
Revealing the Reasons for Degeneration of Resonance-Assisted Hydrogen Bond on the Aromatic Platform: Calculations of Ortho-, Meta-, Para-Disubstituted Benzenes, and (Z)-(E)-Olefins

Abstract
:
1. Introduction
2. Results and Discussion
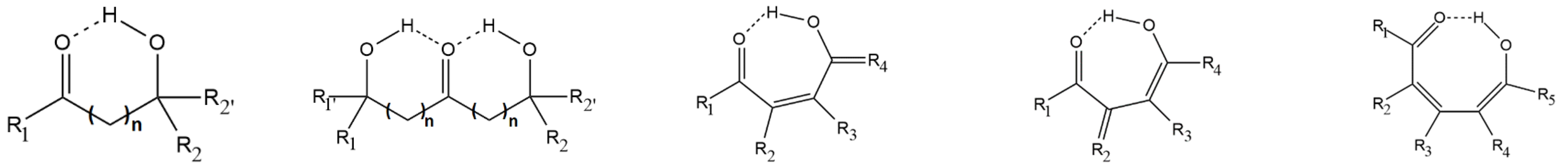
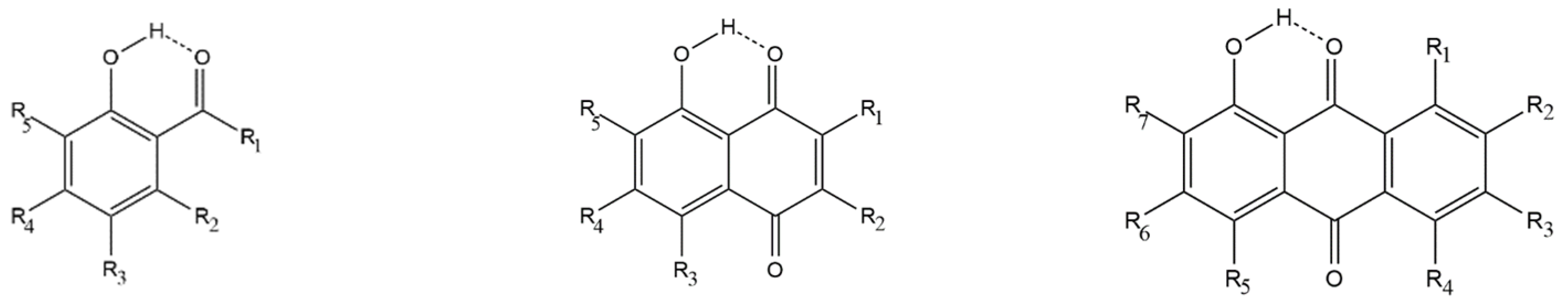
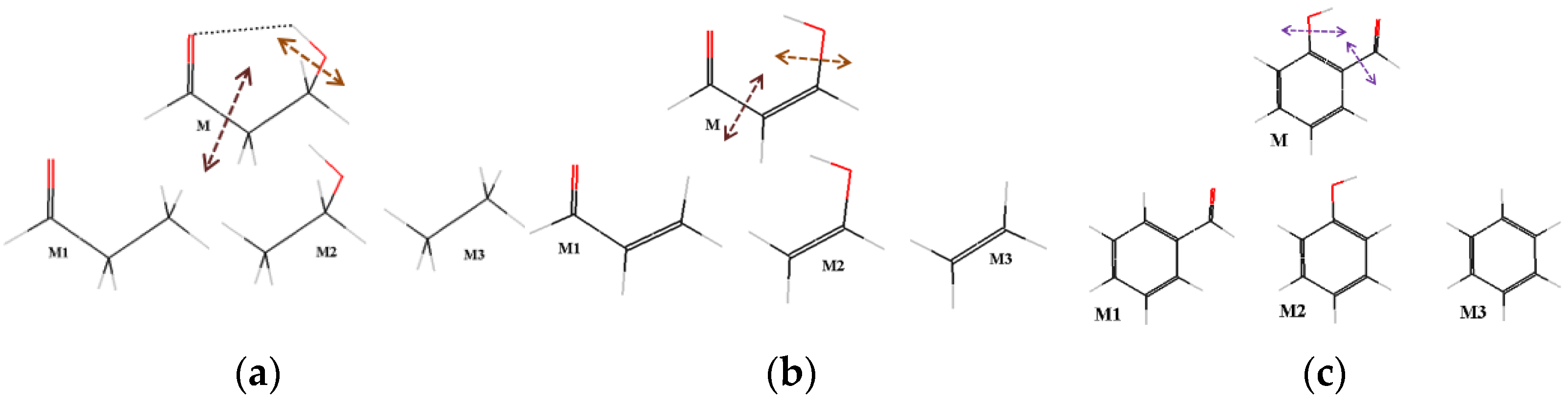
2.1. Specifications of the Studied Compounds
2.2. Previously Studied Compounds with the O−H···O=C Intramolecular Hydrogen Bonding
2.3. Newly Studied Compounds Exhibiting Push-Pull Effect
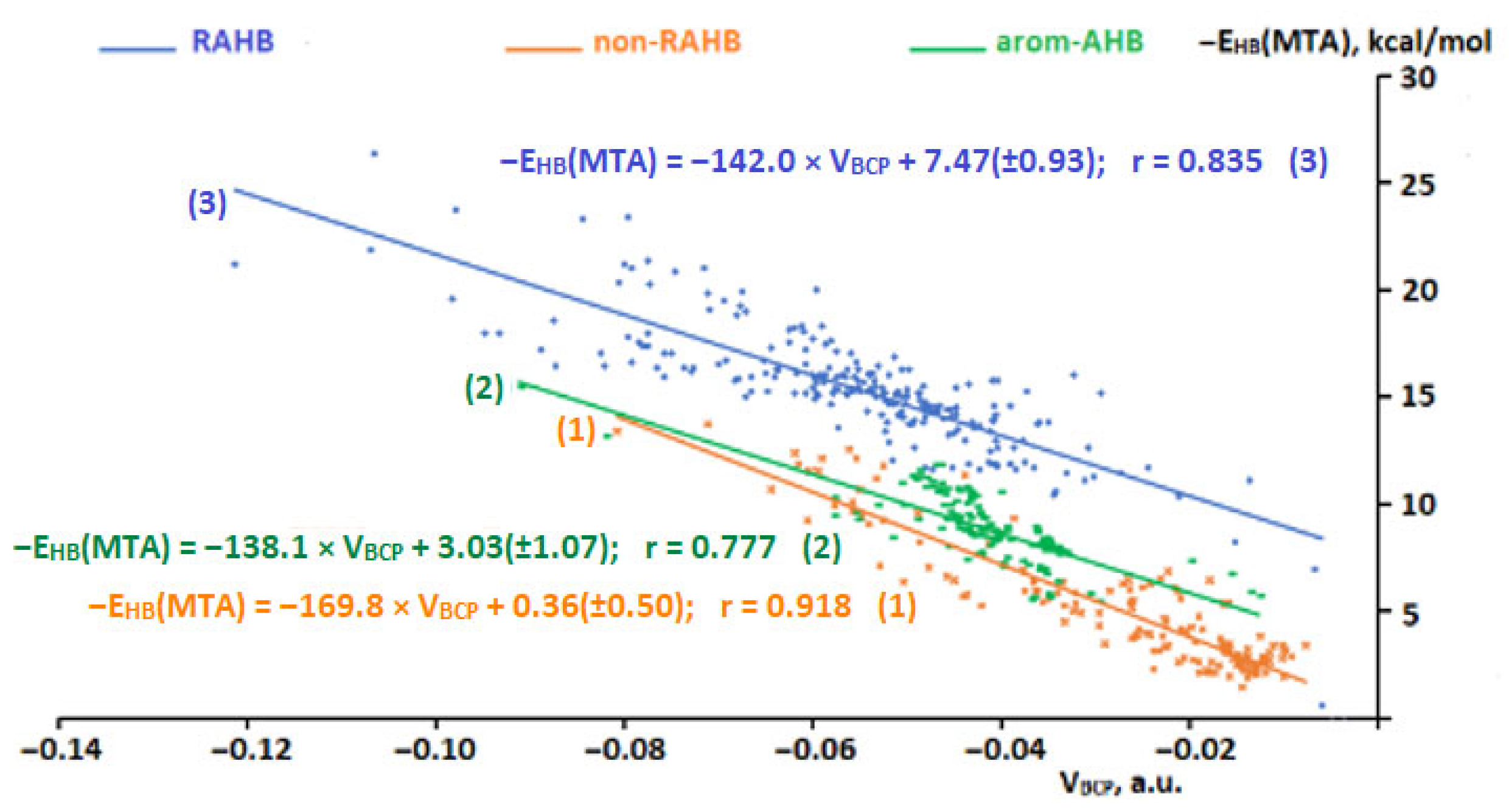
2.4. Dependences of Hydrogen Bond Energy on Magnitudes of Potential Energy Density at Hydrogen Bond Critical Point for the Non-RAHB, RAHB, and Arom-AHB Clusters
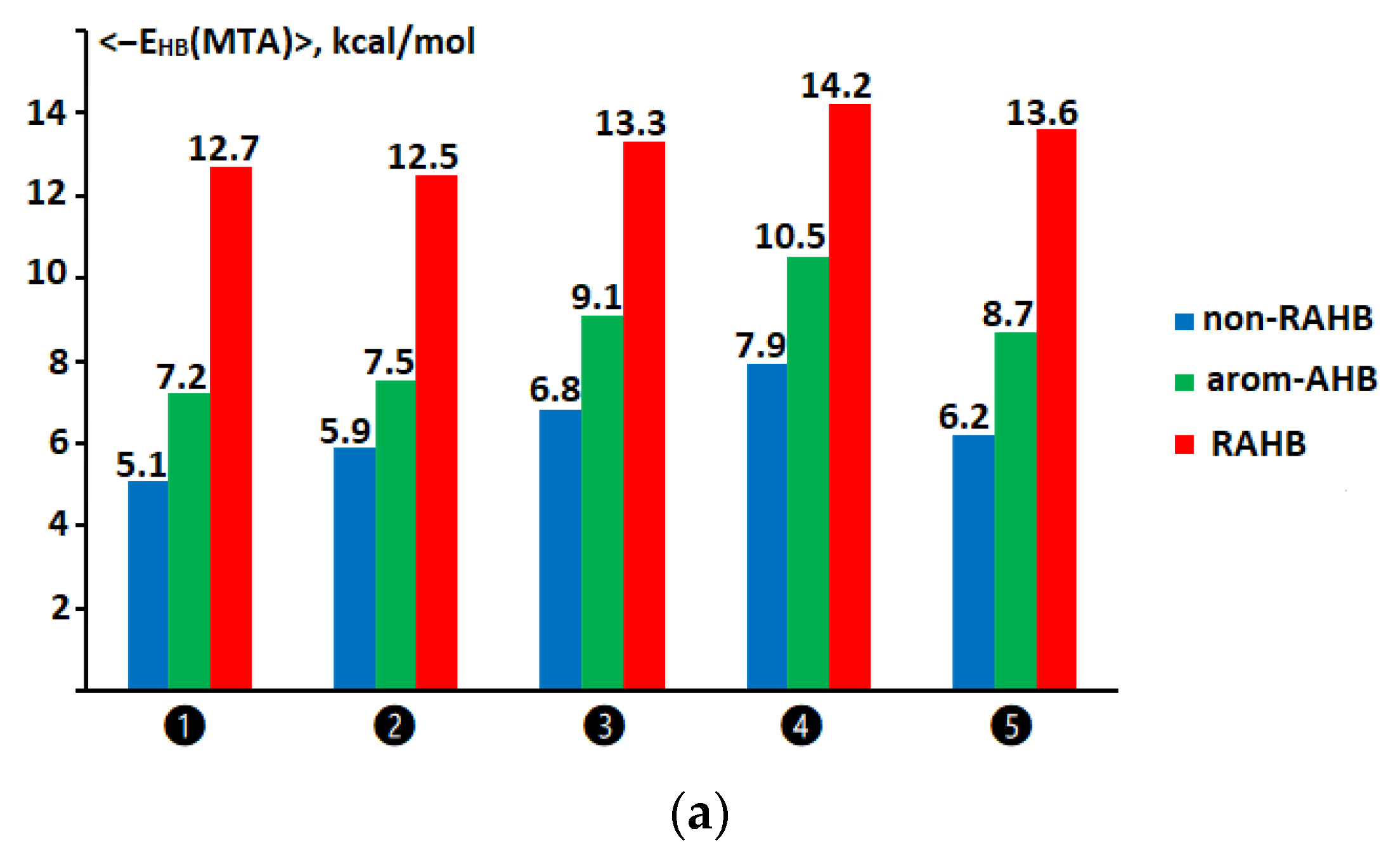
2.5. Comparison of the Hydrogen Bond Energy for Compounds of the Non-RAHB, RAHB, and Arom-AHB Clusters within the Same Range of Hydrogen Bond Descriptors
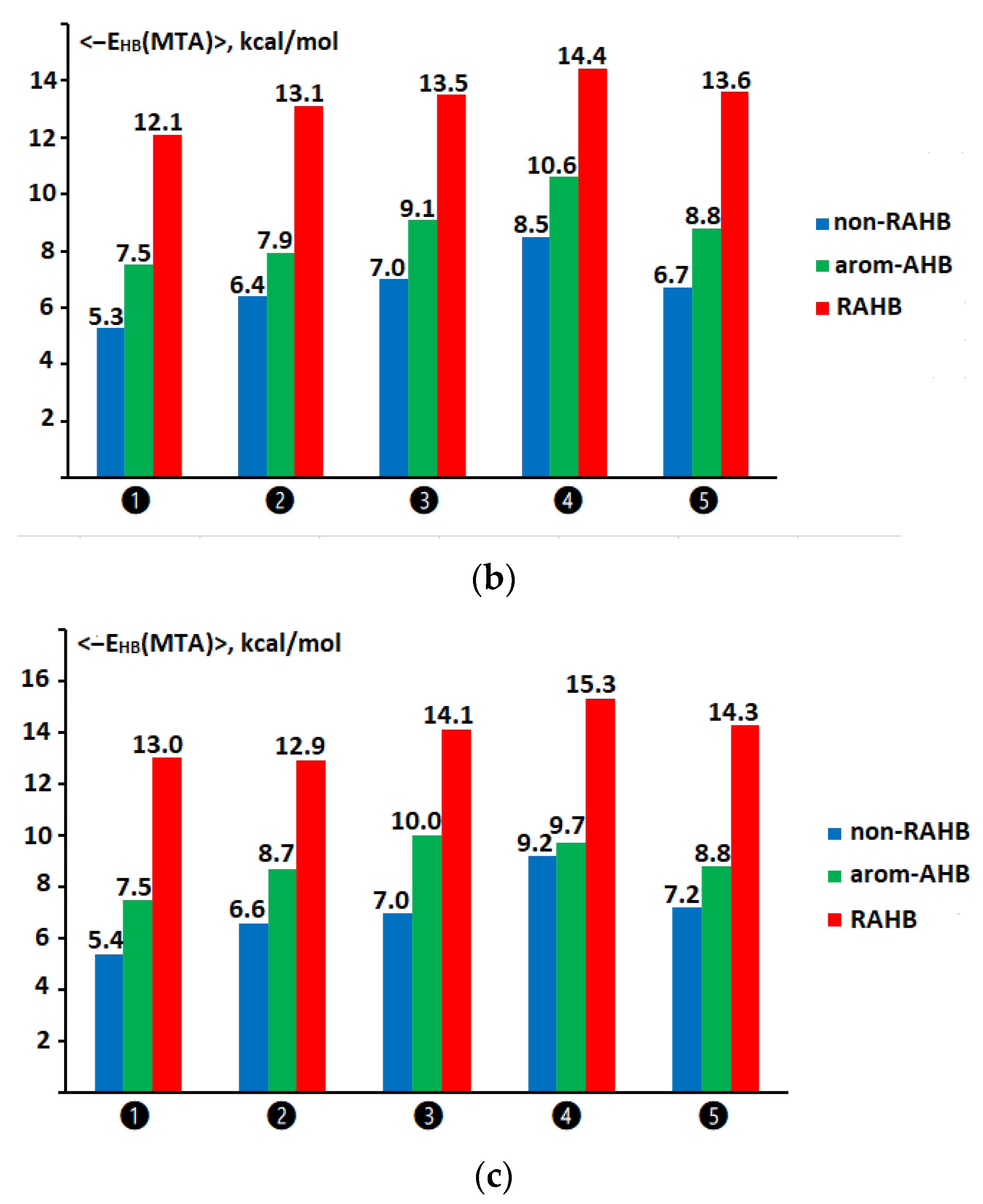
2.6. Comparison of Hydrogen Bond Energy Values for Compounds of the Non-RAHB, RAHB, and Arom-AHB Clusters Assessed via Molecular Tailoring and Function-Based Approaches
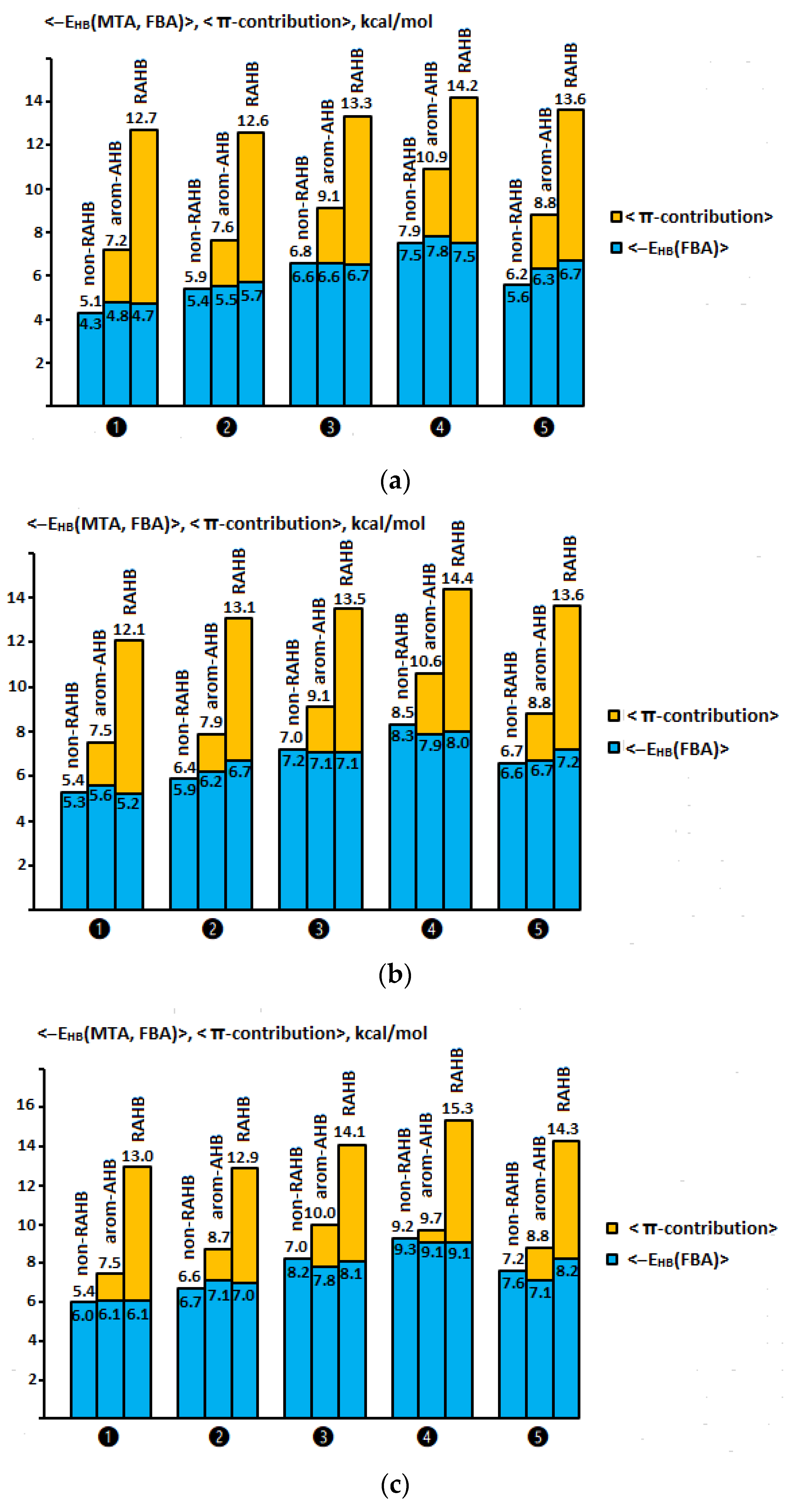
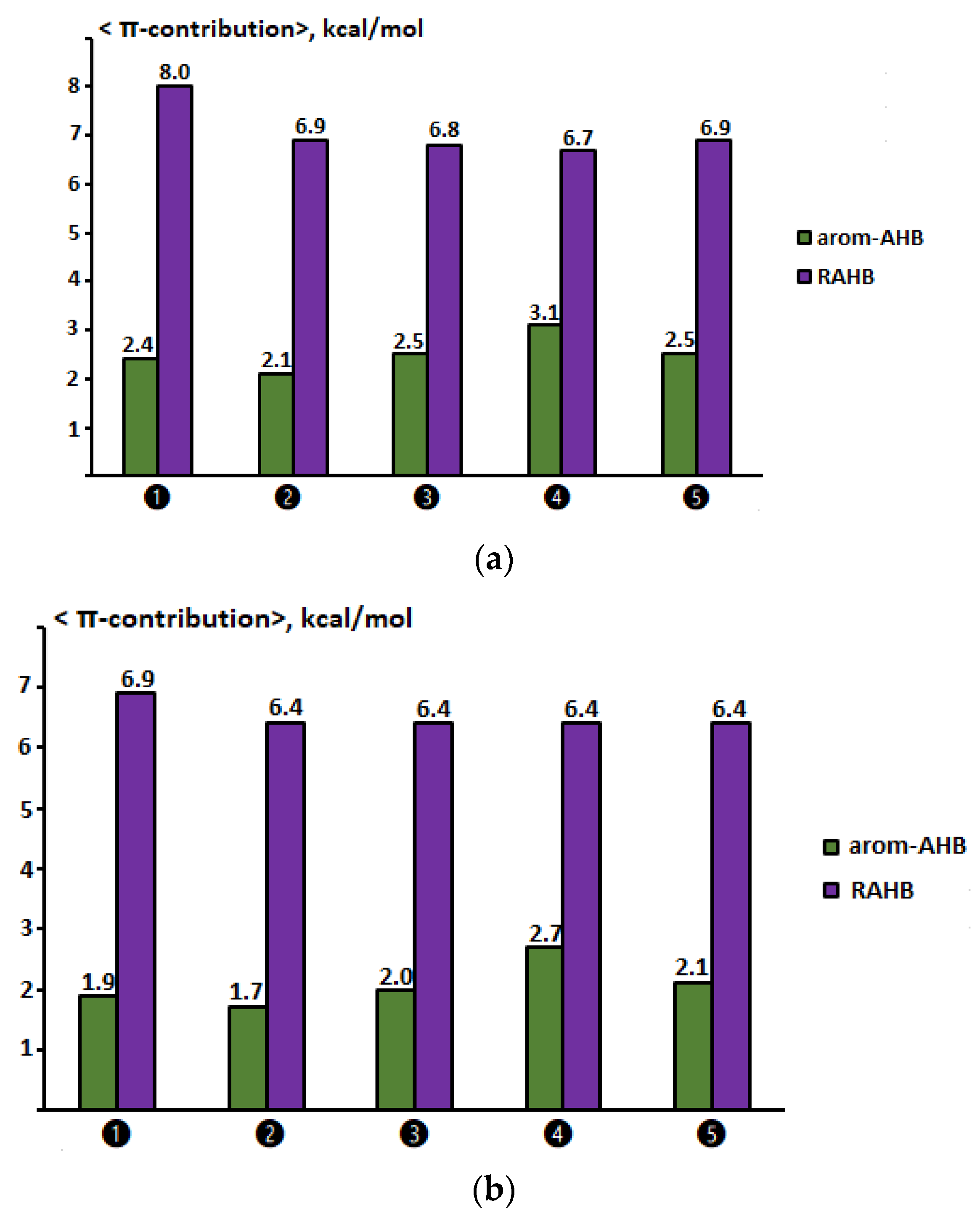
2.7. Comparison of π-Contributions to the Total Energy of Arom-AHB and RAHB Interactions
2.8. Testing the Ability of the Aromatic Ring and the Double Bond to Conduct the Resonance Effect
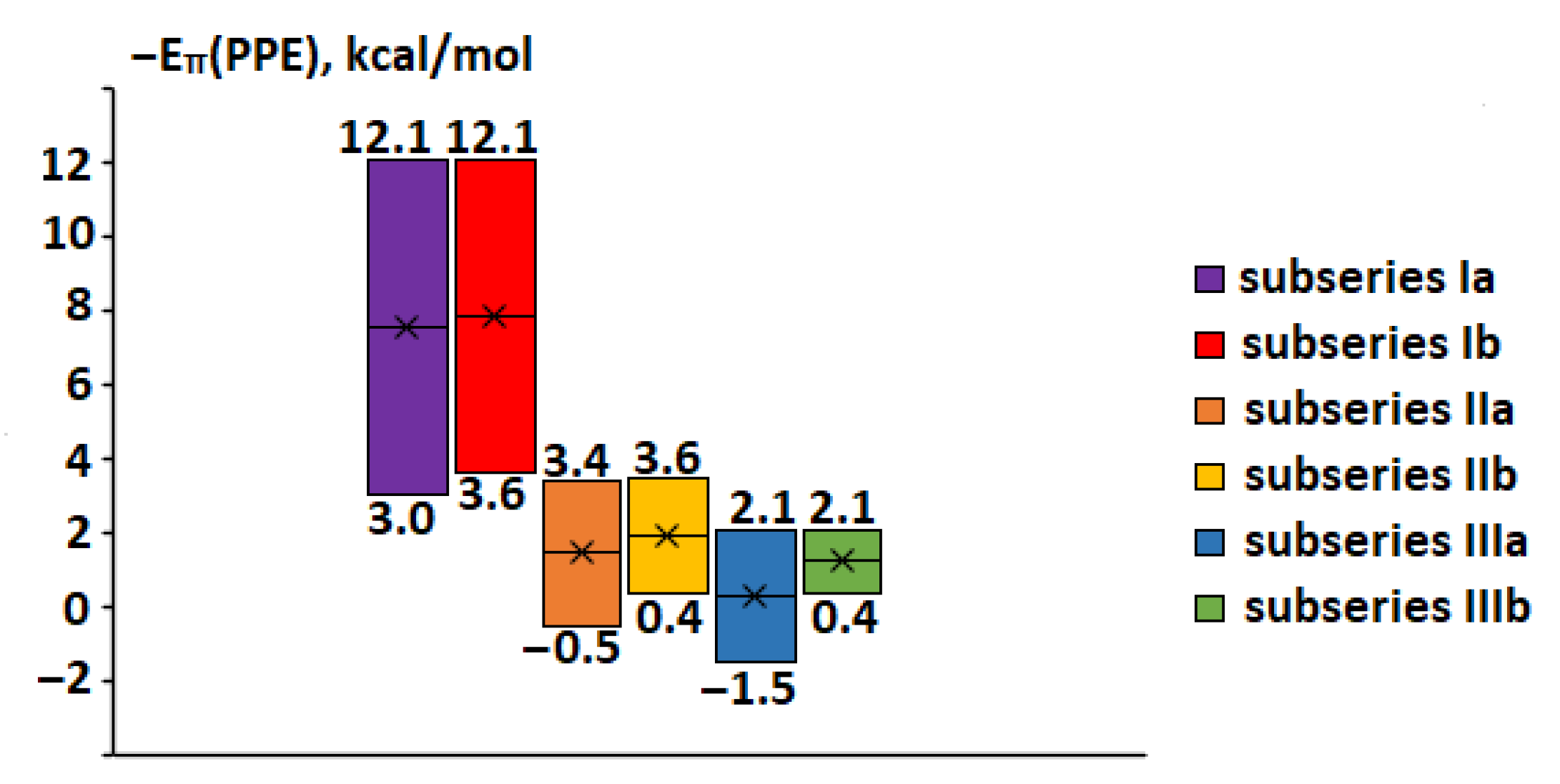
2.9. Relationship of −Eπ(PPE) Values with Structural and Spectral Parameters and Frontier Orbitals Energy
2.9.1. Dependence of π-Spacer Bonds Lengths on −Eπ(PPE) Values
2.9.2. Dependence of C=O Vibration Frequency on −Eπ(PPE) Value
2.9.3. Dependence of HOMO and LUMO Energy on −Eπ(PPE) Value
3. Methods and Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Gilli, G.; Bellucci, F.; Ferretti, V.; Bertolasi, V. Evidence for resonance-assisted hydrogen bonding from crystal-structure correlations on the enol form of the β-diketone fragment. J. Am. Chem. Soc. 1989, 111, 1023–1028. [Google Scholar] [CrossRef]
- Bertolasi, V.; Gilli, P.; Ferretti, V.; Gilli, G. Evidence for resonance-assisted hydrogen bonding. 2. Intercorrelation between crystal structure and spectroscopic parameters in eight intramolecularly hydrogen bonded 1,3-diaryl-1,3-propanedione enols. J. Am. Chem. Soc. 1991, 113, 4917–4925. [Google Scholar] [CrossRef]
- Gilli, P.; Bertolasi, V.; Pretto, L.; Lycka, A.; Gilli, G. The Nature of Solid-State N−H···O/O−H···N Tautomeric Competition in Resonant Systems. Intramolecular Proton Transfer in Low-Barrier Hydrogen Bonds Formed by the ···O=C−C=N−NH··· ⇄ ···HO−C=C−N=N··· Ketohydrazone−Azoenol System. A Variable-Temperature X-ray Crystallographic and DFT Computational Study. J. Am. Chem. Soc. 2002, 124, 13554–13567. [Google Scholar] [PubMed]
