
A Non-Instrumental Green Analytical Method Based on Surfactant-Assisted Dispersive Liquid–Liquid Microextraction–Thin-Layer Chromatography–Smartphone-Based Digital Image Colorimetry(SA-DLLME-TLC-SDIC) for Determining Favipiravir in Biological Samples

 ,
,  , ,
, ,  , and
, and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Screening of TLC Parameters
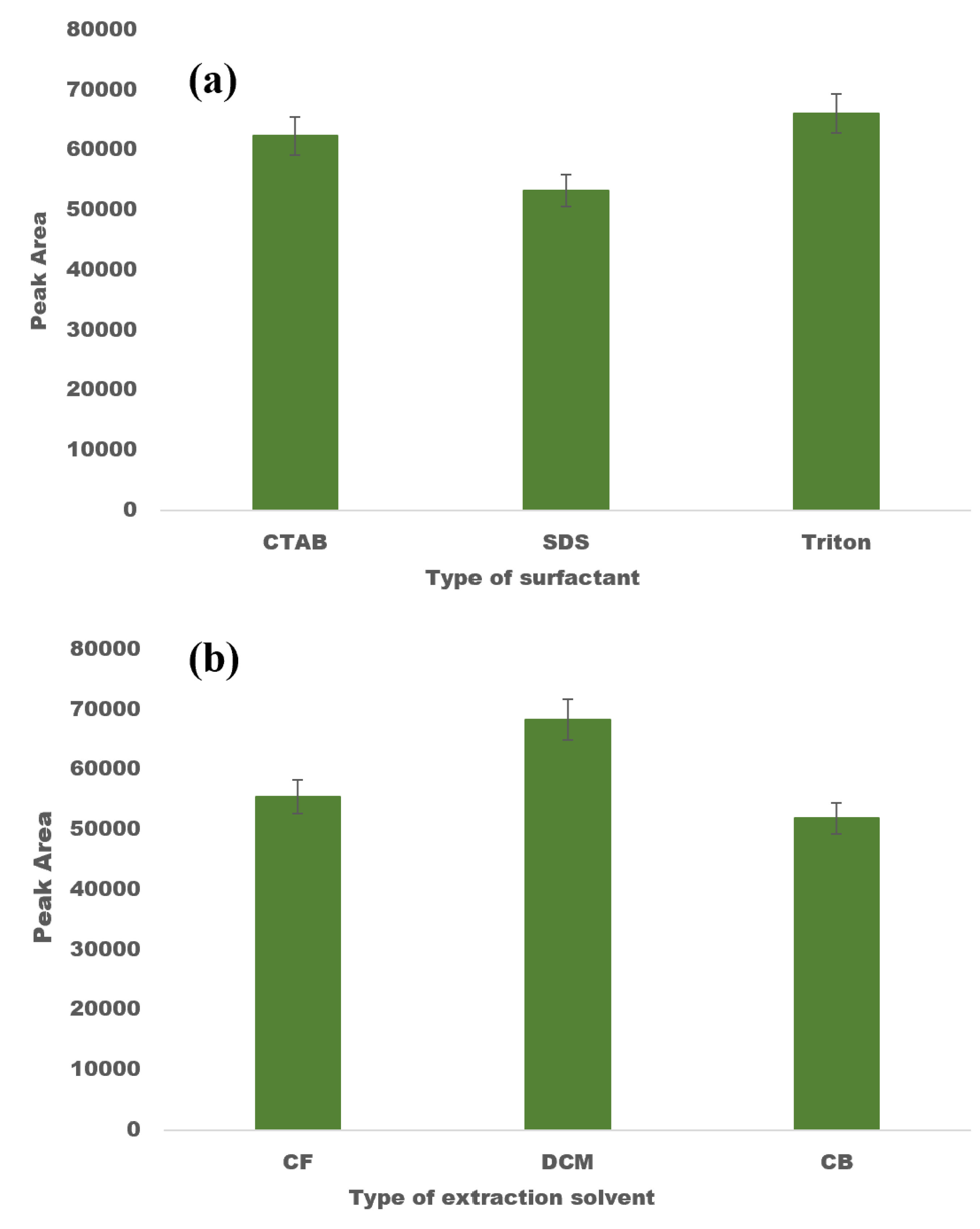
2.2. Screening of Surfactant and Extraction Solvent
2.3. Multivariate Optimisation
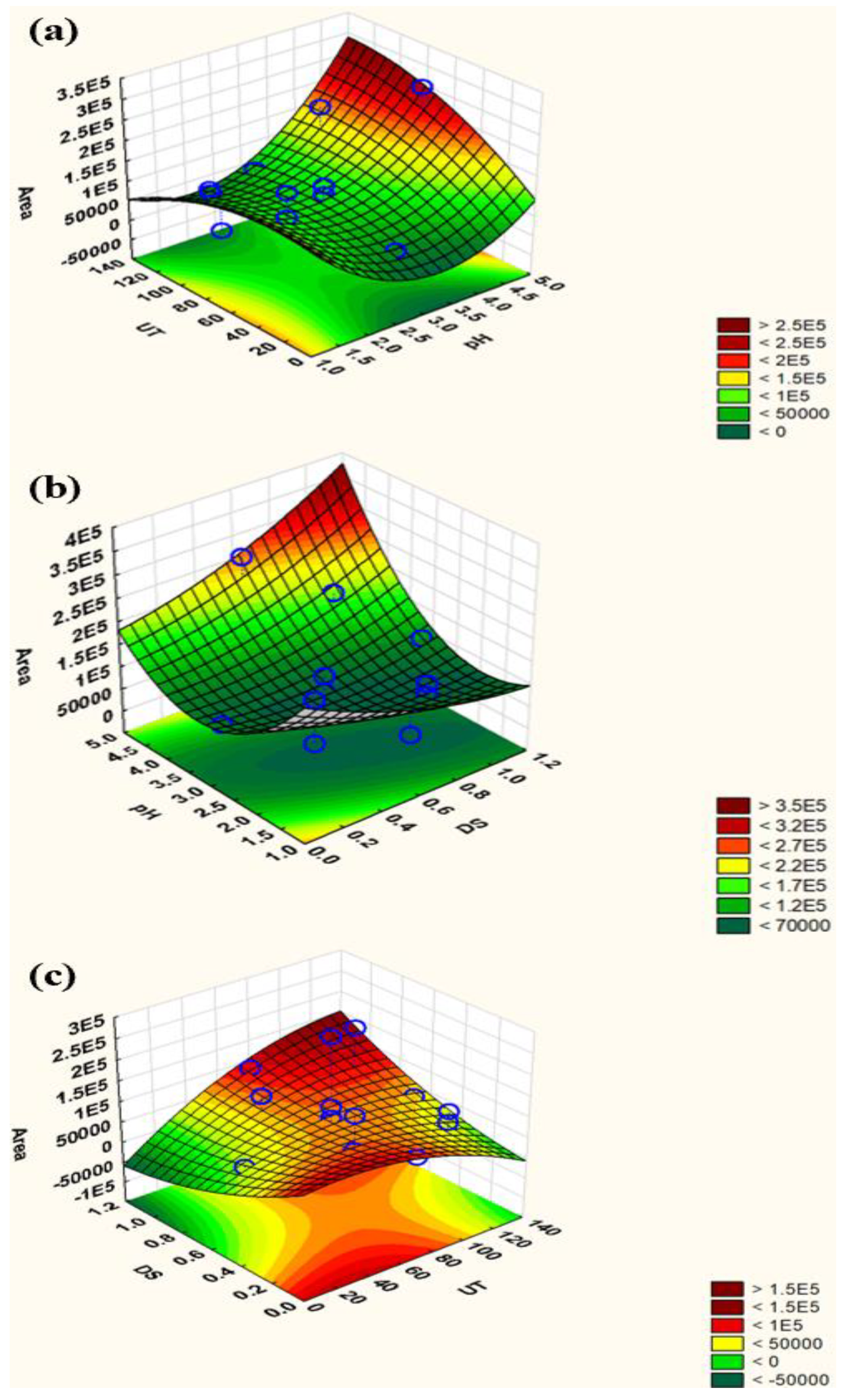
2.3.1. Plackett–Burman Design (PBD)
2.3.2. Central Composite Design (CCD)
2.4. Analytical Performance of the Method
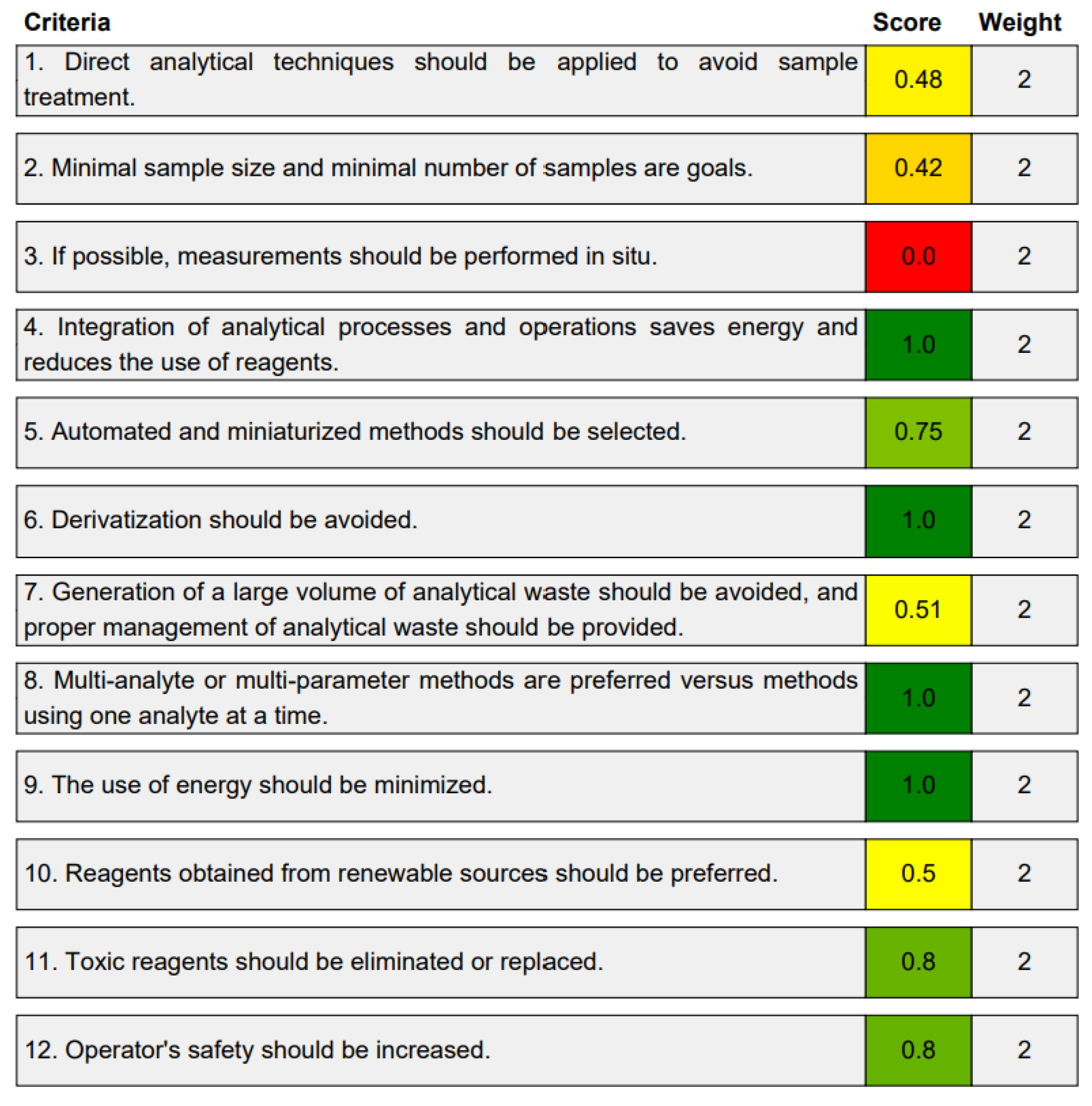
2.5. Assessment of the Green Character of the Developed Procedure
2.6. Application to Real Samples
3. Experimental Section
3.1. Reagents and Materials
3.2. Sample Collection
3.3. Preparation of Standards and Samples
3.4. Multivariate Analysis
3.5. SA-DLLME Procedure
3.5.1. Pharmaceutical Formulations
3.5.2. Biological Samples
3.6. Thin-Layer Chromatography–Smartphone-Based Digital Image Colorimetry (TLC-SDIC) Procedure
3.7. Method Validation
3.8. HPLC Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mikhail, I.E.; Elmansi, H.; Belal, F.; Ibrahim, A.E. Green micellar solvent-free HPLC and spectrofluorimetric determination of favipiravir as one of COVID-19 antiviral regimens. Microchem. J. 2021, 165, 106189. [Google Scholar] [CrossRef] [PubMed]
- Helmy, Y.A.; Fawzy, M.; Elaswad, A.; Sobieh, A.; Kenney, S.P.; Shehata, A.A. The COVID-19 pandemic: A comprehensive review of taxonomy, genetics, epidemiology, diagnosis, treatment, and control. J. Clin. Med. 2020, 9, 1225. [Google Scholar] [CrossRef] [PubMed]
- Noureldeen, D.A.M.; Boushra, J.M.; Lashien, A.S.; Hakiem AF, A.; Attia, T.Z. Novel environment friendly TLC-densitometric method for the determination of anti-coronavirus drugs “Remdesivir and Favipiravir”: Green assessment with application to pharmaceutical formulations and human plasma. Microchem. J. 2022, 174, 107101. [Google Scholar] [CrossRef] [PubMed]
- Morsy, M.I.; Nouman, E.G.; Abdallah, Y.M.; Zainelabdeen, M.A.; Darwish, M.M.; Hassan, A.Y.; Gouda, A.S.; Rezk, M.R.; Abdel-Megied, A.M.; Marzouk, H.M. A novel LC-MS/MS method for determination of the potential antiviral candidate favipiravir for the emergency treatment of SARS-CoV-2 virus in human plasma: Application to a bioequivalence study in Egyptian human volunteers. J. Pharm. Biomed. Anal. 2021, 199, 114057. [Google Scholar] [CrossRef]
- Marzouk, H.M.; Rezk, M.R.; Gouda, A.S.; Abdel-Megied, A.M. A novel stability-indicating HPLC-DAD method for determination of favipiravir, a potential antiviral drug for COVID-19 treatment; application to degradation kinetic studies and in-vitro dissolution profiling. Microchem. J. 2022, 172, 106917. [Google Scholar] [CrossRef]
