

Novel 7-Chloro-4-aminoquinoline-benzimidazole Hybrids as Inhibitors of Cancer Cells Growth: Synthesis, Antiproliferative Activity, in Silico ADME Predictions, and Docking
 , , ,
, , ,
Abstract
:1. Introduction
2. Results and Discussion
2.1. Chemistry
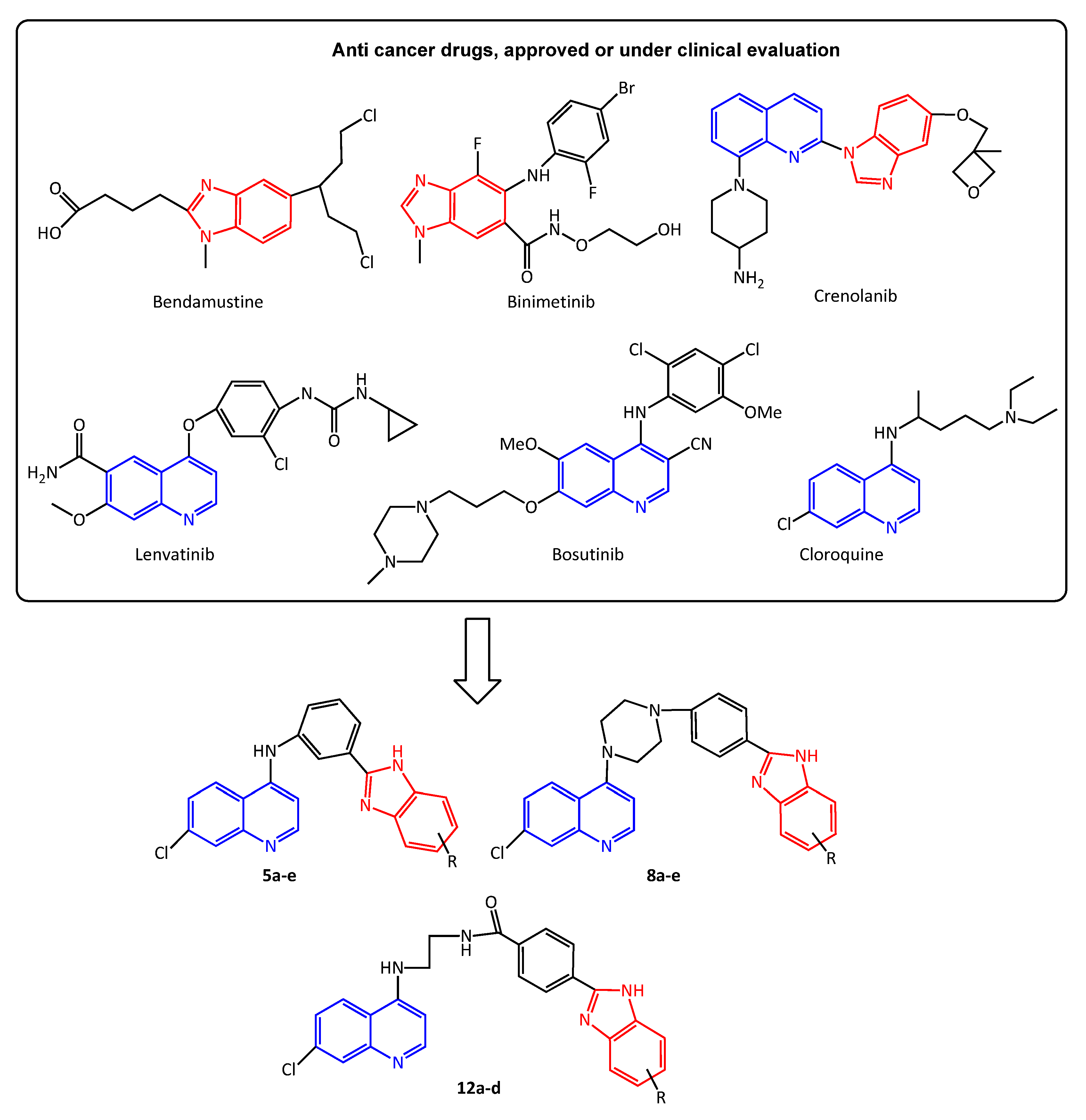
Design and Synthesis of Novel Benzimidazole Derivatives
2.2. Biological Activity
2.2.1. Evaluation of Antiproliferative Activity of the Novel Compounds
2.2.2. Cell Cycle Redistribution
2.2.3. Apoptosis Induction
2.3. Absorption, Distribution, Metabolism, Excretion (ADME), and Toxicity Properties
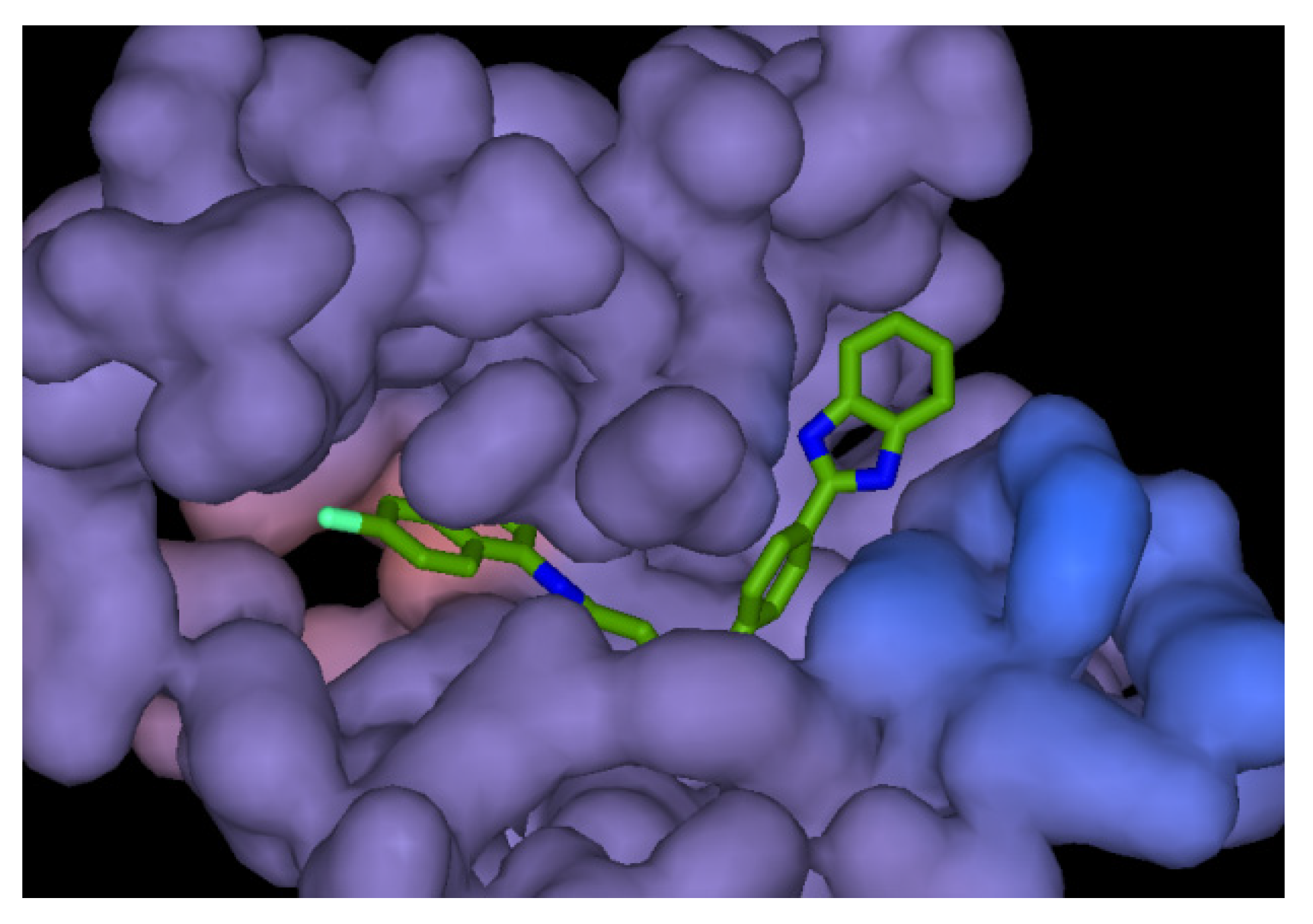
2.4. Molecular Docking
3. Materials and Methods
3.1. Chemistry
3.1.1. General Procedure for the Synthesis of Compounds 5a–e
2-(3-(7-chloroquinolin-4-ylamino)phenyl)-1H-benzo[d]imidazole-5-carboximidamide trihydrochloride (5a)
2-(3-(7-chloroquinolin-4-ylamino)phenyl)-N-isopropyl-1H-benzo[d]imidazole-5-carboximidamide trihydrochloride (5b)
7-chloro-N-(3-(5-(4,5-dihydro-1H-imidazol-2-yl)-1H-benzo[d]imidazol-2-yl)phenyl)quinolin-4-amine trihydrochloride (5c)
N-(3-(1H-benzo[d]imidazol-2-yl)phenyl)-7-chloroquinolin-4-amine (5d)
7-chloro-N-(3-(5-chloro-1H-benzo[d]imidazol-2-yl)phenyl)quinolin-4-amine (5e)
3.1.2. 4-(4-(7-chloroquinolin-4-yl)piperazin-1-yl)benzaldehyde (7)
3.1.3. General Procedure for the Synthesis of Compounds 8a–e
2-(4-(4-(7-chloroquinolin-4-yl)piperazin-1-yl)phenyl)-1H-benzo[d]imidazole-5-carboxamidine dihydrochloride (8a)
2-(4-(4-(7-chloroquinolin-4-yl)piperazin-1-yl)phenyl)-N-isopropyl-1H-benzo[d]imidazole-5-carboxamidine dihydrochloride (8b)
7-chloro-4-(4-(4-(5-(4,5-dihydro-1H-imidazol-2-yl)-1H-benzo[d]imidazol-2-yl)phenyl)piperazin-1-yl)quinoline dihydrochloride (8c)
4-(4-(4-(1H-benzo[d]imidazol-2-yl)phenyl)piperazin-1-yl)-7-chloroquinoline (8d)
7-chloro-4-(4-(4-(5-chloro-1H-benzo[d]imidazol-2-yl)phenyl)piperazin-1-yl)quinoline (8e)
3.1.4. General Procedure for the Synthesis of Compounds 10a–d
4-(5-carbamimidoyl-1H-benzo[d]imidazol-2-yl)benzoic acid dihydrochloride (10a)
4-(5-(N-isopropylcarbamimidoyl)-1H-benzo[d]imidazol-2-yl)benzoic acid dihydrochloride (10b)
4-(5-(4,5-dihydro-1H-imidazol-2-yl)-1H-benzo[d]imidazol-2-yl)benzoic acid hydrochloride (10c)
4-(1H-benzo[d]imidazol-2-yl)benzoic acid (10d)
3.1.5. General Procedure for the Synthesis of Compounds 12a–d
4-(5-carbamimidoyl-1H-benzo[d]imidazol-2-yl)-N-(2-(7-chloroquinolin-4-ylamino)ethyl)benzamide trihydrochloride (12a)
4-(5-(N-isopropylcarbamimidoyl)-1H-benzo[d]imidazol-2-yl)-N-(2-(7-chloroquinolin-4-ylamino)ethyl)benzamide trihydrochloride (12b)
N-(2-(7-chloroquinolin-4-ylamino)ethyl)-4-(5-(4,5-dihydro-1H-imidazol-2-yl)-1H-benzo[d]imidazol-2-yl)benzamide trihydrochloride (12c)
4-(1H-benzo[d]imidazol-2-yl)-N-(2-(7-chloroquinolin-4-ylamino)ethyl)benzamide (12d)
3.2. Biological Activity
3.2.1. Evaluation of the Antiproliferative Effect
Cell Lines
Cell Culturing
Proliferation Assay
3.2.2. Flow Cytometry Analysis of Cell Cycle
3.2.3. Measurement of Mitochondrial Membrane Potential (∆Ψm)
3.2.4. Determination of Apoptosis
3.3. Assessment of Absorption, Distribution, Metabolism, and Excretion (ADME) Properties
3.4. Molecular Docking Study