- Gilli, P.; Bertolasi, V.; Pretto, L.; Ferretti, V.; Gilli, G. Covalent versus Electrostatic Nature of the Strong Hydrogen Bond: Discrimination among Single, Double, and Asymmetric Single-Well Hydrogen Bonds by Variable-Temperature X-ray Crystallographic Methods in β-Diketone Enol RAHB Systems. J. Am. Chem. Soc. 2004, 126, 3845–3855. [Google Scholar] [CrossRef] [PubMed]
- Alkorta, I.; Elguero, J.; Mó, O.; Yáñez, M.; Del Bene, J.E. Are resonance-assisted hydrogen bonds ‘resonance assisted’? A theoretical NMR study. Chem. Phys. Lett. 2005, 411, 411–415. [Google Scholar] [CrossRef]
- Sanz, P.; Mó, O.; Yáñez, M.; Elguero, J. Non-Resonance-Assisted Hydrogen Bonding in Hydroxymethylene and Aminomethylene Cyclobutanones and Cyclobutenones and Their Nitrogen Counterparts. Chem. Phys. Chem. 2007, 8, 1950–1958. [Google Scholar] [CrossRef]
- Sanz, P.; Mó, O.; Yáñez, M.; Elguero, J. Resonance-Assisted Hydrogen Bonds: A Critical Examination. Structure and Stability of the Enols of β-Diketones and β-Enaminones. J. Phys. Chem. A 2007, 111, 3585–3591. [Google Scholar] [CrossRef]
- Fuster, F.; Grabowski, S.J. Intramolecular Hydrogen Bonds: The QTAIM and ELF Characteristics. J. Phys. Chem. A 2011, 115, 10078–10086. [Google Scholar] [CrossRef]
- Guevara-Vela, J.M.; Romero-Montalvo, E.; Costales, A.; Pendás, A.M.; Rocha-Rinza, T. The nature of resonance-assisted hydrogen bonds: A quantum chemical topology perspective. Phys. Chem. Chem. Phys. 2016, 18, 26383–26390. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Zhang, H.; Wu, W.; Mo, Y. A Critical Check for the Role of Resonance in Intramolecular Hydrogen Bonding. A Chem. Eur. J. 2017, 23, 16885–16891. [Google Scholar] [CrossRef]
- Grosch, A.A.; van der Lubbe, S.C.C.; Fonseca Guerra, C. Nature of Intramolecular Resonance Assisted Hydrogen Bonding in Malonaldehyde and Its Saturated Analogue. J. Phys. Chem. A 2018, 122, 1813–1820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanz, P.; Montero-Campillo, M.M.; Mó, O.; Yáñez, M.; Alkorta, I.; Elguero, J. Intramolecular magnesium bonds in malonaldehyde-like systems. A critical view of the resonance-assisted phenomena. Theor. Chem. Acc. 2018, 137, 97. [Google Scholar] [CrossRef] [Green Version]
- Krygowski, T.M.; Bankiewicz, B.; Zarnocki, C.; Palusiak, M. Quasi-aromaticity—What does it mean? Tetrahedron 2015, 71, 4895–4908. [Google Scholar] [CrossRef]
- Nakhaei, E.; Nowroozi, A. On the performance of resonance assisted hydrogen bond theory in malonaldehyde derivatives. Comput. Theor. Chem. 2016, 1096, 27–32. [Google Scholar] [CrossRef]
- Zhou, Y.; Deng, G.; Zheng, Y.-Z.; Xu, J.; Ashraf, H.; Yu, Z.-W. Evidences for Cooperative Resonance-Assisted Hydrogen Bonds in Protein Secondary Structure Analogs. Sci. Rep. 2016, 6, 36932. [Google Scholar] [CrossRef] [Green Version]