- Abdallah, I.A.; Hammad, S.F.; Bedair, A.; Mansour, F.R. Menthol-assisted homogenous liquid-liquid microextraction for HPLC/UV determination of favipiravir as an antiviral for COVID-19 in human plasma. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2022, 1189, 123087. [Google Scholar] [CrossRef]
- Cai, Q.; Yang, M.; Liu, D.; Chen, J.; Shu, D.; Xia, J.; Liao, X.; Gu, Y.; Cai, Q.; Yang, Y.; et al. Experimental Treatment with Favipiravir for COVID-19: An Open-Label Control Study. Engineering 2020, 6, 1192–1198. [Google Scholar] [CrossRef]
- Doi, Y.; Hibino, M.; Hase, R.; Yamamoto, M.; Kasamatsu, Y.; Hirose, M.; Mutoh, Y. A Prospective, Randomized, Open-Label Trial of Early versus Late Favipiravir Therapy in Hospitalized Patients with COVID-19. Antimicrob. Agents Chemother. 2020, 64, 1–9. [Google Scholar] [CrossRef]
- Furuta, Y.; Takahashi, K.; Kuno-Maekawa, M.; Sangawa, H.; Uehara, S.; Kozaki, K.; Nomura, N.; Egawa, H.; Shiraki, K. Mechanism of action of T-705 against influenza virus. Antimicrob. Agents Chemother. 2005, 49, 981–986. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.H.T.; Guedj, J.; Anglaret, X.; Laouénan, C.; Madelain, V.; Taburet, A.M.; Baize, S.; Sissoko, D.; Pastorino, B.; Rodallec, A.; et al. Favipiravir pharmacokinetics in Ebola-Infected patients of the JIKI trial reveals concentrations lower than targeted. PLoS Negl. Trop. Dis. 2017, 11, 1–18. [Google Scholar] [CrossRef]
- Tkach, V.V.; Kushnir, M.V.; de Oliveira, S.C.; Ivanushko, J.G.; Velyka, A.V.; Molodianu, A.F.; Yagodynets, P.I.; Kormosh, Z.O.; dos Reis, L.V.; Luganska, O.V.; et al. Theoretical description for anti-COVID-19 drug Remdesivir electrochemical determination, assisted by squaraine Dye–Ag2O2 composite. Biointerface Res. Appl. Chem. 2021, 11, 9201–9208. [Google Scholar]
- Mohamed, M.A.; Eldin, G.M.G.; Ismail, S.M.; Zine, N.; Elaissari, A.; Jaffrezic Renault, N.; Errachid, A. Innovative electrochemical sensor for the precise determination of the new antiviral COVID-19 treatment Favipiravir in the presence of coadministered drugs. J. Electroanal. Chem. 2021, 895, 115422. [Google Scholar] [CrossRef] [PubMed]
- Allahverdiyeva, S.; Yunusoglu, O.; Yardım, Y.; Sentürk, Z. First electrochemical evaluation of favipiravir used as an antiviral option in the treatment of COVID-19: A study of its enhanced voltammetric determination in cationic surfactant media using a boron-doped diamond electrode. Anal. Chim. Acta 2021, 1159, 338418. [Google Scholar] [CrossRef] [PubMed]