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. C. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2022. CA A Cancer J. C. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Nussinov, R.; Tsai, C.J.; Jang, H. Anticancer Drug Resistance: An Update and Perspective. Drug Resist. Updat. 2021, 59, 100796:1–100796:12. [Google Scholar] [CrossRef] [PubMed]
- Mao, J.; Wang, D.; Xu, P.; Wang, Y.; Zhang, H.; Wang, S.; Xu, F.; Wang, J.; Zhang, F. Structure-Based Drug Design and Synthesis of Novel N-Aryl-2,4-bithiazole-2-amine CYP1B1-Selective Inhibitors in Overcoming Taxol Resistance in A549 Cells. J. Med. Chem. 2022, 65, 16451–16480. [Google Scholar] [CrossRef] [PubMed]
- Labozzetta, M.; Barreca, M.; Spano, V.; Raimondi, M.V.; Poma, P.; Notarbartolo, M.; Barraja, P.; Montalbano, A. Novel insights on [1,2]oxazolo [5,4-e]isoindoles on multidrug resistant acute myeloid leukemia cell line. Drug. Dev. Res. 2022, 83, 1239–1466. [Google Scholar] [CrossRef]
- Moreno, S.; Fickl, M.; Bauer, I.; Brunner, M.; Razkova, A.; Dietmar, R.; Delazer, I.; Micura, R.; Lusser, A. 6-Thioguanosine Monophosphate Prodrugs Display Enhanced Performance against Thiopurine-Resistant Leukemia and Breast Cancer Cells. J. Med. Chem. 2022, 65, 15165–15173. [Google Scholar] [CrossRef]
- Kumar, S.; Kumar, V. Have molecular hybrids delivered effective anti-cancer treatments and what should future drug discovery focus on? Expert Opin. Drug Discov. 2021, 16, 335–363. [Google Scholar] [CrossRef]
- Soltan, O.M.; Shoman, M.E.; Abdel-Aziz, S.A.; Narumi, A.; Konno, H.; Abdel-Aziz, M. Molecular hybrids: A five-year survey on structures of multiple targeted hybrids of protein kinase inhibitors for cancer therapy. Eur. J. Med. Chem. 2021, 225, 113768:1–113768:32. [Google Scholar] [CrossRef]
- Szumilak, M.; Wiktorowska-Owczarek, A.; Stanczak, A. Hybrid Drugs—A Strategy for Overcoming Anticancer Drug Resistance? Molecules 2021, 26, 2601. [Google Scholar] [CrossRef]
- Singh, A.K.; Kumar, A.; Singh, H.; Sonawane, P.; Paliwal, H.; Thareja, S.; Pathak, P.; Grishina, M.; Jaremko, M.; Emwas, A.H.; et al. Concept of Hybrid Drugs and Recent Advancements in Anticancer Hybrids. Pharmaceuticals 2022, 15, 1071. [Google Scholar] [CrossRef]
- Guo, Y.; Hou, X.; Fang, H. Recent Applications of Benzimidazole as a Privileged Scaffold in Drug Discovery. Mini Rev. Med. Chem. 2021, 21, 1367–1379. [Google Scholar] [CrossRef] [PubMed]
- Alzhrani, Z.M.M.; Alam, M.M.; Nazreen, S. Recent Advancements on Benzimidazole: A Versatile Scaffold in Medicinal Chemistry. Mini Rev. Med. Chem. 2022, 22, 365–386. [Google Scholar] [CrossRef] [PubMed]
- Lalic, H.; Aurer, I.; Batinic, D.; Visnjic, D.; Smoljo, T.; Babic, A. Bendamustine: A review of pharmacology, clinical use and immunological effects. Oncol. Rep. 2022, 47, 114. [Google Scholar] [CrossRef] [PubMed]
- Bogeljic-Patekar, M.; Milunović, V.; Mišura-Jakobac, K.; Perica, D.; Mandac-Rogulj, I.; Kursar, M.; Planinc-Peraica, A.; Ostojić-Kolonić, S. Bendamustine: An Old Drug in the New Era for Patients with Non-Hodgkin Lymphomas and Chronic Lymphocytic Leukemia. Acta Clin. Croat. 2018, 57, 542–553. [Google Scholar] [CrossRef] [Green Version]
