
Stereoselective Epoxidation of Triterpenic Allylic Alcohols and Cytotoxicity Evaluation of Synthesized Compounds

Abstract
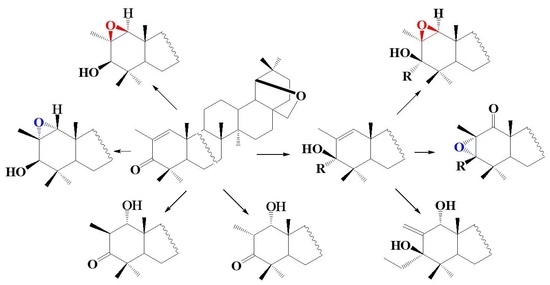
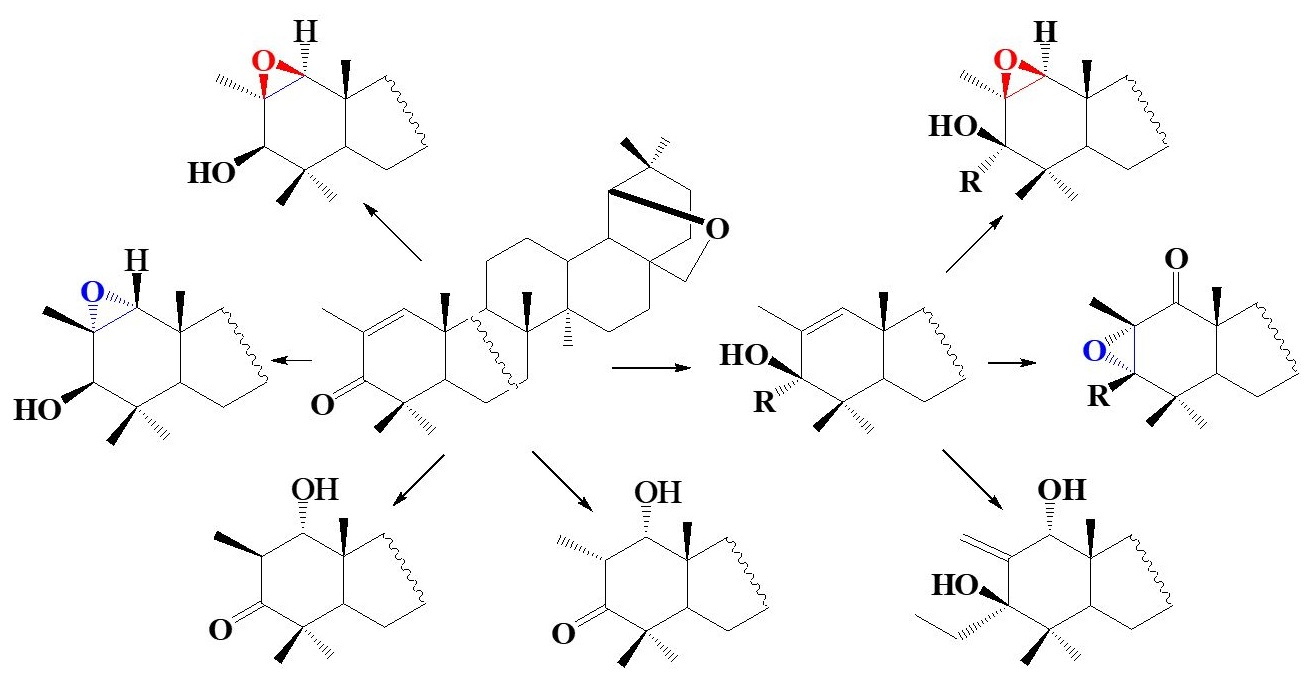
:
1. Introduction
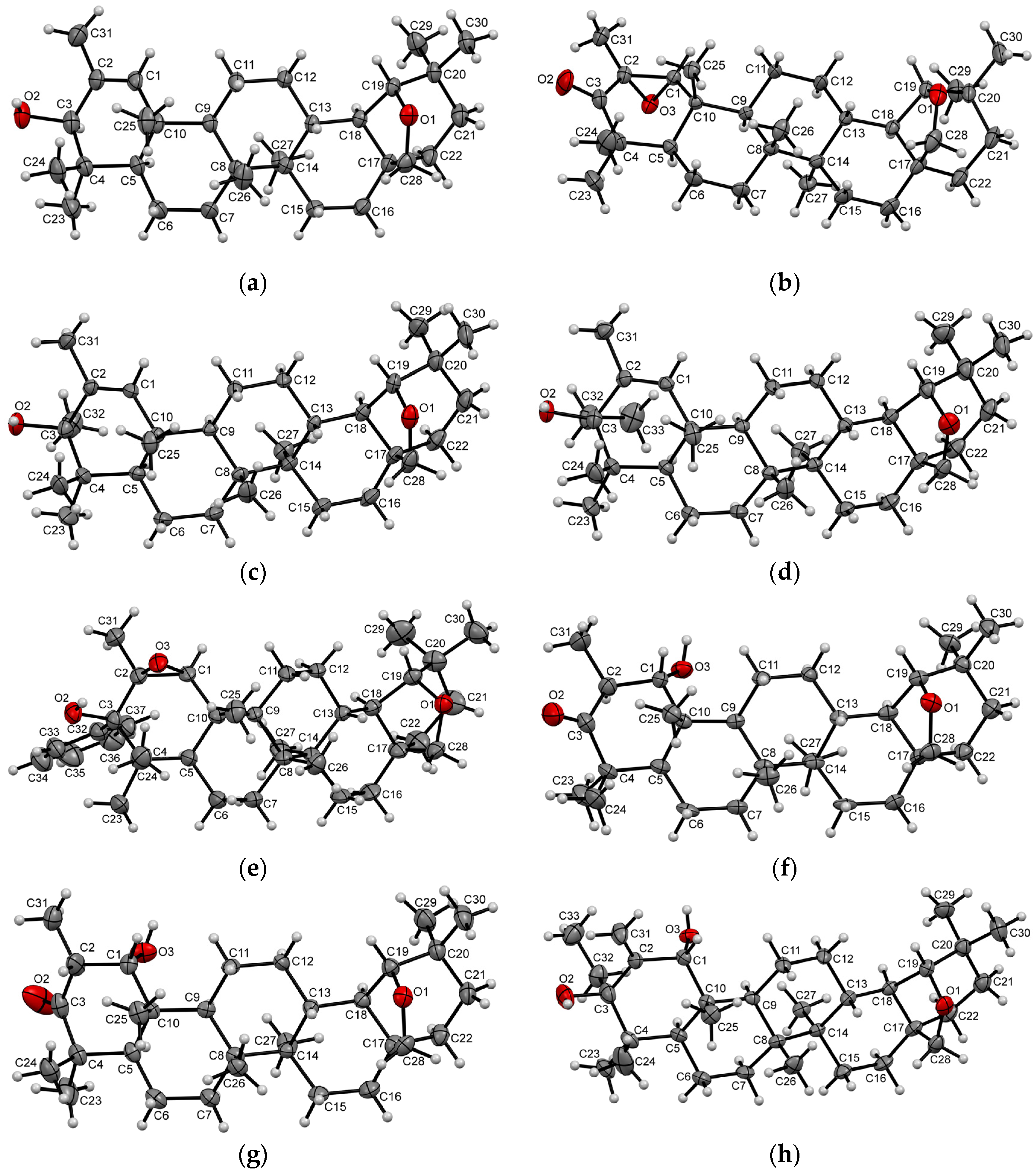
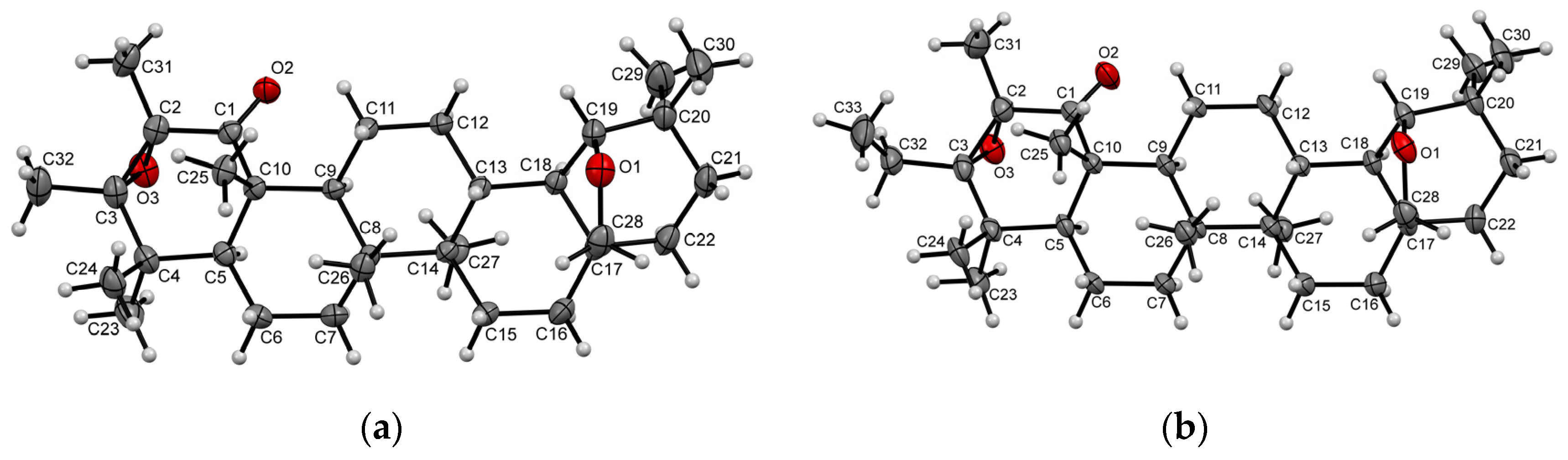
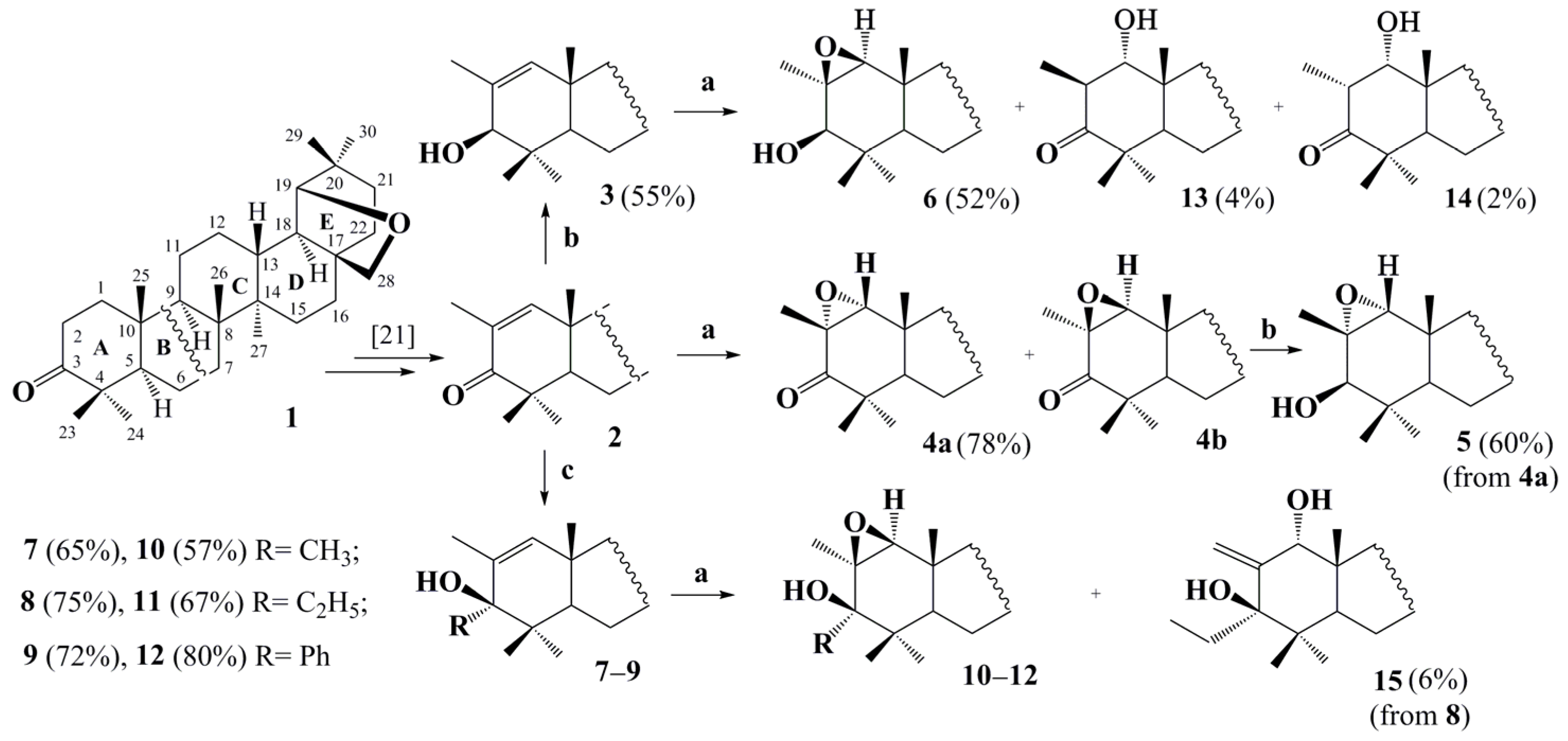
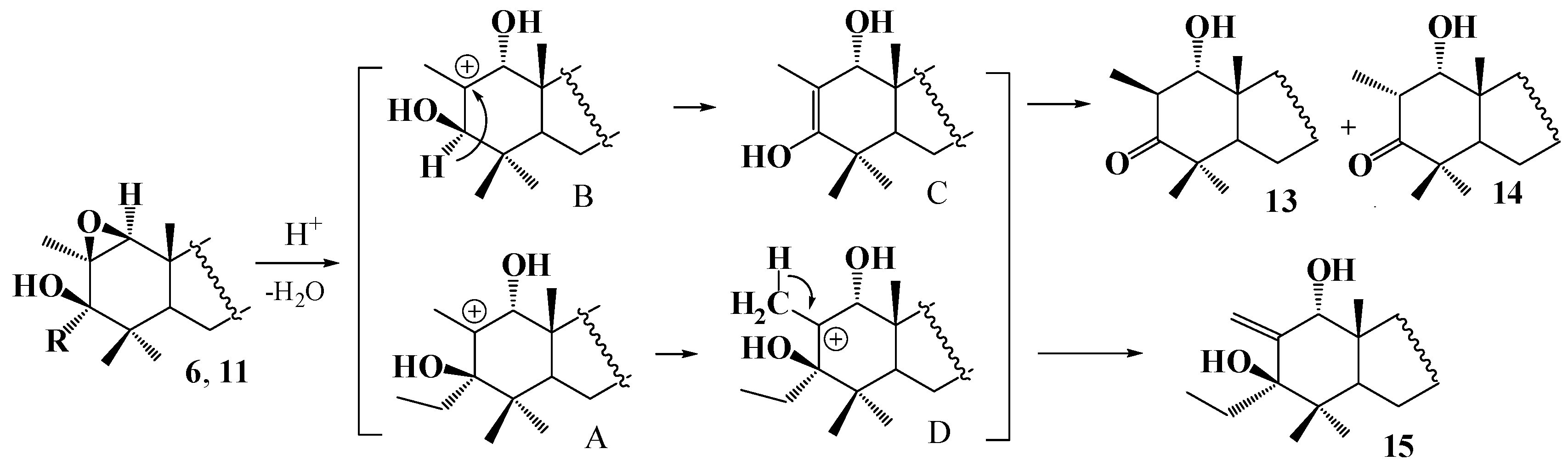
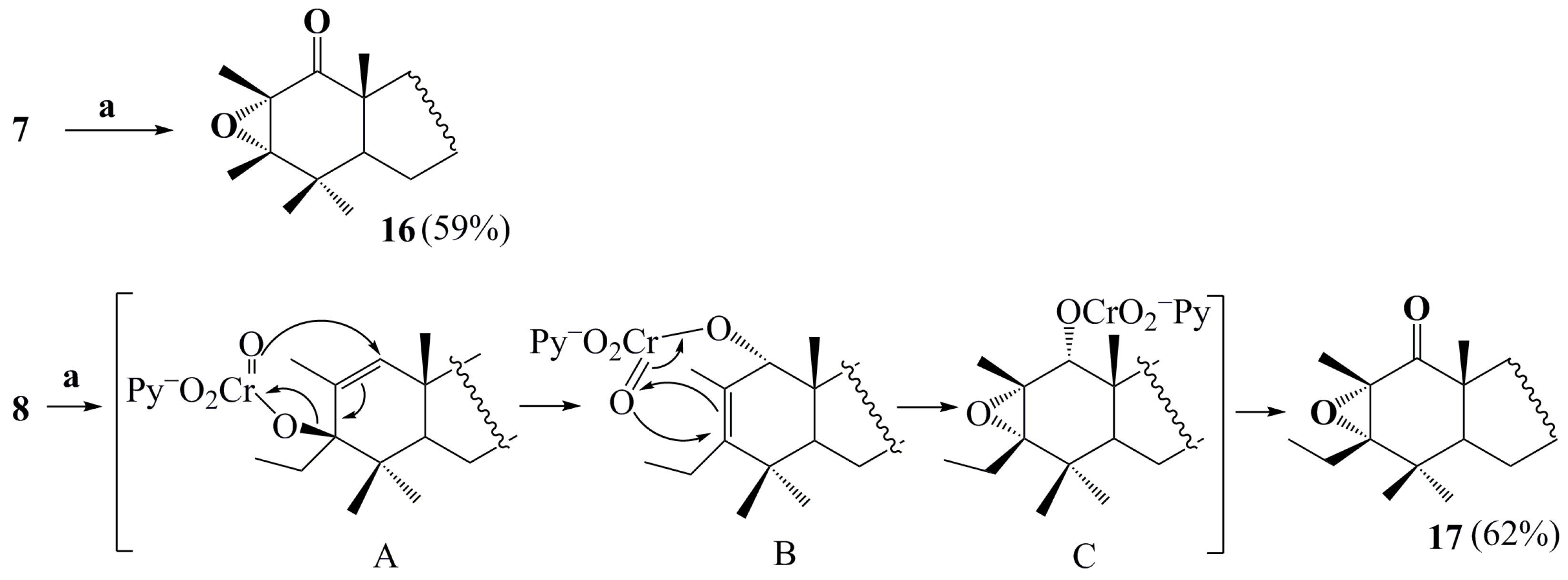
2. Results and Discussion
3. Conclusions
4. Materials and Methods
4.1. General Procedure for Preparing Compounds , , –, –
4.2. General Procedure for Preparing Compounds and
4.3. General Procedure for Preparing Compounds –
4.4. General Procedure for Preparing Compounds and
4.5. Screening for Cytotoxic Activity of Compounds , , , –, , and
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Markov, A.V.; Zenkova, M.A.; Logashenko, E.B. Modulation of tumour-related signaling pathways by natural pentacyclic triterpenoids and their semisynthetic derivatives. Curr. Med. Chem. 2017, 24, 1277–1320. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Zhang, R.H.; Wang, M.; Xu, G.B.; Liao, S.G. Prodrugs of triterpenoids and their derivatives. Eur. J. Med. Chem. 2017, 131, 222–236. [Google Scholar] [CrossRef] [PubMed]
- Miao, Q.; Xu, J.; Lin, A.; Wu, X.; Wu, L.; Xie, W. Recent advances for the synthesis of selenium-containing small molecules as potent antitumor agents. Curr. Med. Chem. 2018, 25, 2009–2033. [Google Scholar] [CrossRef]
- Wu, H.F.; Morris-Natschke, S.L.; Xu, X.D.; Yang, M.H.; Cheng, Y.Y.; Yu, S.S.; Lee, K.H. Recent advances in natural anti-HIV triterpenoids and analogs. Med. Res. Rev. 2020, 40, 2339–2385. [Google Scholar] [CrossRef] [PubMed]