- Pareras, G.; Palusiak, M.; Duran, M.; Solà, M.; Simon, S. Tuning the Strength of the Resonance-Assisted Hydrogen Bond in o-Hydroxybenzaldehyde by Substitution in the Aromatic Ring. J. Phys. Chem. A 2018, 122, 2279–2287. [Google Scholar] [CrossRef] [Green Version]
- Durlak, P.; Latajka, Z. Car–Parrinello and Path Integral Molecular Dynamics Study of the Proton Transfer in the Intramolecular Hydrogen Bonds in the Ketohydrazone–Azoenol System. J. Phys. Chem. B 2018, 122, 7862–7873. [Google Scholar] [CrossRef]
- Nguyen, Y.H.; Lampkin, B.J.; Venkatesh, A.; Ellern, A.; Rossini, A.J.; Van Veller, B. Open-Resonance-Assisted Hydrogen Bonds and Competing Quasiaromaticity. J. Org. Chem. 2018, 83, 9850–9857. [Google Scholar] [CrossRef]
- Lin, X.; Jiang, X.; Wu, W.; Mo, Y. Induction, Resonance, and Secondary Electrostatic Interaction on Hydrogen Bonding in the Association of Amides and Imides. J. Org. Chem. 2018, 83, 13446–13453. [Google Scholar] [CrossRef]
- Rafat, R.; Nowroozi, A. Competition Between the Intramolecular Hydrogen Bond and the π-Electron Delocalization in Some RAHB Systems: A Theoretical Study. J. Struct. Chem. 2019, 60, 755–762. [Google Scholar] [CrossRef]
- Pareras, G.; Szczepanik, D.W.; Duran, M.; Solà, M.; Simon, S. Tuning the Strength of the Resonance-Assisted Hydrogen Bond in Acenes and Phenacenes with Two o-Hydroxyaldehyde Groups—The Importance of Topology. J. Org. Chem. 2019, 84, 15538–15548. [Google Scholar] [CrossRef] [PubMed]
- Shapenova, D.S.; Shiryaev, A.A.; Bolte, M.; Kukułka, M.; Szczepanik, D.W.; Hooper, J.; Babashkina, M.G.; Mahmoudi, G.; Mitoraj, M.P.; Safin, D.A. Resonance Assisted Hydrogen Bonding Phenomenon Unveiled through Both Experiments and Theory: A New Family of Ethyl N-Salicylideneglycinate Dyes. Chem. Eur. J. 2020, 26, 12987–12995. [Google Scholar] [CrossRef]
- Grabowski, S.J. Intramolecular Hydrogen Bond Energy and Its Decomposition—O–H∙∙∙O Interactions. Crystals 2021, 11, 5. [Google Scholar] [CrossRef]
- Gholami, S.; Aarabi, M.; Grabowski, S.J. Coexistence of Intra- and Intermolecular Hydrogen Bonds: Salicylic Acid and Salicylamide and Their Thiol Counterparts. J. Phys. Chem. A 2021, 125, 1526–1539. [Google Scholar] [CrossRef] [PubMed]
- Guevara-Vela, J.M.; Gallegos, M.; Valentín-Rodríguez, M.A.; Costales, A.; Rocha-Rinza, T.; Pendás, A.M. On the Relationship between Hydrogen Bond Strength and the Formation Energy in Resonance-Assisted Hydrogen Bonds. Molecules 2021, 26, 4196. [Google Scholar] [CrossRef]
- Karimi, P.; Sanchooli, M. Tuning the resonance-assisted hydrogen bond (RAHB) of malonaldehyde using π-conjugated substituents and presentation of its energy decomposition. J. Mol. Graph. Mod. 2022, 112, 108142. [Google Scholar] [CrossRef]
- Domagała, M.; Simon, S.; Palusiak, M. Resonance-Assisted Hydrogen Bond—Revisiting the Original Concept in the Context of Its Criticism in the Literature. Int. J. Mol. Sci. 2022, 23, 233. [Google Scholar] [CrossRef] [PubMed]
