
Modeling Light-Induced Chromophore Hydration in the Reversibly Photoswitchable Fluorescent Protein Dreiklang


Abstract
:1. Introduction
2. Results
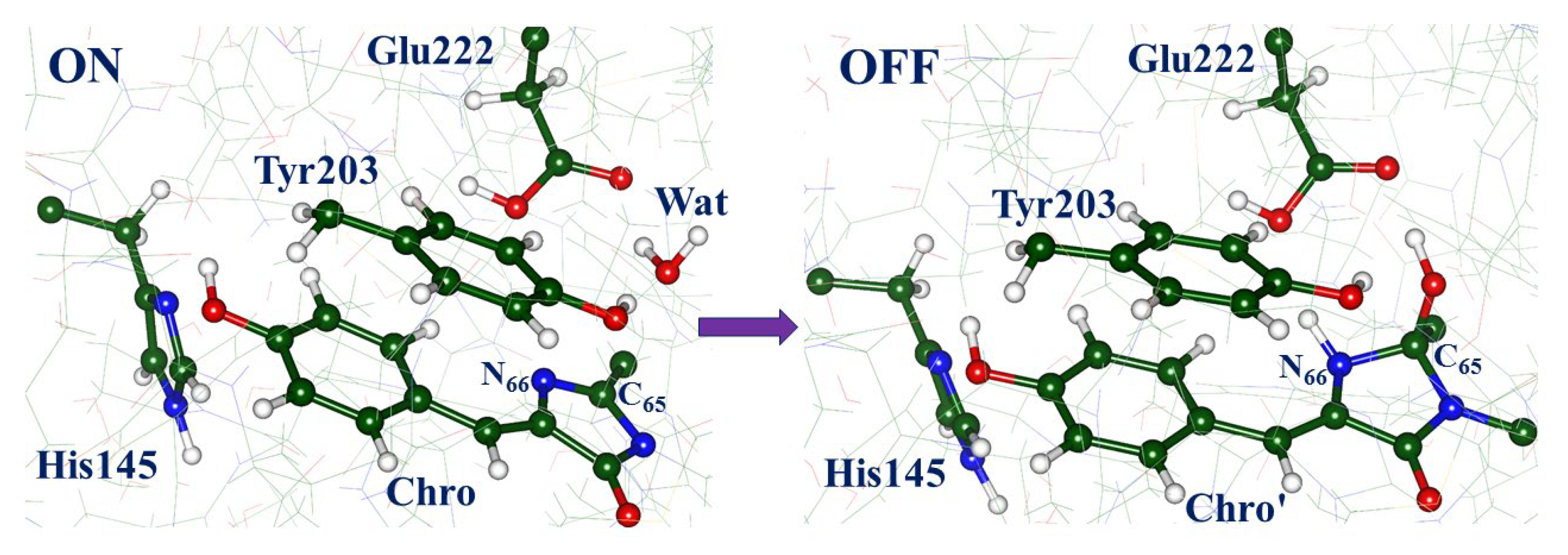
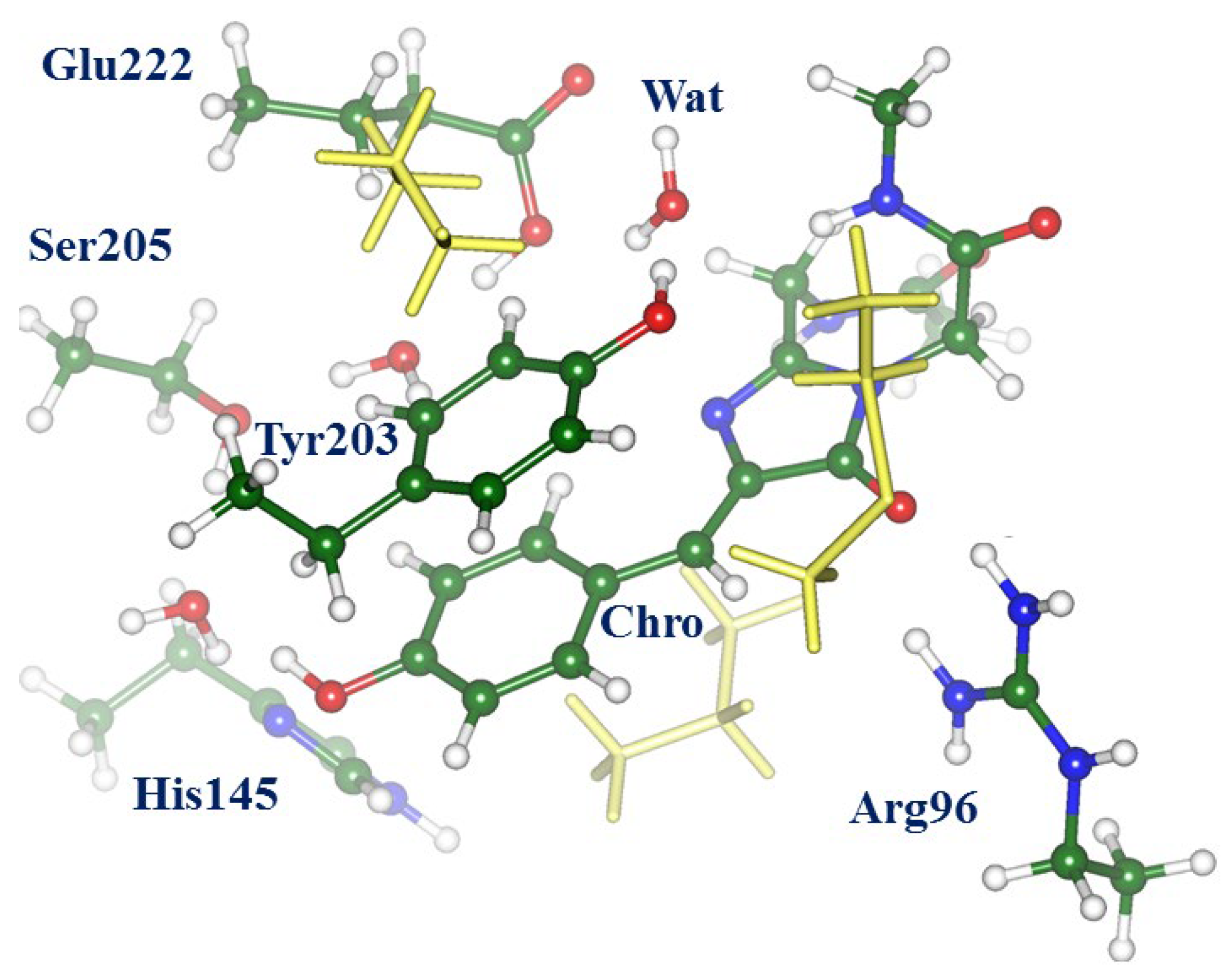
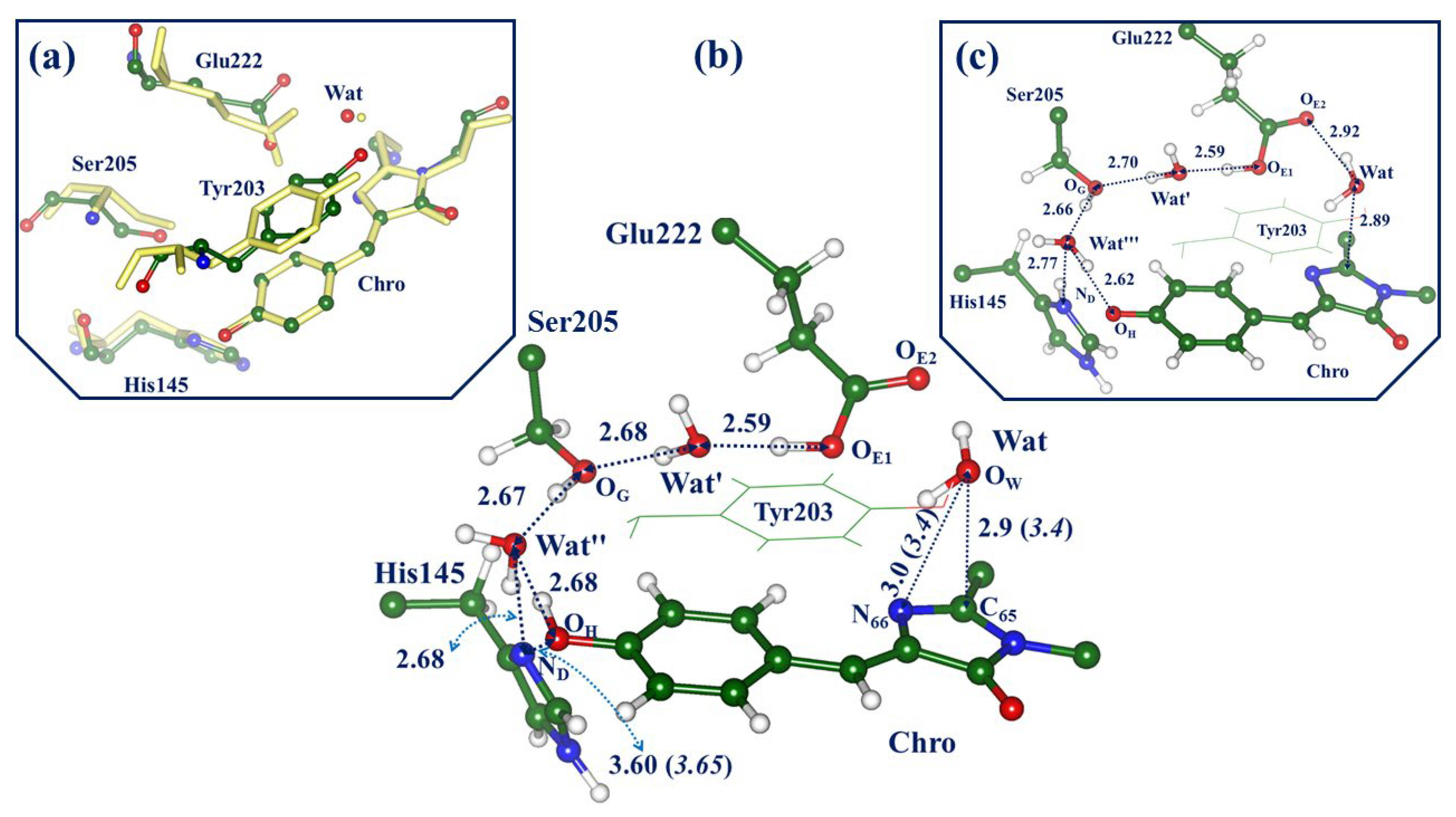
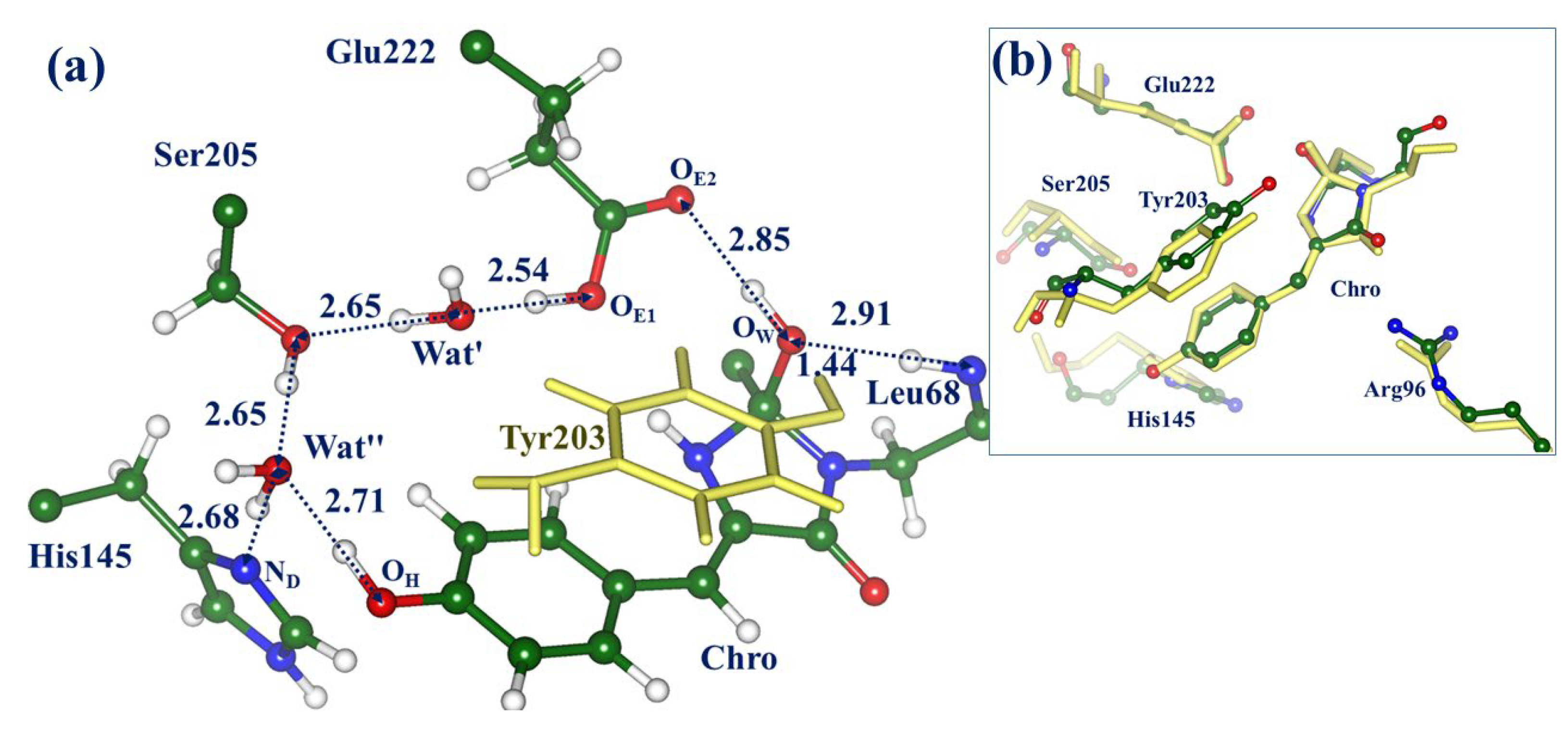
2.1. Structures and Spectra in the ON-State
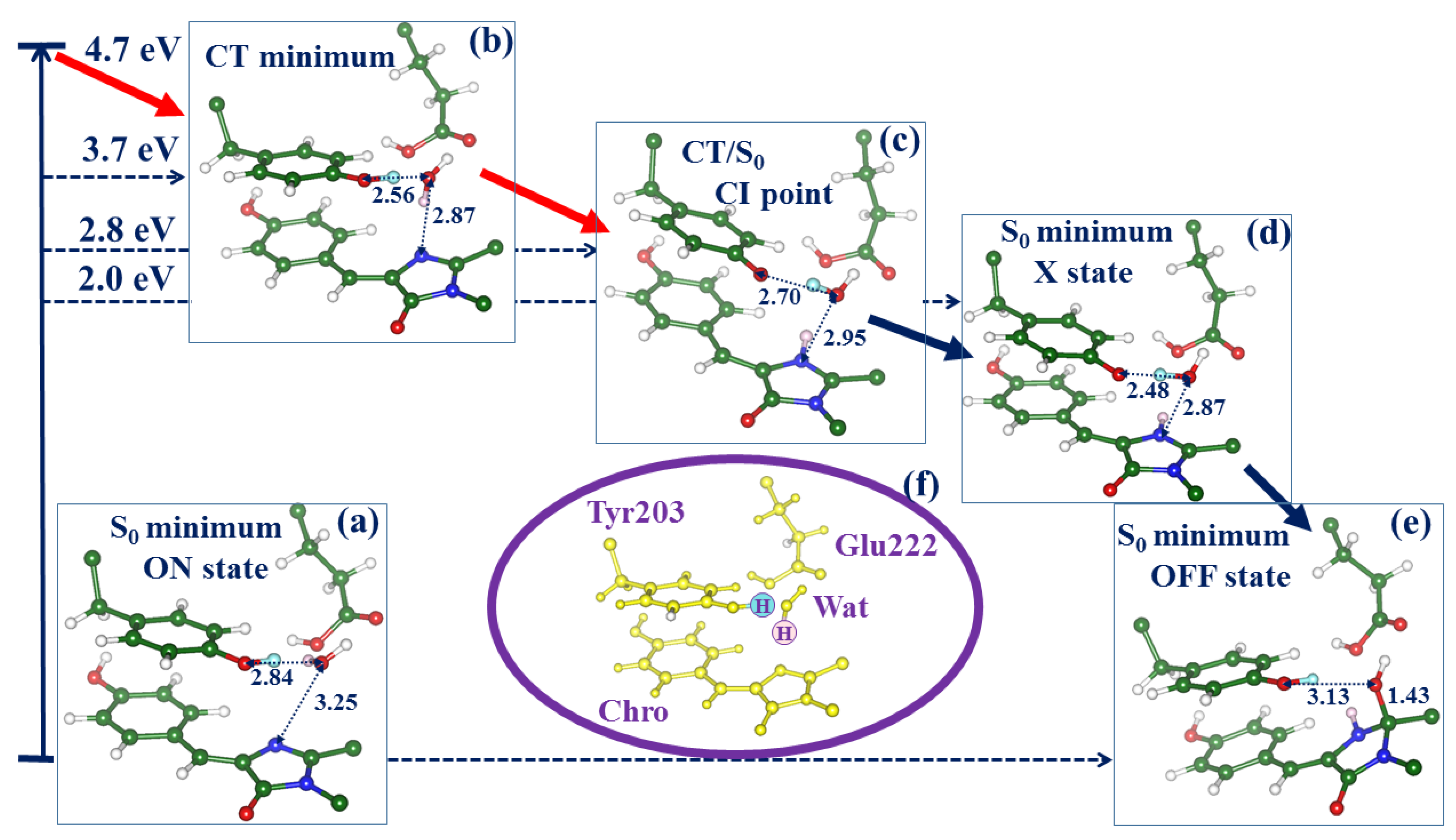
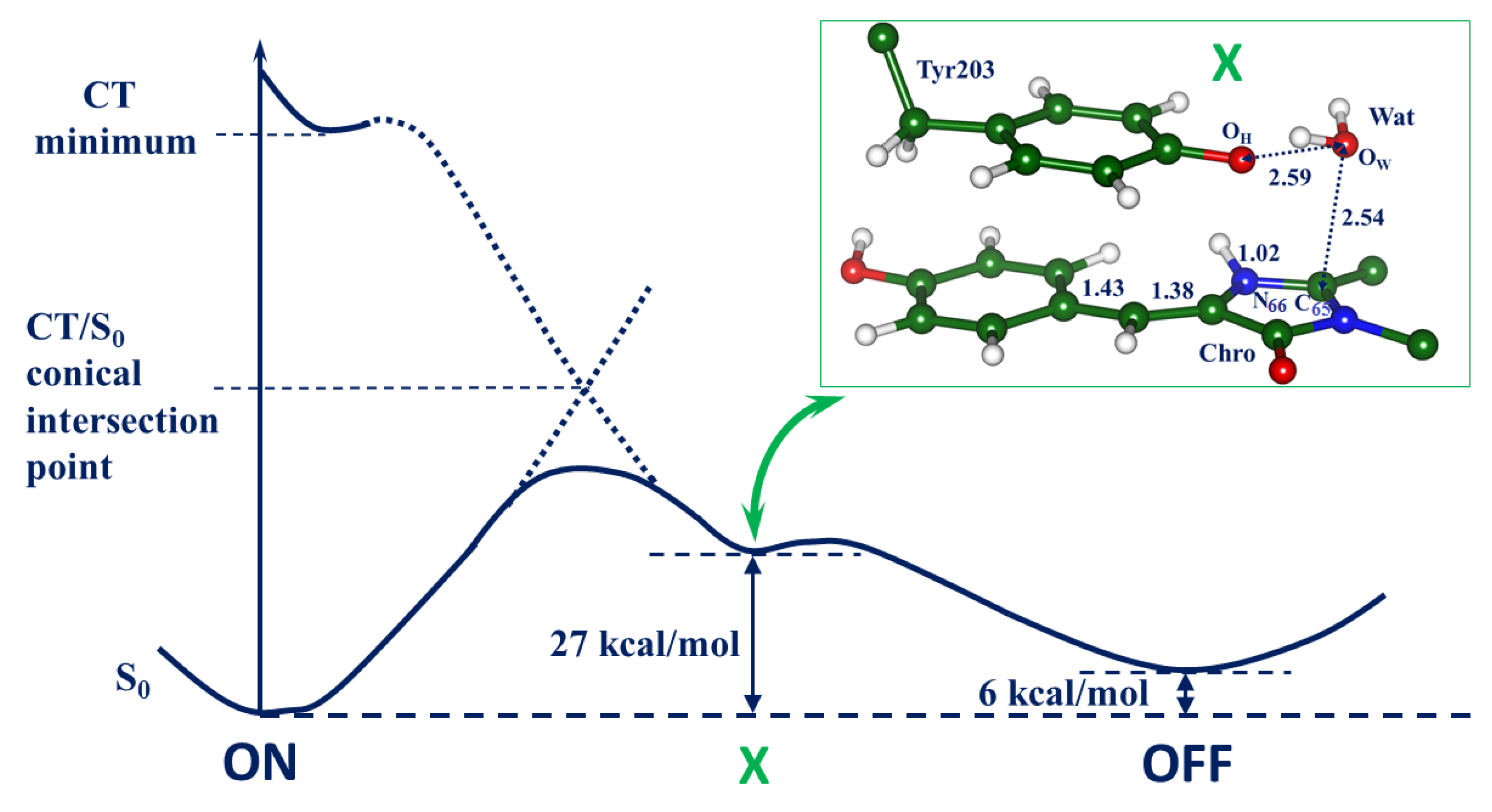
2.2. Evolution of the System in the Charge Transfer Excited State
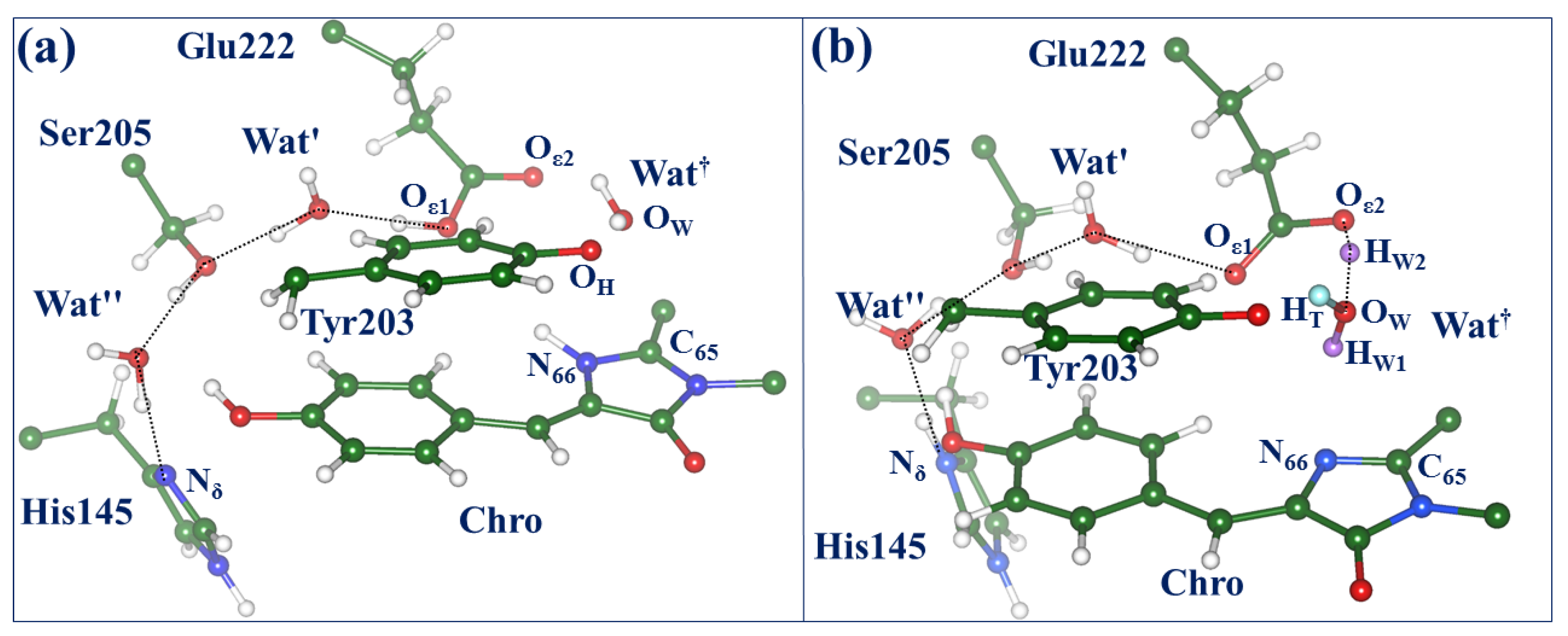
2.3. Back to the Ground State: The X and OFF Structures
2.4. Other Protonation States of Amino Acid Residues in the Active Site
3. Models and Methods