- Amiri, S.; Dastghaib, S.; Ahmadi, M.; Mehrbod, P.; Khadem, F.; Behrouj, H.; Aghanoori, M.-R.; Machaj, F.; Ghamsari, M.; Rosik, J.; et al. Betulin and its derivatives as novel compounds with different pharmacological effects. Biotechnol. Adv. 2020, 38, 107409. [Google Scholar] [CrossRef]
- Şoica, C.; Voicu, M.; Ghiulai, R.; Dehelean, C.; Racoviceanu, R.; Trandafirescu, C.; Roșca, O.-J.; Nistor, G.; Mioc, M.; Mioc, A. Natural compounds in sex hormone-dependent cancers: The role of triterpenes as therapeutic agents. Front. Endocrinol. 2021, 11, 612396. [Google Scholar] [CrossRef]
- Nadendla, R.R. Principles of Organic Medicinal Chemistry; New Age Publishers: New Delhi, India, 2005; 322p. [Google Scholar]
- Urban, M.; Klinot, J.; Tislerova, I.; Biedermann, D.; Hajduch, M.; Cisarova, I.; Sarek, J. Reactions of activated lupane oxo-compounds with diazomethane: An approach to new derivatives of cytotoxic triterpenes. Synthesis 2006, 23, 3979–3986. [Google Scholar] [CrossRef]
- Michaudel, Q.; Journot, G.; Regueiro-Ren, A.; Goswami, A.; Guo, Z.; Tully, T.P.; Zou, L.; Ramabhadran, R.O.; Houk, K.N.; Baran, P.S. Improving physical properties via C-H oxidation: Chemical and enzymatic approaches. Angew. Chem. Int. Ed. 2014, 53, 12091–12096. [Google Scholar] [CrossRef] [Green Version]
- Huang, R.Z.; Jin, L.; Wang, C.G.; Xu, X.J.; Du, Y.; Liao, N.; Ji, M.; Liao, Z.X.; Wang, H.S. A pentacyclic triterpene derivative possessing polyhydroxyl ring A suppresses growth of HeLa cells by reactive oxygen species-dependent NF-κB pathway. Eur. J. Pharmacol. 2018, 838, 157–169. [Google Scholar] [CrossRef]
- Guo, H.; Wang, H.; Huo, Y.X. Engineering critical enzymes and pathways for improved triterpenoid biosynthesis in yeast. ACS Synth. Biol. 2020, 9, 2214–2227. [Google Scholar] [CrossRef]