- Megahed, S.M.; Habib, A.A.; Hammad, S.F.; Kamal, A.H. Experimental design approach for development of spectrofluorimetric method for determination of favipiravir; a potential therapeutic agent against COVID-19 virus: Application to spiked human plasma. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2021, 249, 119241. [Google Scholar] [CrossRef]
- Raasi, K.M. Analytical method development and validation of Remdesivir in bulk and pharmaceutical dosage forms using reverse-phase-high performance liquid chromatography. BR Nahata Smriti Sansthan Int. J. Phram. Sci. Clin. Res. 2021, 1, 282–329. [Google Scholar]
- Nguyen, R.; Goodell, J.C.; Shankarappa, P.S.; Zimmerman, S.; Yin, T.; Peer, C.J.; Figg, W.D. Development and validation of a simple, selective, and sensitive LC-MS/ MS assay for the quantification of remdesivir in human plasma. J. Chromatogr. B 2021, 1171, 122641. [Google Scholar] [CrossRef]
- Rezk, M.R.; Badr, K.A.; Abdel-Naby, N.S.; Ayyad, M.M. A novel, rapid and simple UPLC–MS/MS method for quantification of favipiravir in human plasma: Application to a bioequivalence study. Biomed. Chromatogr. 2021, 35, e5098. [Google Scholar] [CrossRef]
- Eryavuz Onmaz, D.; Abusoglu, S.; Onmaz, M.; Yerlikaya, F.H.; Unlu, A. Development and validation of a sensitive, fast and simple LC-MS/MS method for the quantitation of favipiravir in human serum. J. Chromatogr. B 2021, 1176, 122768. [Google Scholar] [CrossRef]
- Xiao, D.; John Ling, K.H.; Tarnowski, T.; Humeniuk, R.; German, P.; Mathias, A.; Chu, J.; Chen, Y.-S.; van Ingen, E. Validation of LC-MS/MS methods for determination of remdesivir and its metabolites GS-441524 and GS-704277 in acidified human plasma and their application in COVID-19 related clinical studies. Anal. Biochem. 2021, 617, 114118. [Google Scholar] [CrossRef]
- Avataneo, V.; de Nicolo, A.; Cusato, J.; Antonucci, M.; Manca, A.; Palermiti, A.; Waitt, C.; Walimbwa, S.; Lamorde, M.; di Perri, G.; et al. Development and validation of a UHPLC-MS/MS method for quantification of the prodrug remdesivir and its metabolite GS-441524: A tool for clinical pharmacokinetics of SARS-CoV-2/COVID-19 and Ebola virus disease. J. Antimicrob. Chemother. 2020, 75, 1772–1777. [Google Scholar] [CrossRef]
- Jain, B.; Jain, R.; Kabir, A.; Sharma, S. Rapid Determination of Non-Steroidal Anti-Inflammatory Drugs in Urine Samples after In-Matrix Derivatization and Fabric Phase Sorptive Extraction-Gas Chromatography-Mass Spectrometry Analysis. Molecules 2022, 27, 7188. [Google Scholar] [CrossRef] [PubMed]
- Sajid, M.; Płotka-Wasylka, J. Green analytical chemistry metrics: A review. Talanta 2022, 238, 123046. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; Singh, R. Applications of dispersive liquid-liquid micro-extraction in forensic toxicology. TrAC-Trends Anal. Chem. 2016, 75, 227–237. [Google Scholar] [CrossRef]
