 (w); MS (ESI + ve) m/z 566 ([M + H]+, 100%), 588 ([M + Na]+, 70%); (ESI − ve) m/z 564 ([M − H]−, 26%), 600 ([M + Cl]−, 18%), 628 ([M + TFA − H]−, 12%); HRMS (ESI + ve TOF) calcd for C27H27N5O5S2 588.1351, found 582.1357 ([M + Na]+.
- (6R,7S)-7-Methoxy-8-oxo-7-(2-(4-phenyl-1H-1,2,3-triazol-1-yl)acetamido)-3-((phenylthio)methyl)-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid 21h
- This compound was prepared according to the General Procedure using 20 (55.4 mg, 0.114 mmol), Pd2(dba)3·CHCl3 (11.8 mg, 0.0114 mmol, 10 mol%), BIPHEPHOS (18.8 mg, 0.0239 mmol, 21 mol%), and benzenethiol (23.3 μL, 0.227 mmol, 2.0 equiv.) in MeCN (2 mL) with 10 h of reaction time. Work-up and purification via RP-HPLC (MeCN/H2O—17:8–7:10, 15 min, injection volume = 160 μL) as described above gave the titled compound as a pale-yellow oil (12.7 mg, 0.0236 mmol, 21%). Compound purity by HPLC (see Supplementary Materials): 99.7%, 254 nm; +42.4 (c 0.40, MeCN); 1H NMR (400 MHz, CD3CN) δ 8.15 (s, 1H, CH, triazole), 7.86 (d, J = 7.1 Hz, 1H, H2″), 7.78 (s, 1H, NH), 7.48–7.27 (m, 7H, H3″, H4″, H6″ and H7″), 5.26 (s, 2H, CH2-triazole), 4.95 (s, 1H, H6), 4.26 (d, J = 13.4 Hz, 1H, H3′A), 3.89 (d, J = 13.3 Hz, 1H, H3′B), 3.53 (d, 1H, J = 16.8 Hz, H4A), 3.51 (s, 3H, O-CH3), 3.36 (d, J = 16.8 Hz, 1H, H4B); 13C NMR (101 MHz, CD3CN) δ 167.0 (C=O, amide), 162.5 (C2′), 161.0 (C8), 147.8 (Cq, tetrazole), 136.4 (C3), 134.6 (C5″), 132.8 (C6″), 131.4 (C1″), 129.7 (C7″), 129.5 (C3″), 128.7 (C4″), 128.2 (C8″), 126.1 (C2″), 125.2 (C2), 122.9 (CH, triazole), 96.7 (C7), 65.6 (C6), 53.8 (O-CH3), 52.5 (CH2-triazole), 37.2 (C3′), 29.1 (C4); IR (cm−1) max 3531 (w), 3280 (w), 3031 (w), 2943 (w), 2840 (w), 1772 (s, β-lactam C=O), 1704 (s), 1623 (w), 1533 (m), 1482 (w), 1468 (m), 1415 (m), 1371 (m), 1152 (w), 1109 (m), 1087 (m), 1024 (w), 1000 (2), 832 (s), 746 (s), 629 (s); MS (ESI + ve) m/z 538 ([M+H]+, 91%), 560 ([M + Na]+, 42%), 576 ([M + K]+ 37%); (ESI − ve) m/z 536 ([M − H]−, 19%), 650 ([M + TFA − H]−, 30%); HRMS (ESI + ve TOF) calcd for C25H23N5O5S2Na 560.1117, found 560.1122 ([M + Na]+.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Elander, R.P. Industrial production of β-lactam antibiotics. Appl. Microbiol. Biotechnol. 2003, 61, 385–392. [Google Scholar] [CrossRef]
- Giamerellou, H. Anaerobic infection therapy. Int. J. Antimicrob. Agents 2000, 16, 341–346. [Google Scholar]
- Srikhanta, Y.N.; Hutton, M.L.; Awad, M.M.; Drinkwater, N.; Singleton, J.; Day, S.L.; Cunningham, B.A.; McGowan, S.; Lyras, D. Cephamycins inhibit pathogen sporulation and effectively treat recurrent Clostridioides difficile infection. Nat. Microbiol. 2019, 5, 166–180. [Google Scholar]
- Quote from Chemieliva Pharm. Co. Ltd. Available online: https://www.chemieliva.com (accessed on 6 July 2020).