- Shah, S.; Tan, H.; Sultan, S.; Faridz, M.; Shah, M.; Nurfazilah, S.; Hussain, M. Microbial-catalyzed biotransformation of multifunctional triterpenoids derived from phytonutrients. Int. J. Mol. Sci. 2014, 15, 12027–12060. [Google Scholar] [CrossRef]
- Luchnikova, N.A.; Grishko, V.V.; Ivshina, I.B. Biotransformation of oleanane and ursane triterpenic acids. Molecules 2020, 25, 5526. [Google Scholar] [CrossRef]
- Berger, M.; Knittl-Frank, C.; Bauer, S.; Winter, G.; Maulide, N. Application of relay C−H oxidation logic to polyhydroxylated oleanane triterpenoids. Chem 2020, 6, 1183–1189. [Google Scholar] [CrossRef]
- Mu, T.; Wei, B.; Zhu, D.; Yu, B. Site-selective C-H hydroxylation of pentacyclic triterpenoids directed by transient chiral pyridine-imino groups. Nat. Commun. 2020, 11, 4371. [Google Scholar] [CrossRef]
- Borkova, L.; Hodon, J.; Urban, M. Synthesis of betulinic acid derivatives with modified A-ring and their application as potential drug candidates. Asian J. Org. Chem. 2018, 7, 1542–1560. [Google Scholar] [CrossRef]
- Huang, L.; Luo, H.; Li, Q.; Wang, D.; Zhang, J.; Hao, X.; Yang, X. Pentacyclic triterpene derivatives possessing polyhydroxyl ring A inhibit Gram-positive bacteria growth by regulating metabolism and virulence genes expression. Eur. J. Med. Chem. 2015, 95, 64–75. [Google Scholar] [CrossRef]
- Huang, L.R.; Hao, X.J.; Li, Q.J.; Wang, D.P.; Zhang, J.X.; Luo, H.; Yang, X.S. 18β-glycyrrhetinic acid derivatives possessing a trihydroxylated a ring are potent gram-positive antibacterial agents. J. Nat. Prod. 2016, 79, 721–731. [Google Scholar] [CrossRef]