- Jain, R.; Singh, R. Microextraction Techniques in Analytical Toxicology; CRC Press: Boca Raton, FL, USA, 2021. [Google Scholar]
- Jain, R.; Singh, R. Microextraction techniques for analysis of cannabinoids. In TrAC-Trends in Analytical Chemistry; Elsevier: Amsterdam, The Netherlands, 2016; Volume 80, pp. 156–166. [Google Scholar] [CrossRef]
- Jha, R.R.; Thakur, R.S.; Jain, R. Dispersive Liquid-Liquid Microextraction and Its Variants. In Microextraction Techniques in Analytical Toxicology; CRC Press: Boca Raton, FL, USA, 2022; pp. 159–170. [Google Scholar]
- Mudiam MK, R.; Jain, R.; Singh, A.; Khan, H.A.; Parmar, D. Development of ultrasound-assisted dispersive liquid-liquid microextraction-large volume injection-gas chromatography-tandem mass spectrometry method for determination of pyrethroid metabolites in brain of cypermethrin-treated rats. Forensic Toxicol. 2014, 32, 19–29. [Google Scholar] [CrossRef]
- Jain, R.; Singh, M.; Kumari, A.; Tripathi, R.M. A rapid and cost- effective method based on dispersive liquid- liquid microextraction coupled to injection port silylation-gas chromatography- mass spectrometry for determination of morphine in illicit opium. Anal. Sci. Adv. 2021, 2, 387–396. [Google Scholar] [CrossRef]
- Olędzka, I.; Kowalski, P.; Plenis, A.; Bączek, T. Evaluation of various approaches to the isolation of steroid hormones from urine samples prior to FASS-MEKC analysis. Electrophoresis 2017, 38, 1632–1643. [Google Scholar] [CrossRef]
- Primel, E.G.; Caldas, S.S.; Marube, L.C.; Escarrone, A.L.V. An overview of advances in dispersive liquid–liquid microextraction for the extraction of pesticides and emerging contaminants from environmental samples. In Trends in Environmental Analytical Chemistry; Elsevier: Amsterdam, The Netherlands, 2017; Volume 14, pp. 1–18. [Google Scholar] [CrossRef]
- Jain, R.; Jha, R.R.; Kumari, A.; Khatri, I. Dispersive liquid liquid microextraction combined with digital image colorimetry for paracetamol analysis. Microchem. J. 2021, 162, 105870. [Google Scholar] [CrossRef]
- Jain, B.; Jain, R.; Jha, R.R.; Bajaj, A.; Sharma, S. A green analytical approach based on smartphone digital image colorimetry for aspirin and salicylic acid analysis. Green Anal. Chem. 2022, 3, 100033. [Google Scholar] [CrossRef]
- Jain, R.; Singh, R.; Sudhaker, S.; Barik, A.K. Coupling Microextraction with Thin Layer Chromatography-Image Processing Analysis: A New Analytical Platform for Drug Analysis. Toxicol. Forensic Med. 2017, 2, 17–25. [Google Scholar] [CrossRef]
- Jain, R.; Kumari, A.; Khatri, I. Simple and rapid analysis of acetaminophen in human autopsy samples by vortex-assisted dispersive liquid–liquid microextraction-thin layer chromatography-image analysis. Sep. Sci. Plus 2021, 4, 92–100. [Google Scholar] [CrossRef]