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brakemann, T.; Stiel, A.C.; Weber, G.; Andresen, M.; Testa, I.; Grotjohann, T.; Leutenegger, M.; Plessmann, U.; Urlaub, H.; Eggeling, C.; et al. A Reversibly Photoswitchable GFP-Like Protein with Fluorescence Excitation Decoupled from Switching. Nat. Biotechnol. 2011, 29, 942–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, L.; Fang, C. Photoswitchable Fluorescent Proteins: Mechanisms on Ultrafast Timescales. Int. J. Mol. Sci. 2022, 23, 6459. [Google Scholar] [CrossRef] [PubMed]
- Uriarte, L.M.; Vitale, R.; Nizi’nski, S.; Hadjidemetriou, K.; Zala, N.; Lukacs, A.; Greetham, G.M.; Sazanovich, I.V.; Weik, M.; Ruckebusch, C.; et al. Structural information about the trans-to-cis isomerization mechanism of the photoswitchable fluorescent protein rsEGFP2 revealed by multiscale infrared transient absorption. J. Phys. Chem. Lett. 2022, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Grigorenko, B.L.; Domratcheva, T.; Polyakov, I.V.; Nemukhin, A.V. Protonation States of Molecular Groups in the Chromophore Binding Site Modulate Properties of the Reversibly Switchable Fluorescent Protein rsEGFP2. J. Phys. Chem. Lett. 2021, 12, 8263–8271. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Romei, M.G.; Boxer, S.G. Structural evidence of photoisomerization pathways in fluorescent proteins. J. Am. Chem. Soc. 2019, 141, 15504–15508. [Google Scholar] [CrossRef]
- Nemukhin, A.V.; Grigorenko, B.L.; Khrenova, M.G.; Krylov, A.I. Computational Challenges in Modeling of Representative Bioimaging Proteins: GFP-Like Proteins, Flavoproteins, and Phytochromes. J. Phys. Chem. B 2019, 29, 6133–6149. [Google Scholar] [CrossRef]