- Motlhanka, D.; Houghton, P.; Miljkovic-Brake, A.; Habtemariam, S. A novel pentacyclic triterpene glycoside from a resin of Commiphora glandulosa from Botswana. Afr. J. Pharm. Pharmacol. 2010, 4, 549–554. [Google Scholar]
- Liang, Z.M.; Wang, X.H.; Huang, L.R.; Li, Q.J.; Guan, T.Q.; Hao, X.J.; Luo, H.; Yang, X.S. 1α,2α-Epoxy-3β-hydroxy oleanolic acid derivatives regulation of the metabolism, haemolysis and β-lactamase gene expression in vitro and their structure–microbicidal activity relationship. Bioorganic Med. Chem. Lett. 2016, 26, 3870–3875. [Google Scholar] [CrossRef]
- Konysheva, A.V.; Zhukova, A.E.; Dmitriev, M.V.; Grishko, V.V. Synthesis and intramolecular cyclization of a 2,3-seco-oleanane triterpenoid with an ethylketone fragment. Chem. Nat. Compd. 2018, 54, 1094–1099. [Google Scholar] [CrossRef]
- Hanson, J.R.; Hitchcock, P.B.; Kiran, I. The stereochemistry of epoxidation of steroidal 4,6-dienes. J. Chem. Res.—Part S 1999, 3, 198–199. [Google Scholar] [CrossRef]
- Kinot, J.; Krumpolc, M.; Vystrčil, A. Triterpenes. IX. Reaction of isomeric 2,3-epoxides with Grignard reagent. Collect. Czechoslov. Chem. Commun. 1966, 31, 3174–3181. [Google Scholar] [CrossRef]
- Kehrli, A.R.H.; Taylor, D.A.H.; Niven, M. The synthesis of a 1α,2α,3α-triacetoxy limonoid. J. Chem. Soc. Perkin Trans. 1990, 7, 2057–2065. [Google Scholar] [CrossRef]
- García-Granados, A.; López, P.E.; Melguizo, E.; Parra, A.; Simeó, Y. Oxidation of several triterpenic diene and triene systems. Oxidative cleavage to obtain chiral intermediates for drimane and phenanthrene semi-synthesis. Tetrahedron 2004, 60, 3831–3845. [Google Scholar] [CrossRef]