- Jain, R.; Tripathi, R.M.; Negi, A.; Singh, S.P. A simple, cost-effective and rapid method for simultaneous determination of Strychnosnux-vomica alkaloids in blood and Ayurvedic medicines based on ultrasound-assisted dispersive liquid-liquid microextraction-thin-layer chromatography-image analysis. J. Chromatogr. Sci. 2020, 58, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Meisen, I.; Wisholzer, S.; Soltwisch, J.; Dreisewerd, K.; Mormann, M.; Müthing, J.; Karch, H.; Friedrich, A.W. Normal silica gel and reversed phase thin-layer chromatography coupled with UV spectroscopy and IR-MALDI-o-TOF-MS for the detection of tetracycline antibiotics. Anal. Bioanal. Chem. 2010, 398, 2821–2831. [Google Scholar] [CrossRef]
- Parys, W.; Pyka-Pająk, A. TLC–Densitometry for Determination of Omeprazole in Simple and Combined Pharmaceutical Preparations. Pharmaceuticals 2022, 15, 1016. [Google Scholar] [CrossRef]
- Domínguez, C.; Jover, E.; Garde, F.; Bayona, J.M.; Erra, P. Characterization of Supercritical Fluid Extracts from Raw Wool by TLC-FID and GC-MS. J. Am. Oil Chem. Soc. 2003, 80, 717–724. [Google Scholar] [CrossRef]
- Nakamura, K.; Suzuki, Y.; Goto-Inoue, N.; Yoshida-Noro, C.; Suzuki, A. Structural characterization of neutral glycosphingolipids by thin-layer chromatography coupled to matrix-assisted laser desorption/ionization quadrupole ion trap time-of-flight MS/MS. Anal. Chem. 2006, 78, 5736–5743. [Google Scholar] [CrossRef] [PubMed]
- Olech, M.; Komsta, L.; Nowak, R.; Ciesla, L.; Hajnos, M.W. Investigation of antiradical activity of plant material by thin-layer chromatography with image processing. Food Chem. 2012, 132, 549–553. [Google Scholar] [CrossRef] [PubMed]
- Skowron, M.; Zakrzewski, R.; Ciesielski, W. Application of thin-layer chromatography image analysis technique in quantitative determination of Sphingomyelin. J. Anal. Chem. 2016, 71, 808–813. [Google Scholar] [CrossRef]
- Moffat, A.C.; Osselton, M.D.; Widdop, B.; Watts, J. Clarke’s Analysis of Drugs and Poisons; Pharmaceutical Press: London, UK, 2011; Volume 3. [Google Scholar]
- Fried, B. Thin-layer chromatography: Techniques and applications. Pascal Fr. Bibliogr. Databases 1982, 17, 313. [Google Scholar]
- Ghaedi, M.; Roosta, M.; Khodadoust, S.; Daneshfar, A. Application of optimized vortex-assisted surfactant-enhanced DLLME for preconcentration of thymol and carvacrol, and their determination by HPLC-UV: Response surface methodology. J. Chromatogr. Sci. 2015, 53, 1222–1231. [Google Scholar] [CrossRef] [Green Version]