- AK Scientific Inc. Web Site. Available online: https://aksci.com (accessed on 20 September 2023).
- Javed, M.I.; Brewer, M. Diphenyldiazomethane. Org. Synth. 2008, 85, 189–195. [Google Scholar]
- Christensen, B.G.; Ratcliffe, R.W. 7-Acylaminocephalosporanales. DE2365582 A1, 26 June 1975. [Google Scholar]
- Christensen, B.G.; Ratcliffe, R.W. dl-7-Azidocephalosporins. DE2365406 A1, 23 January 1975. [Google Scholar]
- Christensen, B.G.; Ratcliffe, R.W. Cephalosporin. DE2318829 A1, 31 October 1973. [Google Scholar]
- Cimarusti, C.M.; Applegate, H.E.; Chang, H.W.; Floyd, D.M.; Koster, W.H.; Slusarchyk, W.A.; Young, M.G. Monobactams. The conversion of 6-APA to (S)-3-amino-2-oxoazetidine-l-sulfonic acid and its 3(RS)-methoxy derivative. J. Org. Chem. 1982, 47, 179–180. [Google Scholar] [CrossRef]
- Lunn, W.H.W.; Mason, E.V. The synthesis of 7α-methoxy-7β-amidocephalosporanic acids by methoxylation of 7β-(p-nitrobenzyloxycarboxamido) cephalosporanic acid. Tetrahedron Lett. 1974, 15, 1311–1313. [Google Scholar] [CrossRef]
- Koppel, G.A.; Koehler, R.E. Functionalization of C6(7) of penicillins and cephalosporins. One-step stereoselective synthesis of 7-alpha-methoxycephalosporin C. J. Am. Chem. Soc. 1973, 95, 2403–2404. [Google Scholar] [CrossRef]
- Yanagisawa, H.; Fukushima, M.; Ando, A.; Nakao, H. A novel general method for synthesizing 7α-methoxycephalosporins. Tetrahedron Lett. 1975, 16, 2705–2708. [Google Scholar]
- Nakao, H.; Yanagisawa, H.; Ishihara, S.; Nakayama, E.; Ando, A.; Nakazawa, J.-I.; Shimizu, B.; Kaneko, M.; Nagano, M.; Sugawara, S. Semisynthetic cephamycins. II. Structure-activity studies related to cefmetazole (CS-1170). J. Antibiot. 1979, 32, 320–329. [Google Scholar] [CrossRef]
- Yoshida, Y.; Matsuda, K.; Sasaki, H.; Matsumoto, Y.; Matsumoto, S.; Tawara, S.; Takasugi, H. Studies on anti-Helicobacter pylori agents. Part 2: New cephem derivatives. Bioorg. Med. Chem. 2000, 8, 2317–2335. [Google Scholar] [CrossRef]
- Grant, J.W.; Smyth, T.P. Toward the Development of a cephalosporin-based dual-release prodrug for use in ADEPT. J. Org. Chem. 2004, 69, 7965–7970. [Google Scholar] [CrossRef] [PubMed]
- Patterson, L.D.; Miller, M.J. Enzymatic deprotection of the cephalosporin 3′-acetoxy group using Candida antarctica lipase B. J. Org. Chem. 2010, 75, 1289–1292. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Miller, M.J.; Franzblau, S.; Wan, B.; Mollmann, U.; Möllmann, U. Syntheses and studies of quinolone-cephalosporins as potential anti-tuberculosis agents. Bioorg. Med. Chem. Lett. 2006, 16, 5534–5537. [Google Scholar] [CrossRef]
- Ali, I.A.I.; El Ashry, E.S.H.; Schmidt, R.R. Protection of hydroxy groups with diphenylmethyl and 9-fluorenyl trichloroacetimidates—Effect on anomeric stereocontrol. Eur. J. Org. Chem. 2003, 21, 4121–4131. [Google Scholar] [CrossRef]
- Adhikari, A.A.; Shah, J.P.; Howard, K.T.; Russo, C.M.; Wallach, D.R.; Linaburg, M.R.; Chisholm, J.D. Convenient formation of diphenylmethyl esters using diphenylmethyl trichloroacetimidate. Synlett 2014, 25, 283–287. [Google Scholar]