- Amer, H.; Mereiter, K.; Stanetty, C.; Hofinger, A.; Czollner, L.; Beseda, I.; Jordis, U.; Kueenburg, B.; Claßen-Houben, D.; Kosma, P. Synthesis and crystal structures of ring A modified glycyrrhetinic acid derivatives derived from 2,3-oxirane and 2,3-thiirane intermediates. Tetrahedron 2010, 66, 4390–4402. [Google Scholar] [CrossRef]
- Kazakova, O.B.; Khusnutdinova, E.F.; Korlyukov, A.A. Stereospecific epoxidation of an olean-18(19)-ene-type triterpenoid. Chem. Nat. Compd. 2011, 46, 900–901. [Google Scholar] [CrossRef]
- Mikhailova, L.R.; Budaev, A.S.; Spirikhin, L.V.; Baltina, L.A. Oxidation of licorice-root triterpene-acid derivatives by m-chloroperbenzoic acid. Chem. Nat. Compd. 2019, 55, 88–91. [Google Scholar] [CrossRef]
- Pakulski, Z.; Cmoch, P.; Korda, A.; Luboradzki, R.; Gwardiak, K.; Karczewski, R. Rearrangements of the betulin core. Synthesis of terpenoids possessing the bicyclo[3.3.1]nonane fragment by rearrangement of lupane-type epoxides. J. Org. Chem. 2021, 86, 1084–1095. [Google Scholar] [CrossRef]
- Paryzek, Z. Epoxidation of lanost-9(11)-enes. The effect of a β-carbonyl group upon the stereochemistry of epoxidation. J. Chem. Soc. Perkin Trans. 1978, 1, 329–3369. [Google Scholar] [CrossRef]
- Kvasnica, M.; Tislerova, I.; Sarek, J.; Sejbal, J.; Cisarova, I. Preparation of new oxidized 18-α-oleanane derivatives. Collect. Czech. Chem. Commun. 2005, 70, 1447–1464. [Google Scholar] [CrossRef]
- Tolmacheva, I.A.; Shelepen’kina, L.N.; Shashkov, A.S.; Grishko, V.V.; Glushkov, V.A.; Tolstikov, A.G. Reaction of 3-acetoxy-(2,3),(19β,28)-diepoxyoleanane with cyclic and linear amines. Chem. Nat. Compd. 2007, 43, 153–158. [Google Scholar] [CrossRef]
- Csuk, R.; Nitsche, C.; Sczepek, R.; Schwarz, S.; Siewert, B. Synthesis of antitumor-active betulinic acid-derived hydroxypropargylamines by copper-catalyzend mannich reactions. Arch. Pharm. 2013, 346, 232–246. [Google Scholar] [CrossRef]