- Tran, B.; Cohen, M.S. The discovery and development of binimetinib for the treatment of melanoma. Expert Opin. Drug Discov. 2020, 15, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Zager, J.S.; Eroglu, Z. Encorafenib/binimetinib for the treatment of BRAF-mutant advanced, unresectable, or metastatic melanoma: Design, development, and potential place in therapy. OncoTargets Ther. 2018, 11, 9081–9089. [Google Scholar] [CrossRef] [Green Version]
- Roviello, G.; D’Angelo, A.; Petrioli, R.; Roviello, F.; Cianchi, F.; Nobili, S.; Mini, E.; Lavacchi, D. Encorafenib, Binimetinib, and Cetuximab in BRAF V600E-Mutated Colorectal Cancer. Transl. Oncol. 2020, 13, 100795. [Google Scholar] [CrossRef]
- Cutsem, E.V.; Huijberts, S.; Grothey, A.; Yaeger, R.; Cuyle, P.; Elez, E.; Fakih, M.; Montagut, C.; Peeters, M.; Yoshino, T.; et al. Binimetinib, Encorafenib, and Cetuximab Triplet Therapy for Patients With BRAF V600E–Mutant Metastatic Colorectal Cancer: Safety Lead-In Results From the Phase III BEACON Colorectal Cancer Study. J. Clin. Oncol. 2019, 37, 1460–1469. [Google Scholar] [CrossRef]
- Zhang, H.; Savage, S.; Reister Schultz, A.; Bottomly, D.; White, L.; Segerdell, E.; Wilmot, B.; McWeeney, S.K.; Eide, C.A.; Nechiporuk, T.; et al. Clinical resistance to crenolanib in acute myeloid leukemia due to diverse molecular mechanisms. Nat. Commun. 2019, 10, 244. [Google Scholar] [CrossRef] [Green Version]
- Senerovic, L.; Opsenica, D.; Moric, I.; Aleksic, I.; Spasić, M.; Vasiljevic, B. Quinolines and Quinolones as Antibacterial, Antifungal, Anti-virulence, Antiviral and Anti-parasitic Agents. In Advances in Microbiology, Infectious Diseases and Public Health; Donelli, G., Ed.; Springer: Cham, Switzerland, 2019; Volume 1282, ISBN 978-3-030-53646-6. [Google Scholar] [CrossRef]
- Musiol, R. An overview of quinoline as a privileged scaffold in cancer drug discovery. Expert. Opin. Drug. Discov. 2017, 12, 583–597. [Google Scholar] [CrossRef]
- Basavarajaiah, S.M. The Versatile Quinoline and Its Derivatives as anti-Cancer Agents: An Overview. Polycycl. Aromat. Compd. 2022, 1–13. [Google Scholar] [CrossRef]
- Al-Salama, Z.T.; Syed, Y.Y.; Scott, L.J. Lenvatinib: A Review in Hepatocellular Carcinoma. Drugs 2019, 79, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Yang, M.; Ching-Lung, L. Clinical efficacy of lenvatinib for the treatment of radioiodine-refractory thyroid carcinoma: A systematic review and meta-analysis of clinical trials. Clin. Endocrin. 2021, 95, 478–488. [Google Scholar] [CrossRef] [PubMed]
- Gambacorti-Passerini, C.; Le Coutre, P.; Piazza, R. The role of bosutinib in the treatment of chronic myeloid leukemia. Future Oncol. 2019, 16, 4395–4408. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Aziz, A.K.; Saadeldin, M.K.; Salem, A.H.; Ibrahim, S.A.; Shouman, S.; Abdel-Naim, A.B.; Orecchia, R. A Critical Review of Chloroquine and Hydroxychloroquine as Potential Adjuvant Agents for Treating People with Cancer. Future Pharmacol. 2022, 2, 28. [Google Scholar] [CrossRef]
- Fereira, P.M.P.; de Sousa, R.W.R.; de Oliviera Fereira, J.R.; Militao, G.C.G.; Bezerra, D.P. Chloroquine and hydroxychloroquine in antitumor therapies based on autophagy-related mechanisms. Pharmacol. Res. 2021, 168, 105582. [Google Scholar] [CrossRef]