- Smyrnova, D.; del Carmen Marín, M.; Olivucci, M.; Ceulemans, A. Systematic Excited State Studies of Reversibly Switchable Fluorescent Proteins. J. Chem. Theory Comput. 2018, 14, 3163–3172. [Google Scholar] [CrossRef]
- Acharya, A.; Bogdanov, A.M.; Grigorenko, B.L.; Bravaya, K.B.; Nemukhin, A.V.; Lukyanov, K.A.; Krylov, A.I. Photoinduced Chemistry in Fluorescent Proteins: Curse or Blessing? Chem. Rev. 2017, 117, 758–795. [Google Scholar] [CrossRef]
- Gozem, S.; Luk, H.L.; Schapiro, I.; Olivucci, M. Theory and Simulation of the Ultrafast Double-Bond Isomerization of Biological Chromophores. Chem. Rev. 2017, 117, 13502–13565. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.X.; Lin, M.Z. Photoswitchable fluorescent proteins: Ten years of colorful chemistry and exciting applications. Curr. Opin. Chem. Biol. 2013, 17, 682–690. [Google Scholar] [CrossRef]
- Bourgeois, D.; Adam, V. Reversible photoswitching in fluorescent proteins: A mechanistic view. IUBMB Life 2012, 64, 482–491. [Google Scholar] [CrossRef]
- Lacombat, F.; Plaza, P.; Plamont, M.-A.; Espagne, A. Photoinduced Chromophore Hydration in the Fluorescent Protein Dreiklang Is Triggered by Ultrafast Excited-State Proton Transfer Coupled to a Low-Frequency Vibration. J. Phys. Chem. Lett. 2017, 8, 1489–1495. [Google Scholar] [CrossRef]
- Grigorenko, B.L.; Polyakov, I.; Krylov, A.I.; Nemukhin, A.V. Computational modeling reveals the mechanism of fluorescent state recovery in the reversibly photoswitchable protein Dreiklang. J. Phys. Chem. B 2019, 123, 8901–8909. [Google Scholar] [CrossRef]