- Pereslavtseva, A.V.; Tolmacheva, I.A.; Slepukhin, P.A.; El’tsov, O.S.; Kucherov, I.I.; Eremin, V.F.; Grishko, V.V. Synthesis of A-pentacyclic triterpene α,β-alkenenitriles. Chem. Nat. Compd. 2014, 49, 1059–1066. [Google Scholar] [CrossRef]
- Konysheva, A.V.; Nebogatikov, V.O.; Tolmacheva, I.A.; Dmitriev, M.V.; Grishko, V.V. Synthesis of cytotoxically active derivatives based on alkylated 2,3-seco-triterpenoids. Eur. J. Med. Chem. 2017, 140, 74–83. [Google Scholar] [CrossRef]
- Konysheva, A.V.; Eroshenko, D.V.; Grishko, V.V. Synthesis, Cyclization, and cytotoxic activity of 2,3-secolupane triterpenoids with an ethylketone fragment. Nat. Prod. Commun. 2019, 14, 1–7. [Google Scholar] [CrossRef]
- Carey, F.A.; Sundberg, R.J. Advanced Organic Chemistry Part A: Structure and Mechanisms, 5th ed.; Springer: New York, NY, USA, 2007; 1199p. [Google Scholar]
- Kočovský, P. Stereochemistry of epoxidation of allylic and homoallylic cyclohexene alcohols. J. Chem. Soc. Perkin Trans. 1994, 13, 1759–1763. [Google Scholar] [CrossRef]
- Rosatella, A.A.; Afonso, C.A.M. Brønsted acid-catalyzed dihydroxylation of olefins in aqueous medium. Adv. Synth. Catal. 2011, 353, 2920–2926. [Google Scholar] [CrossRef]
- Jat, J.L.; Kumar, G. Isomerization of Epoxides. Adv. Synth. Catal. 2019, 361, 4426–4441. [Google Scholar] [CrossRef]
- Chandrasekaran, S.; Ganesh, V. Oxidation adjacent to oxygen of alcohols by chromium reagents. In Comprehensive Organic Synthesis, 2nd ed.; Knochel, P., Ed.; Elsevier: Amsterdam, The Netherlands, 2014; Volume 7, pp. 277–294. [Google Scholar]