- Mahmudov, K.T.; Pombeiro, A.J.L. Resonance-Assisted Hydrogen Bonding as a Driving Force in Synthesis and a Synthon in the Design of Materials. Chem. Eur. J. 2016, 22, 16356–16398. [Google Scholar] [CrossRef] [PubMed]
- Karas, L.J.; Wu, C.-H.; Das, R.; Wu, J. I-C. Hydrogen bond design principles. WIREs Comput. Mol. Sci. 2020, 10, 1477. [Google Scholar] [CrossRef]
- Hansen, P.E.; Spanget-Larsen, J. NMR and IR Investigations of Strong Intramolecular Hydrogen Bonds. Molecules 2017, 22, 552. [Google Scholar] [CrossRef]
- Wu, J.I.; Jackson, J.E.; Schleyer, P.R. Reciprocal hydrogen bonding–aromaticity relationships. J. Am. Chem. Soc. 2014, 136, 13526–13529. [Google Scholar] [CrossRef]
- Kakeshpour, T.; Wu, J.I.; Jackson, J.E. AMHB:(anti) aromaticity-modulated hydrogen bonding. J. Am. Chem. Soc. 2016, 138, 3427–3432. [Google Scholar] [CrossRef]
- Kakeshpour, T.; Bailey, J.P.; Jenner, M.R.; Howell, D.E.; Staples, R.J.; Holmes, D.; Wu, J.I.; Jackson, J.E. High-field NMR spectroscopy reveals aromaticity-modulated hydrogen bonding in heterocycles. Angew. Chem. Int. Ed. 2017, 56, 9842–9846. [Google Scholar] [CrossRef] [PubMed]
- Nekoei, A.-R.; Vatanparast, M. π-Hydrogen bonding and aromaticity: A systematic interplay study. Phys. Chem. Chem. Phys. 2019, 21, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Arunan, E.G.; Desiraju, R.; Klein, R.A.; Sadlej, J.; Scheiner, S.; Alkorta, I.; Clary, D.C.; Crabtree, R.H.; Dannenberg, J.J.; Hobza, P.; et al. Definition of the Hydrogen Bond. Pure Appl. Chem. 2011, 83, 1637–1641. [Google Scholar] [CrossRef]
- Arunan, E.G.; Desiraju, R.; Klein, R.A.; Sadlej, J.; Scheiner, S.; Alkorta, I.; Clary, D.C.; Crabtree, R.H.; Dannenberg, J.J.; Hobza, P.; et al. Definition of the Hydrogen Bond: An Account. Pure Appl. Chem. 2011, 83, 1619–1636. [Google Scholar] [CrossRef]
- Afonin, A.V.; Vashchenko, A.V. Benchmark calculations of intramolecular hydrogen bond energy based on molecular tailoring and function-based approaches: Developing hybrid approach. Int. J. Quantum Chem. 2019, 119, e26001. [Google Scholar] [CrossRef]
- Afonin, A.V.; Vashchenko, A.V. Quantitative decomposition of resonance-assisted hydrogen bond energy in β-diketones into resonance and hydrogen bonding (π- and σ-) components using molecular tailoring and function-based approaches. J. Comp. Chem. 2020, 41, 1285–1298. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, T. Relation between hydroxyl proton chemical shifts and torsional frequencies in some ortho-substituted phenol derivatives. J. Phys. Chem. 1975, 79, 1888–1890. [Google Scholar] [CrossRef]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Iogansen, A.V. Direct proportionality of the hydrogen bonding energy and the intensification of the stretching ν(XH) vibration in infrared spectra. Spectrochim. Acta Part A 1999, 55, 1585–1612. [Google Scholar] [CrossRef]