- Krstulović, L.; Stolić, I.; Jukić, M.; Opačak-Bernardi, T.; Starčević, K.; Bajić, M.; Glavaš-Obrovac, L. New quinoline-arylamidine hybrids: Synthesis, DNA/RNA binding and antitumor activity. Eur. J. Med. Chem. 2017, 137, 196–210. [Google Scholar] [CrossRef]
- Rastija, V.; Jukić, M.; Opačak-Bernardi, T.; Krstulović, L.; Stolić, I.; Glavaš-Obrovac, L.J.; Bajić, M. Investigation of the structural and physicochemical requirements of quinoline-arylamidine hybrids for the growth inhibition of K562 and Raji leukemia cells. Turk. J. Chem. 2019, 43, 251–265. [Google Scholar] [CrossRef]
- Medlen, C.E.; Chibale, K.; Soares de Melo, C. Inhibition of the Growth of Tumour Cells. International Patent WO 2008/135886 A2, November 2008. [Google Scholar]
- Abou-Elkhair, R.A.I.; Hassan, A.E.A.; Boykin, D.W.; Wilson, W.D. Lithium Hexamethyldisilazane Transformation of Transiently Protected 4-Aza/Benzimidazole Nitriles to Amidines and their Dimethyl Sulfoxide Mediated Imidazole Ring Formation. Org. Lett. 2016, 18, 4714–4717. [Google Scholar] [CrossRef]
- Hranjec, M.; Starčević, K.; Zamola, B.; Mutak, S.; Đerek, M.; Karminski-Zamola, G. New amidino-benzimidazolyl derivatives of tylosin and desmycosin. J. Antibiot. 2002, 55, 308–314. [Google Scholar] [CrossRef] [Green Version]
- Fairley, T.A.; Tidwell, R.R.; Donkor, I.; Naiman, V.; Ohemeng, K.A.; Lombardy, R.J.; Bentley, J.A.; Cory, M. Structure, DNA minor groove binding, and base pair specificity of alkyl- and aryl-linked bis(amidinobenzimidazoles) and bis(amidinoindoles). J. Med. Chem. 1993, 36, 1746–1753. [Google Scholar] [CrossRef] [PubMed]
- Salahuddin, A.; Inam, A.; van Zyl, R.L.; Heslop, D.C.; Chen, C.T.; Avecilla, F.; Agarwal, S.M.; Azam, A. Synthesis and evaluation of 7-chloro-4-(piperazin-1-yl)quinoline-sulfonamide as hybrid antiprotozoal agents. Bioorg. Med. Chem. 2013, 21, 3080–3089. [Google Scholar] [CrossRef]
- Solomon, V.R.; Puri, S.K.; Srivastava, K.; Katti, S.B. Design and synthesis of new antimalarial agents from 4-aminoquinoline. Bioorg. Med. Chem. 2005, 13, 2157–2165. [Google Scholar] [CrossRef] [PubMed]
- Pešić, D.; Starčević, K.; Toplak, A.; Herreros, E.; Vidal, J.; Almela, M.J.; Jelić, D.; Alihodžić, S.; Spaventi, R.; Perić, M. Design, Synthesis, and in Vitro Activity of Novel 2′-O-Substituted 15-Membered Azalides. J. Med. Chem. 2012, 55, 3216–3227. [Google Scholar] [CrossRef]
- Bistrović, A.; Krstulović, L.; Harej, A.; Grbčić, P.; Sedić, M.; Koštrun, S.; Pavelić, S.K.; Bajić, M.; Raić-Malić, S. Design, synthesis and biological evaluation of novel benzimidazole amidines as potent multi-target inhibitors for the treatment of non-small cell lung cancer. Eur. J. Med. Chem. 2018, 1, 1616–1634. [Google Scholar] [CrossRef]
- Bistrović, A.; Krstulović, L.; Stolić, I.; Drenjančević, D.; Talapko, J.; Taylor, M.C.; Kelly, J.M.; Bajić, M.; Raić-Malić, S. Synthesis, anti-bacterial and anti-protozoal activities of amidinobenzimidazole derivatives and their interactions with DNA and RNA. J. Enzyme Inhib. Med. Chem. 2018, 33, 1323–1334. [Google Scholar] [CrossRef]
- Zuo, D.; Jiang, X.; Han, M.; Shen, J.; Lang, B.; Guan, Q.; Bai, Z.; Han, C.; Li, Z.; Zhang, W.; et al. Methyl 5-[(1H-indol-3-yl)selanyl]-1H-benzoimidazol-2-ylcarbamate (M-24), a novel tubulin inhibitor, causes G2/M arrest and cell apoptosis by disrupting tubulin polymerization in human cervical and breast cancer cells. Toxicol. Vitr. 2017, 42, 139–149. [Google Scholar] [CrossRef]