- Luzzio, F.A. 1,3-Oxidative transpositions of allylic alcohols in organic synthesis. Tetrahedron 2012, 68, 5323–5339. [Google Scholar] [CrossRef]
- Papillaud, B.; Tiffon, F.; Taran, M.; Miguel, B.A.S.; Delmond, B. Part I. epoxydes diterpeniques: Synthese et reactivite d’epoxydes derives d’acides resiniques. Tetrahedron 1985, 41, 1845–1857. [Google Scholar] [CrossRef]
- Ley, S.V.; Madin, A. Oxidation adjacent to oxygen of alcohols by chromium reagents. In Comprehensive Organic Synthesis; Trost, B.M., Ed.; Plenum: New York, NY, USA, 1991; Volume 7, 251p. [Google Scholar]
- Kharitonov, Y.V.; Shul’ts, E.E.; Shakirov, M.M. Synthetic transformations of higher terpenoids. XXXIII.*Preparation of 15,16-dihydroisopimaric acid and methyl dihydroisopimarate and their transformations. Chem. Nat. Compd. 2014, 49, 2857–2899. [Google Scholar] [CrossRef]
- Singha, R.; Ghosh, P. Phytochemical investigation of Sapium baccatum: Identification of 3α-hydroxy-1α, 2α-epoxy lupan. J. Indian Chem. Soc. 2018, 95, 549–552. [Google Scholar] [CrossRef]
- Salomatina, O.V.; Sen’kova, A.V.; Moralev, A.D.; Savin, I.A.; Komarova, N.I.; Salakhutdinov, N.F.; Zenkova, M.A.; Markov, A.V. Novel Epoxides of Soloxolone Methyl: An Effect of the Formation of Oxirane Ring and Stereoisomerism on Cytotoxic Profile, Anti-Metastatic and Anti-Inflammatory Activities In Vitro and In Vivo. Int. J. Mol. Sci. 2022, 23, 6214. [Google Scholar] [CrossRef] [PubMed]
- Marco-Contelles, J.; Molina, M.T.; Anjum, S. Naturally occurring cyclohexane epoxides: Sources, biological activities and synthesis. Chem. Rev. 2004, 104, 2857–2899. [Google Scholar] [CrossRef] [PubMed]
- Way2Drug Predictive Services. PASS Online. Available online: http://www.pharmaexpert.ru/passonline/index.php (accessed on 6 April 2022).