- Ohi, N.; Aoki, B.; Shinazaki, T.; Moro, K.; Kuroki, T.; Noto, T.; Nehashi, T.; Matsumoto, M.; Okazaki, H.; Matsunaga, I. Semisynthetic beta-lactam antibiotics. IV. Synthesis and antibacterial activity of new ureidocephalosporin and ureidocephamycin derivatives containing a catechol moiety or its acetate. Chem. Pharm. Bull. 1987, 35, 1903–1909. [Google Scholar] [CrossRef]
- Kamachi, H.; Okita, T.; Yamasaki, T.; Naito, T. Direct Introduction of a formamido group into the 7 alpha (6 alpha)-position of cephalosporins (penicillins). J. Antibiot. 1990, 43, 820–829. [Google Scholar] [CrossRef] [PubMed]
- Cama, L.D.; Leanza, W.J.; Beattie, T.R.; Christensen, B.G. Substituted penicillin and cephalosporin derivatives. I. Stereospecific introduction of the C-6 (7) methoxy group. J. Am. Chem. Soc. 1972, 94, 1408–1410. [Google Scholar] [CrossRef]
- Cun, W.Y.; Bate, C.E.; Srikhanta, Y.N.; Hutton, M.L.; Webb, C.T.; Revitt-Mills, S.; Lyras, D.; McGowan, S.; Yu, H.; Keller, P.A.; et al. Design, synthesis, and evaluation of cephamycin-based anti-sporulation agents targeting Clostridioides difficile. J. Med. Chem. 2023. [Google Scholar]
- Hein, J.E.; Fokin, V.V. Copper-catalyzed azide–alkyne cycloaddition (cuaac) and beyond: New reactivity of copper(i) acetylides. Chem. Soc. Rev. 2010, 39, 1302–1315. [Google Scholar] [CrossRef]
- Hatfield, L.D. 3-(Thiomethyl)cephalosporins. DE2809058 A1, 14 September 1978. [Google Scholar]
- Schlatzer, T.; Schröder, H.; Trobe, M.; Lembacher-Fadum, C.; Stangl, S.; Schlögl, C.; Weber, H.; Breinbauer, R. Pd/BIPHEPHOS is an efficient catalyst for the Pd-catalyzed S-allylation of thiols with high n-selectivity. Adv. Synth. Catal. 2020, 362, 331–336. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
| Entry | Ligand | Ligand Equiv. | Pd Equiv. | Reaction Time | Ratio of 20:21a a |
|---|---|---|---|---|---|
| 1 | BIPHEPHOS | 2 mol% | 2 mol% | 5 d | 1:0 |
| 2 | P(OPh)3 | 2 mol% | 2 mol% | 5 d | 1:0 |
| 3 | dppf | 2 mol% | 2 mol% | 5 d | 1:0 |
| 4 b | BIPHEPHOS | 2 mol% | 2 mol% | 12 h | 1:2.3 |
| 5 c | BIPHEPHOS | 20 mol% | 21 mol% | 10 h | 0:1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cun, W.Y.; Keller, P.A.; Pyne, S.G. Synthesis of 7α-Methoxy-7-(4-phenyl-1H-1,2,3-triazol-1-yl)acetamino-3′-arylthio-cephalosporic Acid Derivatives from 7-Aminocephalosporic Acid. Molecules 2023, 28, 7338. https://doi.org/10.3390/molecules28217338
Cun WY, Keller PA, Pyne SG. Synthesis of 7α-Methoxy-7-(4-phenyl-1H-1,2,3-triazol-1-yl)acetamino-3′-arylthio-cephalosporic Acid Derivatives from 7-Aminocephalosporic Acid. Molecules. 2023; 28(21):7338. https://doi.org/10.3390/molecules28217338
Chicago/Turabian StyleCun, Wendy Y., Paul A. Keller, and Stephen G. Pyne. 2023. "Synthesis of 7α-Methoxy-7-(4-phenyl-1H-1,2,3-triazol-1-yl)acetamino-3′-arylthio-cephalosporic Acid Derivatives from 7-Aminocephalosporic Acid" Molecules 28, no. 21: 7338. https://doi.org/10.3390/molecules28217338