- Zhang, L.; Bochkur Dratver, M.; Yazal, T.; Dong, K.; Nguyen, A.; Yu, G.; Dao, A.; Bochkur Dratver, M.; Duhacek-Muggy, S.; Bhat, K.; et al. Mebendazole Potentiates Radiation Therapy in Triple-Negative Breast Cancer. Int. J. Radiat. Oncol. Biol. Phys. 2019, 103, 195–207. [Google Scholar] [CrossRef]
- Feng, L.; Su, W.; Cheng, J.; Xiao, T.; Li, H.; Chen, D.; Zhang, Z. Benzimidazole Hybrids as Anticancer Drugs: An Updated Review on Anticancer Properties, Structure–Activity Relationship, and Mechanisms of Action (2019–2021). Arch. Pharm. 2022, 355, 2200051. [Google Scholar] [CrossRef]
- Amend, S.R.; Torga, G.; Lin, K.; Kostecka, L.G.; de Marzo, A.; Austin, R.; Austin, R.H.; Pienta, K.J. Polyploid giant cancer cells: Unrecognized actuators of tumorigenesis, metastasis, and resistance. Prostate 2019, 79, 1489–1497. [Google Scholar] [CrossRef]
- Niu, N.; Zhang, J.; Zhang, N.; Mercado-Uribe, I.; Tao, F.; Han, Z.; Pathak, S.; Multani, A.S.; Kuang, J.; Yao, J.; et al. Linking genomic reorganization to tumor initiation via the giant cell cycle. Oncogenesis 2016, 5, e281. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, T.; Wakefield, L.; Peters, A.; Peto, M.; Spellman, P.; Grompe, M. Proliferative Polyploid Cells Give Rise to Tumors via Ploidy Reduction. Nat. Commun. 2021, 12, 646. [Google Scholar] [CrossRef]
- Was, H.; Borkowska, A.; Olszewska, A.; Klemba, A.; Marciniak, M.; Synowiec, A.; Kieda, C. Polyploidy Formation in Cancer Cells: How a Trojan Horse Is Born. Semin. Cancer Biol. 2022, 81, 24–36. [Google Scholar] [CrossRef]
- Carneiro, B.A.; El-Deiry, W.S. Targeting apoptosis in cancer therapy. Nat. Rev. Clin. Oncol. 2020, 17, 395–417. [Google Scholar] [CrossRef]
- Pfeffer, C.M.; Singh, A.T.K. Apoptosis: A Target for Anticancer Therapy. Int. J. Mol. Sci. 2018, 19, 448. [Google Scholar] [CrossRef] [Green Version]
- Nazreen, S.; Almalki, A.S.A.; Elbehairi, S.E.I.; Shati, A.A.; Alfaifi, M.Y.; Elhenawy, A.A.; Alsenani, N.I.; Alfarsi, A.; Alhadhrami, A.; Alqurashi, E.A.; et al. Cell Cycle Arrest and Apoptosis-Inducing Ability of Benzimidazole Derivatives: Design, Synthesis, Docking, and Biological Evaluation. Molecules 2022, 27, 6899. [Google Scholar] [CrossRef]
- Ma, K.; Chen, G.; Li, W.; Kepp, O.; Zhu, Y.; Chen, Q. Mitophagy, Mitochondrial Homeostasis, and Cell Fate. Front. Cell Dev. Biol. 2020, 8, 467. [Google Scholar] [CrossRef]
- Gharbaran, R.; Shi, C.; Onwumere, O.; Redenti, S. Plumbagin Induces Cytotoxicity via Loss of Mitochondrial Membrane Potential and Caspase Activation in Metastatic Retinoblastoma. Anticancer Res. 2021, 41, 4725–4732. [Google Scholar] [CrossRef]
- Li, M.H.; Yang, P.; Yang, T.; Zhang, K.; Liu, Y.; Liu, J.; Li, L.M.; Luo, X.Y.; Yang, S.X.; Zou, Q.; et al. A Novel Water-Soluble Benzothiazole Derivative BD926 Triggers ROS-Mediated B Lymphoma Cell Apoptosis via Mitochondrial and Endoplasmic Reticulum Signaling Pathways. Int. J. Oncol. 2016, 49, 2127–2134. [Google Scholar] [CrossRef]