- Poroikov, V.V. Computer-aided drug design: From discovery of novel pharmaceutical agents to systems pharmacology. Biomed. Chem. 2020, 66, 30–41. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Keil, B. Laboratory Technika Organicke Chemie; Nakladatelstvi ČSAV: Praha, Czech Republic, 1963; 751p. [Google Scholar]
- CrysAlisPro, Version 1.171.37.33 (Release 27-03-2014 CrysAlis171.NET); Agilent Technologies: Yarnton, UK, 2014.
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Palatinus, L.; Chapuis, G. SUPERFLIP - A computer program for the solution of crystal structures by charge flipping in arbitrary dimensions. J. Appl. Crystallogr. 2007, 40, 786–790. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Spek, A.L. PLATON SQUEEZE: A tool for the calculation of the disordered solvent contribution to the calculated structure factors. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 9–18. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tested Compound | Estimated Activity, Pa | ||||||
|---|---|---|---|---|---|---|---|
| Antineoplastic | Colorectal Cancer | Colon Cancer | Lung Cancer | Ovarian Cancer | Carcinoma | Thyroid Cancer | |
| 2 | 0.940 | 0.914 | 0.911 | 0.786 | 0.780 | - | - |
| 3 | 0.961 | 0.900 | 0.898 | 0.819 | 0.774 | - | 0.703 |
| 4a, 4b | 0.983 | 0.902 | 0.900 | 0.819 | 0.758 | 0.742 | - |
| 5 | 0.967 | 0.898 | 0.898 | 0.845 | 0.820 | 0.778 | 0.710 |
| 6 | 0.967 | 0.898 | 0.895 | 0.845 | 0.820 | 0.778 | 0.778 |
| 7 | 0.911 | 0.886 | 0.884 | 0.786 | 0.710 | - | - |
| 8 | 0.911 | 0.886 | 0.884 | 0.769 | - | - | - |
| 9 | 0.911 | 0.870 | 0.868 | 0.747 | 0.744 | - | - |
| 10 | 0.959 | 0.899 | 0.896 | 0.827 | 0.748 | 0.736 | - |
| 11 | 0.960 | 0.901 | 0.898 | 0.826 | 0.755 | 0.726 | - |
| 12 | 0.954 | 0.837 | 0.833 | 0.756 | - | - | - |
| 13, 14 | 0.959 | 0.888 | 0.881 | 0.826 | 0.822 | - | - |
| 15 | 0.934 | 0.890 | 0.887 | 0.832 | 0.701 | - | - |
| 16 | 0.938 | 0.898 | 0.895 | 0.852 | 0.735 | - | 0.716 |
| 17 | 0.950 | 0.856 | 0.852 | 0.810 | - | - | - |
| Tested Compound | IC50 (Mean ± SD), μM | |||||
|---|---|---|---|---|---|---|
| HEpG2 | HCT116 | MS | RD TE32 | A549 | MCF-7 | |
| 2 ** | 132.1 ± 15.61 | >200 | 156.1 ± 24.83 | >200 | 136.8 ± 48.1 | 60.94 ± 2.32 |
| 3 | 135.6 ± 13.21 | 128.2 ± 15.66 | 52.56 ± 4.58 | 145.3 ± 35.3 | 96.42 ± 12.96 | 115.8 ± 6.14 |
| 4a | 129.7 ± 13.52 | 129.1 ± 15.16 | 44.02 ± 6.80 | 142.3 ± 24.8 | 39.17 ± 5.11 | 97.27 ± 10.93 |
| 5 | >200 | >200 | >200 | >200 | >200 | 78.72 ± 13.55 |
| 9 | >200 | >200 | >200 | >200 | >200 | 45.27 ± 4.18 |
| 10 | >200 | >200 | >200 | >200 | >200 | >200 |
| 11 | >200 | >200 | >200 | >200 | >200 | 45.88 ± 7.43 |
| 12 | >200 | >200 | >200 | >200 | >200 | 37.08 ± 5.05 |
| 17 | >200 | >200 | >200 | >200 | >200 | 117.00 ± 4.24 |
| DOX * | 1.78 ± 0.31 | 1.96 ± 0.19 | 1.29 ± 0.16 | 1.27 ± 0.03 | 2.04 ± 0.22 | 0.14 ± 0.03 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krainova, G.; Beloglazova, Y.; Dmitriev, M.; Grishko, V. Stereoselective Epoxidation of Triterpenic Allylic Alcohols and Cytotoxicity Evaluation of Synthesized Compounds. Molecules 2023, 28, 550. https://doi.org/10.3390/molecules28020550
Krainova G, Beloglazova Y, Dmitriev M, Grishko V. Stereoselective Epoxidation of Triterpenic Allylic Alcohols and Cytotoxicity Evaluation of Synthesized Compounds. Molecules. 2023; 28(2):550. https://doi.org/10.3390/molecules28020550
Chicago/Turabian StyleKrainova, Gulnaz, Yulia Beloglazova, Maksim Dmitriev, and Victoria Grishko. 2023. "Stereoselective Epoxidation of Triterpenic Allylic Alcohols and Cytotoxicity Evaluation of Synthesized Compounds" Molecules 28, no. 2: 550. https://doi.org/10.3390/molecules28020550