- Moradi, M.; Yamini, Y.; Esrafili, A.; Seidi, S. Application of surfactant assisted dispersive liquid-liquid microextraction for sample preparation of chlorophenols in water samples. Talanta 2010, 82, 1864–1869. [Google Scholar] [CrossRef]
- Hemdan, A.; Magdy, R.; Farouk, M.; Fares, N.v. Central composite design as an analytical optimization tool for the development of eco-friendly HPLC-PDA methods for two antihypertensive mixtures containing the angiotensin receptor blocker Valsartan: Greenness assessment by four evaluation tools. Microchem. J. 2022, 183, 108105. [Google Scholar] [CrossRef]
- Jain, B.; Jain, R.; Jha, R.R.; Ghosh, A.; Basu, D.; Abourehab MA, S.; Bajaj, A.; Chauhan, V.; Kaur, S.; Sharma, S. Cellulose paper sorptive extraction (CPSE): A simple and affordable microextraction method for analysis of basic drugs in blood as a proof of concept. J. Chromatogr. B 2023, 1214, 123551. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; Jain, B.; Chauhan, V.; Deswal, B.; Kaur, S.; Sharma, S.; Abourehab, M.A.S. Simple determination of dichlorvos in cases of fatal intoxication by gas Chromatography-Mass spectrometry. J. Chromatogr. B 2023, 1215, 123582. [Google Scholar] [CrossRef] [PubMed]
- Feng, G.; Ding, W.; Deng, Y.; Zhao, R.; Gong, Y.; Duan, C.; Sun, J. A Kind of Favipiravir Has the HPLC Assay Method of Related Substance. CN104914185B, 10 June 2015. [Google Scholar]
- Bulduk, I. HPLC-UV method for quantification of favipiravir in pharmaceutical formulations. Acta Chromatogr. 2021, 33, 209–215. [Google Scholar]
- Habler, K.; Brügel, M.; Teupser, D.; Liebchen, U.; Scharf, C.; Schönermarck, U.; Vogeser, M.; Paal, M. Simultaneous quantification of seven repurposed COVID-19 drugs remdesivir (plus metabolite GS-441524), chloroquine, hydroxychloroquine, lopinavir, ritonavir, favipiravir and azithromycin by a two-dimensional isotope dilution LC–MS/MS method in human serum. J. Pharm. Biomed. Anal. 2021, 196, 113935. [Google Scholar] [CrossRef] [PubMed]
- Timofeeva, I.; Kanashina, D.; Kirsanov, D.; Bulatov, A. A heating-assisted liquid-liquid microextraction approach using menthol: Separation of benzoic acid in juice samples followed by HPLC-UV determination. J. Mol. Liq. 2018, 261, 265–270. [Google Scholar] [CrossRef]
- Akça, Z.; Özok H, İ.; Yardim, Y.; Şentürk, Z. Electroanalytical investigation and voltammetric quantification of antiviral drug favipiravir in the pharmaceutical formulation and urine sample using a glassy carbon electrode in anionic surfactant media. Turk. J. Chem. 2022, 46, 869–880. [Google Scholar] [CrossRef]
- Urso, A.D.; Rudge, J.; Patsalos, P.N.; De Grazia, U. Volumetric Absorptive Microsampling: A New Sampling Tool for Therapeutic Drug Monitoring of Antiepileptic Drugs. Ther. Drug Monit. 2019, 41, 681–692. [Google Scholar] [CrossRef]