- Wendler, K.; Thar, J.; Zahn, S.; Kirchner, B. Estimating the Hydrogen Bond Energy. J. Phys. Chem. A 2010, 114, 9529–9536. [Google Scholar] [CrossRef] [PubMed]
- Nikolaienko, T.Y.; Bulavin, L.A.; Hovorun, D.M. Bridging QTAIM with vibrational spectroscopy: The energy of intramolecular hydrogen bonds in DNA-related biomolecules. Phys. Chem. Chem. Phys. 2012, 14, 7441–7447. [Google Scholar] [CrossRef] [PubMed]
- Brovarets’, O.O.; Yurenko, Y.P.; Hovorun, D.M. Intermolecular CH⋯O/N H-bonds in the biologically important pairs of natural nucleobases: A thorough quantum-chemical study. J. Biomol. Struct. Dyn. 2014, 32, 993–1022. [Google Scholar] [CrossRef] [PubMed]
- Afonin, A.V.; Vashchenko, A.V.; Sigalov, M.V. Estimating the energy of intramolecular hydrogen bonds from 1H NMR and QTAIM calculations. Org. Biomol. Chem. 2016, 14, 11199–11211. [Google Scholar] [CrossRef]
- Afonin, A.V.; Sterkhova, I.V.; Vashchenko, A.V.; Sigalov, M.V. Estimating the energy of intramolecular bifurcated (three-centered) hydrogen bond by X-ray, IR and 1H NMR spectroscopy, and QTAIM calculations. J. Mol. Struct. 2018, 1163, 185–196. [Google Scholar] [CrossRef]
- Jabłoński, M. A Critical Overview of Current Theoretical Methods of Estimating the Energy of Intramolecular Interactions. Molecules 2020, 25, 5512. [Google Scholar] [CrossRef]
- Deshmukh, M.M.; Gadre, S.R.; Bartolotti, L.J. Estimation of Intramolecular Hydrogen Bond Energy via Molecular Tailoring Approach. J. Phys. Chem. A 2006, 110, 12519–12523. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, M.M.; Suresh, C.H.; Gadre, S.R. Intramolecular Hydrogen Bond Energy in Polyhydroxy Systems: A Critical Comparison of Molecular Tailoring and Isodesmic Approaches. J. Phys. Chem. A 2007, 111, 6472–6480. [Google Scholar] [CrossRef]
- Deshmukh, M.M.; Bartolotti, L.J.; Gadre, S.R. Intramolecular Hydrogen Bonding and Cooperative Interactions in Carbohydrates via the Molecular Tailoring Approach. J. Phys. Chem. A 2008, 112, 312–321. [Google Scholar] [CrossRef]
- Deshmukh, M.M.; Gadre, S.R. Estimation of N−H···O=C Intramolecular Hydrogen Bond Energy in Polypeptides. J. Phys. Chem. A 2009, 113, 7927–7932. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, M.M.; Bartolotti, L.J.; Gadre, S.R. Intramolecular hydrogen bond energy and cooperative interactions in α-, β-, and γ-cyclodextrin conformers. J. Comput. Chem. 2011, 32, 2996–3004. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Ibnusaud, I.; Gadre, S.R.; Deshmukh, M.M. Fragmentation method reveals a wide spectrum of intramolecular hydrogen bond energies in antioxidant natural products. New J. Chem. 2020, 44, 5841–5849. [Google Scholar] [CrossRef]