- Pathak, N.; Rathi, E.; Kumar, N.; Kini, S.G.; Rao, C.M. A Review on Anticancer Potentials of Benzothiazole Derivatives. Mini-Rev. Med. Chem. 2020, 20, 12–23. [Google Scholar] [CrossRef]
- Doogue, M.P.; Polasek, T.M. The ABCD of clinical pharmacokinetics. Ther. Adv. Drug Saf. 2013, 4, 5–7. [Google Scholar] [CrossRef] [Green Version]
- Kassel, D.B. Applications of high-throughput ADME in drug discovery. Curr. Opin. Chem. Biol. 2004, 8, 339–345. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Yan, A.; Cai, Z. Prediction of human intestinal absorption by GA feature selection and support vector machine regression. Int. J. Mol. Sci. 2008, 10, 1961–1976. [Google Scholar] [CrossRef]
- Carpenter, T.S.; Kirshner, D.A.; Lau, E.Y.; Wong, S.E.; Nilmeier, J.P.; Lightstone, F.C. A Method to predict blood-brain barrier permeability of drug-like compounds using molecular dynamics simulations. Biophys. J. 2014, 107, 630–641. [Google Scholar] [CrossRef] [Green Version]
- Toutain, P.L.; Bousquet-Mélou, A. Plasma clearance. J. Vet. Pharmacol. Ther. 2004, 27, 415–425. [Google Scholar] [CrossRef]
- Paul, M.K.; Mukhopadhyay, A.K. Tyrosine kinase—Role and significance in cancer. Int. J. Med. Sci. 2004, 1, 101–115. [Google Scholar] [CrossRef] [Green Version]
- Basuroy, S.; Sheth, P.; Kuppuswamy, D.; Balasubramanian, S.; Ray, R.M.; Rao, R.K. Expression of kinase-inactive c-Src delays oxidative stress-induced disassembly and accelerates calciummediated reassembly of tight junctions in the Caco-2 cell monolayer. J. Biol. Chem. 2003, 278, 11916–11924. [Google Scholar] [CrossRef]
- Seeliger, M.A.; Ranjitkar, P.; Kasap, C.; Shan, Y.; Shaw, D.E.; Shah, N.P.; Kuriyan, J.; Maly, D.J. Equally Potent Inhibition of c-Src and Abl by Compounds that Recognize Inactive Kinase Conformations. Cancer Res. 2009, 69, 2384–2392. [Google Scholar] [CrossRef] [Green Version]
- Roskoski, R. Src protein–tyrosine kinase structure and regulation. Biochem. Biophys. Res. Commun. 2004, 324, 1155–1164. [Google Scholar] [CrossRef] [PubMed]
- Azam, M.; Seeliger, M.A.; Gray, N.S.; Kuriyan, J.K.; Daley, G.Q. Activation of tyrosine kinases by mutation of the gatekeeper threonine. Nat. Struct. Mol. Biol. 2008, 15, 1109–1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Sharkawy, E.R.; Almalki, F.; Hadda, T.B.; Rastija, V.; Lafridi, H.; Zgou, H. DFT calculations and POM analyses of cytotoxicity of some flavonoids from aerial parts of Cupressus sempervirens: Docking and Identification of Pharmacophore Sites. Bioorg. Chem. 2020, 100, 103850. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.; Chen, S.; Wang, Y.; Gao, C.; Chen, Y.; Tan, C.; Jiang, Y. Design, synthesis and evaluation of azaacridine derivatives as dual-target EGFR and Src kinase inhibitors for antitumor treatment. Eur. J. Med. Chem. 2016, 136, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Boyd, M.R.; Paull, K.D. Some practical considerations and applications of the national cancer institute in vitro anticancer drug discovery screen. Drug Dev. Res. 1995, 34, 91–109. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Meth. 1983, 65, 55–63. [Google Scholar] [CrossRef]
- MedCalc® Statistical Software version 20.112, MedCalc Software Ltd.: Ostend, Belgium, 2022. Available online: https://www.medcalc.org(accessed on 20 July 2022).