- Teasdale, A.; Elder, D.; Nims, R.W. ICH Topic Q2(R), Validation of Analytical Procedure: Methodology. In ICH Harmonized Tripartite Guidelines; John Wiley & Sons: Hoboken, NJ, USA, 2005. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | LOD (µg/Spot) | LOQ (µg/Spot) | R2 | Linearity (µg/Spot) | Precision (%RSD) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Calibration Curve | Intraday (µg/Spot) | Interday (µg/Spot) | |||||||||
| 20 | 60 | 100 | 20 | 60 | 100 | ||||||
| Pharmaceutical formulation | 1.2 | 3.96 | 0.9914 | 5–100 | y = (471.92 ± 17.2) x + (32,100 ± 1022.70) | 0.37 | 0.28 | 0.12 | 5.5 | 8.78 | 8.9 |
| Urine | 1.3 | 4.29 | 0.991 | 5–100 | y = (610.88 ± 19.4) x + (40,665 ± 1151.152) | 1.3 | 0.59 | 0.44 | 6.3 | 8.1 | 6.5 |
| Plasma | 1.5 | 4.95 | 0.994 | 5–100 | y = (555.58 ± 12.00) x + (57,800 ± 710.39) | 1.5 | 0.73 | 0.51 | 5.4 | 6.0 | 5.5 |
| Drug | Accuracy% | RR% | ||||||
|---|---|---|---|---|---|---|---|---|
| 20 µg/Spot | 60 µg/Spot | 100 µg/Spot | EF | ER% | 20 µg/Spot | 60 µg/Spot | 100 µg/Spot | |
| Pharmaceutical formulation | 102 | 102.3 | 96.6 | 53.9 | 107.8 | 90.1 | 96.2 | 98.2 |
| Urine | 97.1 | 103 | 102.3 | 46.0 | 92.0 | 92.5 | 89.5 | 97.5 |
| Plasma | 91.6 | 98.4 | 100.8 | 35.1 | 70.2 | 87.6 | 94.4 | 96 |
| Reagents | Penalty Points |
|---|---|
| DCM | 1 × 4 = 4 |
| MeOH | 1 × 6 = 6 |
| CF | 1 × 6 = 6 |
| Instrument | |
| Energy used | 0 |
| Occupational hazard | 0 |
| Waste | 0 |
| Total | 16 |
| Score | 84 |
| Methods | Eco-Scale | GAPI | AGREE | Ref. | |
|---|---|---|---|---|---|
| HPLC-UV | Reagents | Penalty Points |  |  | [10] |
| MeOH | 6 | ||||
| Instrument | |||||
| Energy used | 2 | ||||
| Waste | 8 | ||||
| Total | 16 | ||||
| Score | 84 | ||||
| HPLC-DAD | Reagents | Penalty Points |  |  | [49] |
| ACN | 8 | ||||
| Instrument | |||||
| Energy used | 2 | ||||
| Waste | 8 | ||||
| Total | 18 | ||||
| Score | 82 | ||||
| HPLC-UV | Reagents | Penalty Points |  |  | [50] |
| ACN | 8 | ||||
| Instrument | |||||
| Energy used | 2 | ||||
| Waste | 8 | ||||
| Total | 18 | ||||
| Score | 82 | ||||
| LC-MS/MS | Reagents | Penalty Points |  |  | [4] |
| MeOH Acetic acid | 6 4 | ||||
| Instrument | |||||
| Energy used | 2 | ||||
| Waste | 3 | ||||
| Total | 15 | ||||
| Score | 85 | ||||
| LC-MS/MS | Reagents | Penalty Points |  |  | [20] |
| ACN MeOH | 8 6 | ||||
| Instrument | |||||
| Energy used | 2 | ||||
| Waste | 3 | ||||
| Total | 19 | ||||
| Score | 81 | ||||
| SA-DLLME-TLC-DIC | Reagents | Penalty Points |  |  | Present study |
| MeOH DCM CF | 6 4 6 | ||||
| Instrument | |||||
| Energy used | 0 | ||||
| Waste | 0 | ||||
| Total | 16 | ||||
| Score | 84 | ||||