- Ahluwalia, D.; Kumar, A.; Warkar, S.G.; Deshmukh, M.M. Effect of substitutions on the geometry and intramolecular hydrogen bond strength in meta-benziporphodimethenes: A new porphyrin analogue. J. Mol. Struct. 2020, 1220, 128773. [Google Scholar] [CrossRef]
- Deshmukh, M.M.; Gadre, S.R. Molecular Tailoring Approach for the Estimation of Intramolecular Hydrogen Bond Energy. Molecules 2021, 26, 2928. [Google Scholar] [CrossRef]
- Rusinska–Roszak, D.; Sowinski, G. Estimation of the intramolecular O-H···O=C hydrogen bond energy via the molecular tailoring approach. Part I: Aliphatic structures. J. Chem. Inf. Model. 2014, 54, 1963–1977. [Google Scholar] [CrossRef]
- Rusinska–Roszak, D. Intramolecular O–H···O=C Hydrogen Bond Energy via the Molecular Tailoring Approach to RAHB Structures. J. Phys. Chem. A 2015, 119, 3674–3687. [Google Scholar] [CrossRef]
- Rusinska–Roszak, D. Energy of Intramolecular Hydrogen Bonding in ortho-Hydroxybenzaldehydes, Phenones and Quinones. Transfer of Aromaticity from ipso-Benzene Ring to the Enol System(s). Molecules 2017, 22, 481. [Google Scholar] [CrossRef] [Green Version]
- Lozynski, M.; Rusinska–Roszak, D. Finding the direct energy-structure correlations in intramolecular aromaticity assisted hydrogen bonding (AAHB). J. Mol. Graph. Model. 2021, 105, 107884. [Google Scholar] [CrossRef]
- Afonin, A.V.; Rusinska-Roszak, D. Molecular tailoring approach as tool for revealing resonance-assisted hydrogen bond: Case study of Z-pyrrolylenones with the N–H∙∙∙O=C intramolecular hydrogen bond. J. Comput. Chem. 2022, 43, 1596–1607. [Google Scholar] [CrossRef]
- Martyniak, A.; Majerz, I.; Filarowski, A. Peculiarities of quasi-aromatic hydrogen bonding. RSC Adv. 2012, 2, 8135–8144. [Google Scholar] [CrossRef]
- Houjou, H.; Motoyama, T.; Banno, S.; Yoshikawa, I.; Araki, K. Experimental and Theoretical Studies on Constitutional Isomers of 2,6-Dihydroxynaphthalene Carbaldehydes. Effects of Resonance-Assisted Hydrogen Bonding on the Electronic Absorption Spectra. J. Org. Chem. 2009, 74, 520–529. [Google Scholar] [CrossRef]
- Houjou, H.; Shingai, H.; Yagi, K.; Yoshikawa, I.; Araki, K. Mutual Interference between Intramolecular Proton Transfer Sites through the Adjoining π-Conjugated System in Schiff Bases of Double-Headed, Fused Salicylaldehydes. J. Org. Chem. 2013, 78, 9021–9031. [Google Scholar] [CrossRef] [PubMed]
- Lenain, P.; Mandado, M.; Mosquera, R.A.; Bultinck, P. Interplay between Hydrogen-Bond Formation and Multicenter π-Electron Delocalization: Intramolecular Hydrogen Bonds. J. Phys. Chem. A 2008, 112, 10689–10696. [Google Scholar] [CrossRef] [PubMed]
- Palusiak, M.; Simon, S.; Solá, M. Interplay between Intramolecular Resonance-Assisted Hydrogen Bonding and Aromaticity in o-Hydroxyaryl Aldehydes. J. Org. Chem. 2006, 71, 5241–5248. [Google Scholar] [CrossRef] [PubMed]