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, druglikeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Hsu, K.C.; Chen, Y.F.; Lin, S.R.; Yang, J.M. iGEMDOCK: A graphical environment of enhancing GEMDOCK using pharmacological interactions and post-screening analysis. BMC Bioinformatics 2011, 12, S33. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| GI50 (μM) | |||||||||
| Compd. | R | MDCK1 | HeLa | CaCo-2 | MCF-7 | CCRF-CEM | THP1 | Raji | HuT78 |
| 5a |  | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
| 5b |  | >100 | >100 | >100 | >100 | >100 | >100 | >100 | 41.8 ± 8.2 |
| 5c |  | >100 | >100 | >100 | >100 | >100 | >100 | >100 | 66.2 ± 14.6 |
| 5d | H | 20.4 ± 2.8 | 0.7 ± 0.1 | 1.0 ± 0.1 | 33.6 ± 8.3 | 8.2 ± 6.2 | 0.6 ± 0.3 | 4.3 ± 1.2 | 0.4 ± 0.3 |
| 5e | Cl | 11.4 ± 6.2 | 11.8 ± 1.2 | 1.1 ± 0.1 | 11.1 ± 1.6 | 15.6 ± 3.4 | 2.1 ± 0.9 | 5.4 ± 2.0 | 6.7 ± 0.5 |
| 8a |  | 42.7 ± 10.8 | 36.3 ± 9.6 | >100 | 20.7 ± 8.6 | 32.1 ± 3.8 | >100 | 37.7 ± 8.6 | 20.2 ± 6.1 |
| 8b |  | 58.8 ± 9.8 | 21.0 ± 1.9 | 100 | 30.9 ± 2.7 | 39.6 ± 9.2 | >100 | 42.9 ± 8.8 | 26.2 ± 1.6 |
| 8c |  | 30.1 ± 5.0 | 41.8 ± 8.4 | 100 | 24.8 ± 2.2 | 25.4 ± 11.5 | 71.3 ± 11.4 | 43.9 ± 16.2 | 27.6 ± 4.7 |
| 8d | H | >100 | >100 | >100 | >100 | 5.0 ± 2.6 | 3.2 ± 1.1 | 3.8 ± 0.5 | 8.1 ± 4.7 |
| 8e | Cl | 2.7 ± 0.3 | 100 | >100 | 84.0 ± 7.7 | 41.0 ± 0.5 | 22.3 ± 7.4 | 13.4 ± 2.6 | 15.6 ± 9.8 |
| 12a |  | 21.6 ± 3.2 | 11.1 ± 3.8 | 24.7 ± 6.2 | 39.1 ± 5.6 | 20.0 ± 6.8 | >100 | 8.9 ± 3.3 | 4.1 ± 2.0 |
| 12b |  | 42.9 ± 5.1 | 100 | 38.7 ± 12.4 | 100 | 43.3 ± 17.5 | 100 | 12.8 ± 3.7 | 5.6 ± 2.7 |
| 12c |  | 39.6 ± 2.2 | 33.2 ± 5.8 | 52.0 ± 9.1 | 100 | 56.3 ± 9.0 | >100 | 20.8 ± 1.8 | 4.4 ± 1.0 |
| 12d | H | 100 | 100 | 16.4 ± 5.6 | 20.0 ± 7.2 | 17.2 ± 7.7 | 4.0 ± 0.8 | 57.7 ± 23.8 | 3.5 ± 1.5 |
| 5-FU | 74.0 ± 3.1 | 8.2 ± 1.9 | 5.9 ± 0.7 | 30.2 ± 4.7 | >50 | 76.3 ± 0.5 | >100 | >100 | |
| ID Com. | MW * | Rot. b. | H-b.acc. | H-b. Donors | Fraction Csp3 | TPSA | MLOGP | log S | GI | BBB |
|---|---|---|---|---|---|---|---|---|---|---|
| 5a | 412.87 | 4 | 3 | 4 | 0 | 103.47 | 3.7 | −5.62 | High | No |
| 5b | 454.95 | 6 | 3 | 4 | 0.12 | 89.48 | 4.31 | −6.45 | Low | No |
| 5c | 438.91 | 4 | 3 | 3 | 0.08 | 77.99 | 4.11 | −5.74 | High | No |
| 5d | 370.83 | 3 | 2 | 2 | 0 | 53.6 | 4.12 | −6.15 | High | No |
| 5e | 405.28 | 3 | 2 | 2 | 0 | 53.6 | 4.6 | −6.73 | High | No |
| 8a | 481.98 | 4 | 3 | 3 | 0.15 | 97.92 | 3.71 | −6.07 | High | No |
| 8b | 524.06 | 6 | 3 | 3 | 0.23 | 83.93 | 4.29 | −6.92 | High | No |
| 8c | 508.02 | 4 | 3 | 2 | 0.21 | 72.44 | 4.1 | −6.2 | High | Yes |
| 8d | 439.94 | 3 | 2 | 1 | 0.15 | 48.05 | 4.11 | −6.58 | High | Yes |
| 8e | 474.38 | 3 | 2 | 1 | 0.15 | 48.05 | 4.58 | −7.17 | High | Yes |
| 12a | 483.95 | 8 | 4 | 5 | 0.08 | 132.57 | 2.53 | −5.34 | Low | No |
| 12b | 526.03 | 10 | 4 | 5 | 0.17 | 118.58 | 3.11 | −6.19 | Low | No |
| 12c | 509.99 | 8 | 4 | 4 | 0.14 | 107.09 | 3.46 | −5.47 | High | No |
| 12d | 441.91 | 7 | 3 | 3 | 0.08 | 82.7 | 3.44 | −5.85 | High | No |
| HIA | VDss | BBB Per. | CNS Per. | Total Clear. | Oral Rat | Minnow Tox. | |
|---|---|---|---|---|---|---|---|
| 5a | 95.43 | 0.08 | −1.20 | −1.81 | 0.87 | 1.68 | 2.09 |
| 5b | 76.62 | 0.30 | −0.94 | −1.53 | 0.86 | 1.25 | 2.42 |
| 5c | 84.81 | 0.08 | 0.25 | −0.85 | 1.01 | 2.66 | −0.47 |
| 5d | 82.53 | −0.03 | 0.33 | −1.42 | 1.00 | 1.90 | −1.66 |
| 5e | 81.58 | −0.02 | 0.28 | −1.30 | 0.94 | 1.81 | −2.05 |