- Sen, T.; Ma, Y.; Polyakov, I.V.; Grigorenko, B.L.; Nemukhin, A.V.; Krylov, A.I. Interplay between Locally Excited and Charge Transfer States Governs the Photoswitching Mechanism in the Fluorescent Protein Dreiklang. J. Phys. Chem. B 2021, 125, 757–770. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Lambert, G.G.; Depernet, H.; Gotthard, G.; Schultz, D.T.; Navizet, I.; Lambert, T.; Adams, S.R.; Torreblanca-Zanca, A.; Chu, M.; Daphne, S.; et al. Aequorea’s Secrets Revealed: New Fluorescent Proteins with Unique Properties for Bioimaging and Biosensing. PLoS Biol. 2020, 18, e3000936. [Google Scholar] [CrossRef]
- Sugiura, K.; Nagai, T. Extension of the short wavelength side of fluorescent proteins using hydrated chromophores, and its application. Commun. Biol. 2022, 5, 1172. [Google Scholar] [CrossRef]
- Valiev, M.; Bylaska, E.J.; Govind, N.; Kowalski, K.; Straatsma, T.P.; Van Dam, H.J.J.; Wang, D.; Nieplocha, J.; Apra, E.; Windus, T.L.; et al. NWChem: A Comprehensive and Scalable Open-Source Solution for Large Scale Molecular Simulations. Comput. Phys. Commun. 2010, 181, 1477–1489. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Truhlar, D.G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 other Functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef] [Green Version]
- Ponder, J.W.; Case, D.A. Force fields for protein simulations. Adv. Prot. Chem. 2003, 66, 27–85. [Google Scholar] [CrossRef]
- Granovsky, A.A. Firefly, Version 8. Available online: http://classic.chem.msu.su/gran/gamess (accessed on 21 November 2022).