- Palusiak, M.; Simon, S.; Solá, M. Interplay between Intramolecular Resonance-Assisted Hydrogen Bonding and Local Aromaticity. II. 1,3-Dihydroxyaryl-2-aldehydes J. Org. Chem. 2009, 74, 2059–2066. [Google Scholar]
- Krygowski, T.M. Crystallographic studies of inter- and intramolecular interactions reflected in aromatic character of π-electron systems. J. Chem. Inf. Comput. Sci. 1993, 33, 70–78. [Google Scholar] [CrossRef]
- Mayer, I. Bond order and valence indices: A personal account. J. Comput. Chem. 2007, 28, 204–221. [Google Scholar] [CrossRef]
- Lee, L.P.; Limas, N.G.; Cole, D.J.; Payne, M.C.; Skylaris, C.-K.; Manz, T.A. Expanding the Scope of Density Derived Electrostatic and Chemical Charge Partitioning to Thousands of Atoms. J. Chem. Theory Comput. 2014, 10, 5377–5390. [Google Scholar] [CrossRef] [Green Version]
- Afonin, A.V.; Rusinska-Roszak, D. A molecular tailoring approach—A new guide to quantify the energy of push–pull effects: A case study on (E)-3-(1H-pyrrol-2-yl)prop-2-enones. Phys. Chem. Chem. Phys. 2020, 22, 22190–22194. [Google Scholar] [CrossRef]
- Afonin, A.V.; Rusinska-Roszak, D. Guide to tuning the chalcone molecular properties based on the push-pull effect energy scale created via the molecular tailoring approach. J. Comput. Chem. 2022, 43, 631–643. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Vallejos, M.M.; Angelina, E.L.; Peruchena, N.M. Bifunctional Hydrogen Bonds in Monohydrated Cycloether Complexes. J. Phys. Chem. A 2010, 114, 2855–2863. [Google Scholar] [CrossRef] [PubMed]
- Afonin, A.V.; Ushakov, I.A.; Vashchenko, A.V.; Kondrashov, E.V.; Rulev, A.Y. GIAO, DFT, AIM and NBO analysis of the N–H· · ·O intramolecular hydrogen-bond influence on the 1J(N,H) coupling constant in push–pull diaminoenones. Magn. Reson. Chem. 2010, 48, 661–670. [Google Scholar] [CrossRef]
- Marder, S.R.; Cheng, L.T.; Tiemann, B.G.; Friedli, A.C.; Blanchard-Desce, M.; Perry, J.W.; Skindhoj, J. Large first hyperpolarizabilities in push-pull polyenes by tuning of the bond length alternation and aromaticity. Science 1994, 263, 511–514. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Xie, Q.; Zhao, L.; Zhu, J. Probing the Most Aromatic and Antiaromatic Pyrrolium Rings by Maximizing Hyperconjugation and Push-Pull Effect. Chem Asian J. 2018, 13, 1419–1423. [Google Scholar] [CrossRef] [PubMed]
- Shen, T.; Chen, D.; Lin, L.; Zhu, J. Dual Aromaticity in Both the T0 and S1 States: Osmapyridinium with Phosphonium Substituents. J. Am. Chem. Soc. 2019, 141, 5720–5727. [Google Scholar] [CrossRef]
- Hansch, C.; Leo, A.; Taft, R.W. A survey of Hammett substituent constants and resonance and field parameters. Chem. Rev. 1991, 91, 165–195. [Google Scholar] [CrossRef]