| 8a | 95.68 | 0.25 | −0.97 | −1.88 | 0.88 | 3.98 | −1.67 |
| 8b | 86.29 | 0.29 | −0.97 | −1.61 | 0.88 | 3.96 | −2.53 |
| 8c | 87.35 | 0.02 | −0.71 | −1.77 | 0.88 | 2.62 | −1.95 |
| 8d | 85.08 | −0.13 | 0.53 | −0.54 | 0.87 | 1.98 | −2.49 |
| 8e | 84.13 | −0.12 | 0.49 | −0.59 | 0.81 | 1.86 | −2.92 |
| 12a | 78.14 | 0.33 | −1.36 | −2.34 | 0.95 | 3.68 | 0.89 |
| 12b | 80.06 | 0.35 | −1.36 | −2.07 | 0.90 | 3.56 | −0.17 |
| 12c | 86.16 | 0.15 | −1.11 | −2.23 | 0.96 | 2.32 | 0.40 |
| 12d | 80.03 | −0.03 | −1.06 | −1.90 | 0.95 | 1.82 | 0.08 |
| ID Com. (Pose Numb.) | Total Energy | Van der Waals Interaction | H-Bond | Electrostatic Interactions |
|---|---|---|---|---|
| 12c (1) | −150.08 | −137.48 | −12.59 | 0.00 |
| G6H (2) | −133.74 | −120.60 | −13.14 | 0.00 |
| 5b (2) | −130.47 | −119.97 | −10.50 | 0.00 |
| 5c (0) | −129.58 | −119.08 | −10.50 | 0.00 |
| 12b (2) | −122.66 | −103.09 | −19.57 | 0.00 |
| 8b (1) | −121.09 | −107.15 | −13.93 | 0.00 |
| 12d (1) | −119.99 | −107.95 | −12.04 | 0.00 |
| 5a (1) | −119.86 | −108.54 | −10.36 | −0.97 |
| 12a (1) | −114.60 | −106.34 | −8.26 | 0.00 |
| 8d (2) | −114.42 | −114.42 | 0.00 | 0.00 |
| 5d (2) | −113.67 | −103.65 | −10.03 | 0.00 |
| 8c (2) | −112.40 | −104.21 | −8.19 | 0.00 |
| 5e (1) | −108.71 | −101.71 | −7.00 | 0.00 |
| 8a (1) | −104.96 | −88.97 | −15.99 | 0.00 |
| 8e (1) | −103.62 | −94.59 | −9.03 | 0.00 |
| H-Bond | Energy/kcal mol−1 (Distance, Å) | H-Bond | Energy/kcal mol−1 (Distance, Å) |
|---|---|---|---|
| H-M-LYS-295 | −3.50 (3.10) | H-M-VAL-402 | −3.26 (3.10) |
| H-S-GLU-310 | −3.50 (2.77) | H-M-ASP-404 | −1.79 (3.34) |
| van der Waals int. | Energy/kcal mol−1 | van der Waals int. | Energy/kcal mol−1 |
| V-LEU-273 | −5.03 | S-THR-338 | −4.71 |
| S-VAL-281 | −7.97 | M-GLY-344 | −2.12 |
| M-ALA-293 | −3.29 | S-SER-345 | −0.88 |
| M-ALA-293 | −3.29 | M-VAL-383 | −0.34 |
| S-LYS-295 | −1.92 | M-HIS-384 | −0.46 |
| S-GLU-310 | −5.08 | S-HIS-384 | −1.55 |
| S-VAL-313 | −0.36 | S-LEU-393 | −3.42 |
| S-MET-314 | −8.07 | M-ALA-403 | −6.65 |
| S-LEU-317 | −0.62 | M-ASP-404 | −10.65 |
| S-LEU-322 | −2.67 | S-ASP-404 | −5.95 |
| M-VAL-323 | −4.06 | M-PHE-405 | −0.55 |
| S-VAL-323 | −0.70 | S-PHE-405 | −10.41 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krstulović, L.; Leventić, M.; Rastija, V.; Starčević, K.; Jirouš, M.; Janić, I.; Karnaš, M.; Lasić, K.; Bajić, M.; Glavaš-Obrovac, L. Novel 7-Chloro-4-aminoquinoline-benzimidazole Hybrids as Inhibitors of Cancer Cells Growth: Synthesis, Antiproliferative Activity, in Silico ADME Predictions, and Docking. Molecules 2023, 28, 540. https://doi.org/10.3390/molecules28020540
Krstulović L, Leventić M, Rastija V, Starčević K, Jirouš M, Janić I, Karnaš M, Lasić K, Bajić M, Glavaš-Obrovac L. Novel 7-Chloro-4-aminoquinoline-benzimidazole Hybrids as Inhibitors of Cancer Cells Growth: Synthesis, Antiproliferative Activity, in Silico ADME Predictions, and Docking. Molecules. 2023; 28(2):540. https://doi.org/10.3390/molecules28020540
Chicago/Turabian StyleKrstulović, Luka, Marijana Leventić, Vesna Rastija, Kristina Starčević, Maja Jirouš, Ivana Janić, Maja Karnaš, Kornelija Lasić, Miroslav Bajić, and Ljubica Glavaš-Obrovac. 2023. "Novel 7-Chloro-4-aminoquinoline-benzimidazole Hybrids as Inhibitors of Cancer Cells Growth: Synthesis, Antiproliferative Activity, in Silico ADME Predictions, and Docking" Molecules 28, no. 2: 540. https://doi.org/10.3390/molecules28020540