| Sample Matrix | Sample Pre-Treatment and Extraction Method | Technique | Linearity Range | LOD | LOQ | Ref. |
|---|---|---|---|---|---|---|
| Spiked human plasma | Protein precipitation | Spectrofluorimetric method | 40–280 ng mL−1 | 9.44 ng mL−1 | 28.60 ng mL−1 | [14] |
| Human serum | SPE | LC-MS/MS | 3291–20,790 μg L−1 | - | 3291 μg L−1 | [51] |
| Human plasma | LLE | HPLC/UV | 0.5–50 mg L−1 | 0.15 mg L−1 | 0.45 mg L−1 | [52] |
| Human plasma | Protein precipitation | UPLC–MS/MS | 0.25–16 μg mL−1 | - | 0.25 μg mL−1 | [17] |
| Human urine | - | SW-AdSV | 1.0–100.0 μg mL–1 | 0.26 μg mL–1 | 0.87 μg mL–1 | [53] |
| Human plasma and urine | SA-DLLME | TLC-DIC | 5–100 µg/spot | 1.2–1.5 µg/spot | 3.96–4.29 µg/spot | Present work |
| Samples | Concentration Prepared (µg/mL) | Concentration Found by HPLC Method (µg/mL) | Accuracy (%) by HPLC Method | Concentration Found by SA-DLLME TLC-DIC Method (µg/mL) | Accuracy (%) by SA-DLLME TLC-DIC Method |
|---|---|---|---|---|---|
| Urine | 10 | 9.1 | 91 | 9.4 | 94 |
| Plasma | 10 | 9.6 | 96 | 9.8 | 98 |
| Samples (Claimed FAV) | Concentration Prepared (µg/mL) | Concentration Found (µg/mL) | Accuracy with Respect to Claimed FAV (%) | Amount of FAV Found b (SA-DLLME TLC-DIC Method) b | Amount of ASA/SA Found b (HPLC Method) |
|---|---|---|---|---|---|
| 800 mg | 10 | 9.3 | 99.4 | 795.2 ± 1.0 | 798.9 ± 2.4 |
| 400 mg | 10 | 9.1 | 99.2 | 396.8 ± 1.3 | 398.9 ± 1.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jain, B.; Jain, R.; Jaiswal, P.K.; Zughaibi, T.; Sharma, T.; Kabir, A.; Singh, R.; Sharma, S. A Non-Instrumental Green Analytical Method Based on Surfactant-Assisted Dispersive Liquid–Liquid Microextraction–Thin-Layer Chromatography–Smartphone-Based Digital Image Colorimetry(SA-DLLME-TLC-SDIC) for Determining Favipiravir in Biological Samples. Molecules 2023, 28, 529. https://doi.org/10.3390/molecules28020529
Jain B, Jain R, Jaiswal PK, Zughaibi T, Sharma T, Kabir A, Singh R, Sharma S. A Non-Instrumental Green Analytical Method Based on Surfactant-Assisted Dispersive Liquid–Liquid Microextraction–Thin-Layer Chromatography–Smartphone-Based Digital Image Colorimetry(SA-DLLME-TLC-SDIC) for Determining Favipiravir in Biological Samples. Molecules. 2023; 28(2):529. https://doi.org/10.3390/molecules28020529
Chicago/Turabian StyleJain, Bharti, Rajeev Jain, Prashant Kumar Jaiswal, Torki Zughaibi, Tanvi Sharma, Abuzar Kabir, Ritu Singh, and Shweta Sharma. 2023. "A Non-Instrumental Green Analytical Method Based on Surfactant-Assisted Dispersive Liquid–Liquid Microextraction–Thin-Layer Chromatography–Smartphone-Based Digital Image Colorimetry(SA-DLLME-TLC-SDIC) for Determining Favipiravir in Biological Samples" Molecules 28, no. 2: 529. https://doi.org/10.3390/molecules28020529