- Clayden, J.; Greeves, N.; Warren, S. Organic Chemistry, 2nd ed.; Oxford University Press Inc.: New York, NY, USA, 2012. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision B.01; Gaussian Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Oxford University Press: New York, NY, USA, 1990. [Google Scholar]
- Keith, T.A. AIMAll, version 13.01.27; TK Gristmill Software: Overland Park, KS, USA, 2013. [Google Scholar]




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 |  |  | ||||||
| subseries Ia,b | subseries IIa,b | subseries IIIa,b | ||||||
| compounds 1–18 | compounds 19–36 | compounds 37–54 | ||||||
| subseries Ia, R2 = NO2, R1 varies | subseries IIa, R2 = NO2, R1 varies | subseries IIIa, R2 = NO2, R1 varies | ||||||
| No | R1 | −Eπ(PPE) | No | R1 | −Eπ(PPE) | No | R1 | −Eπ(PPE) |
| 1 | N(CH3)2 | 12.06 | 19 | N(CH3)2 | 3.44 | 37 | N(CH3)2 | 2.12 |
| 2 | NH2 | 10.60 | 20 | NH2 | 2.71 | 38 | NH2 | 1.84 |
| 3 | OH | 8.79 | 21 | OH | 2.43 | 39 | OH | 1.39 |
| 4 | CH3 | 7.98 | 22 | CH3 | 2.22 | 40 | CH3 | 1.50 |
| 5 | H | 6.92 | 23 | H | 1.56 | 41 | H | 1.09 |
| 6 | Cl | 6.31 | 24 | Cl | 1.11 | 42 | Cl | 0.51 |
| 7 | CHO | 4.31 | 25 | CHO | –0.05a | 43 | CHO | –0.26 a |
| 8 | CN | 3.73 | 26 | CN | –0.30a | 44 | CN | –0.82 a |
| 9 | NO2 | 3.04 | 27 | NO2 | –0.51a | 45 | NO2 | –1.50 a |
| subseries Ib, R1 = N(CH3)2, R2 varies | subseries IIb, R1 = N(CH3)2, R2 varies | subseries IIIb, R1 = N(CH3)2, R2 varies | ||||||
| No | R2 | −Eπ(PPE) | No | R2 | −Eπ(PPE) | No | R2 | −Eπ(PPE) |
| 10 | N(CH3)2 | 3.59 | 28 | N(CH3)2 | 0.40 | 46 | N(CH3)2 | 0.37 |
| 11 | NH2 | 3.42 | 29 | NH2 | 0.59 | 47 | NH2 | 0.50 |
| 12 | OH | 5.17 | 30 | OH | 1.05 | 48 | OH | 1.02 |
| 13 | CH3 | 4.96 | 31 | CH3 | 1.01 | 49 | CH3 | 1.19 |
| 14 | H | 5.88 | 32 | H | 1.30 | 50 | H | 1.19 |
| 15 | Cl | 8.90 | 33 | Cl | 2.38 | 51 | Cl | 1.39 |
| 16 | CHO | 8.77 | 34 | CHO | 2.28 | 52 | CHO | 1.15 |
| 17 | CN | 10.22 | 35 | CN | 3.15 | 53 | CN | 1.56 |
| 18 | NO2 | 12.06 | 36 | NO2 | 3.45 | 54 | NO2 | 2.12 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Afonin, A.V.; Rusinska-Roszak, D. Revealing the Reasons for Degeneration of Resonance-Assisted Hydrogen Bond on the Aromatic Platform: Calculations of Ortho-, Meta-, Para-Disubstituted Benzenes, and (Z)-(E)-Olefins. Molecules 2023, 28, 536. https://doi.org/10.3390/molecules28020536
Afonin AV, Rusinska-Roszak D. Revealing the Reasons for Degeneration of Resonance-Assisted Hydrogen Bond on the Aromatic Platform: Calculations of Ortho-, Meta-, Para-Disubstituted Benzenes, and (Z)-(E)-Olefins. Molecules. 2023; 28(2):536. https://doi.org/10.3390/molecules28020536
Chicago/Turabian StyleAfonin, Andrei V., and Danuta Rusinska-Roszak. 2023. "Revealing the Reasons for Degeneration of Resonance-Assisted Hydrogen Bond on the Aromatic Platform: Calculations of Ortho-, Meta-, Para-Disubstituted Benzenes, and (Z)-(E)-Olefins" Molecules 28, no. 2: 536. https://doi.org/10.3390/molecules28020536