- Granovsky, A.A. Extended Multi-Configuration Quasi-Degenerate Perturbation Theory: The New Approach to Multi-State Multi-Reference Perturbation Theory. J. Chem. Phys. 2011, 134, 214113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmer, M. Green Fluorescent Protein (GFP): Applications, Structure, and Related Photophysical Behavior. Chem. Rev. 2002, 102, 759–782. [Google Scholar] [CrossRef] [PubMed]
- Nadal-Ferret, M.; Gelabert, R.; Moreno, M.; Lluch, J.M. Transient Low-Barrier Hydrogen Bond in the Photoactive State of Green Fluorescent Protein. Phys. Chem. Chem. Phys. 2015, 17, 30876–30888. [Google Scholar] [CrossRef] [PubMed]
- Shinobu, A.; Palm, G.J.; Schierbeek, A.J.; Agmon, N. Visualizing Proton Antenna in a High-Resolution Green Fluorescent Protein Structure. J. Am. Chem. Soc. 2010, 132, 11093–11102. [Google Scholar] [CrossRef]
- Shinobu, A.; Agmon, N. Proton Wire Dynamics in the Green Fluorescent Protein. J. Chem. Theory Comput. 2017, 13, 353–369. [Google Scholar] [CrossRef]
- Shao, Y.; Gan, Z.; Epifanovsky, E.; Gilbert, A.T.B.; Wormit, M.; Kussmann, J.; Lange, A.W.; Behn, A.; Deng, J.; Feng, X.; et al. Advances in molecular quantum chemistry contained in the Q-Chem 4 program package. Mol. Phys. 2015, 113, 184–215. [Google Scholar] [CrossRef] [Green Version]
- Krylov, A.I.; Gill, P.M.W. Q-Chem: An engine for innovation. WIREs Comput. Mol. Sci. 2013, 3, 317–326. [Google Scholar] [CrossRef]
- Chung, L.W.; Sameera, W.M.C.; Ramozzi, R.; Page, A.J.; Hatanaka, M.; Petrova, G.P.; Harris, T.V.; Li, X.; Ke, Z.; Liu, F.; et al. The ONIOM Method and Its Applications. Chem. Rev. 2015, 115, 5678–5796. [Google Scholar] [CrossRef]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.M.; Mittal, J.; Feig, M.; MacKerell, A.D.J., Jr. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone ϕ, ψ and side-chain χ1 and χ2 dihedral angles. Chem. Theor. Comput. 2012, 8, 3257–3273. [Google Scholar]
- Reuter, N.; Lin, H.; Thiel, W. Green Fluorescent Proteins: Empirical Force Field for the Neutral and Deprotonated Forms of the Chromophore. Molecular Dynamics Simulations of the Wild Type and S65T Mutant. J. Phys. Chem. B 2002, 106, 6310–6321. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | Excitation Energy, eV | Wavelength, nm | Oscillator Strength | Experimental Data [1,12], eV/nm |
|---|---|---|---|---|
| ON-A | 2.80 | 442 | 0.30 | 3.01/411–413 |
| ON-B | 2.46 | 503 | 0.78 | 2.43/511 |
| X | 2.79 | 444 | 0.20 | 2.76/450 |
| OFF | 3.57 | 347 | 0.46 | 3.65/340 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grigorenko, B.L.; Polyakov, I.V.; Nemukhin, A.V. Modeling Light-Induced Chromophore Hydration in the Reversibly Photoswitchable Fluorescent Protein Dreiklang. Molecules 2023, 28, 505. https://doi.org/10.3390/molecules28020505
Grigorenko BL, Polyakov IV, Nemukhin AV. Modeling Light-Induced Chromophore Hydration in the Reversibly Photoswitchable Fluorescent Protein Dreiklang. Molecules. 2023; 28(2):505. https://doi.org/10.3390/molecules28020505
Chicago/Turabian StyleGrigorenko, Bella L., Igor V. Polyakov, and Alexander V. Nemukhin. 2023. "Modeling Light-Induced Chromophore Hydration in the Reversibly Photoswitchable Fluorescent Protein Dreiklang" Molecules 28, no. 2: 505. https://doi.org/10.3390/molecules28020505