
Organoboron Complexes as Thermally Activated Delayed Fluorescence (TADF) Materials for Organic Light-Emitting Diodes (OLEDs): A Computational Study
 , , , and
, , , and
Abstract
:
1. Introduction
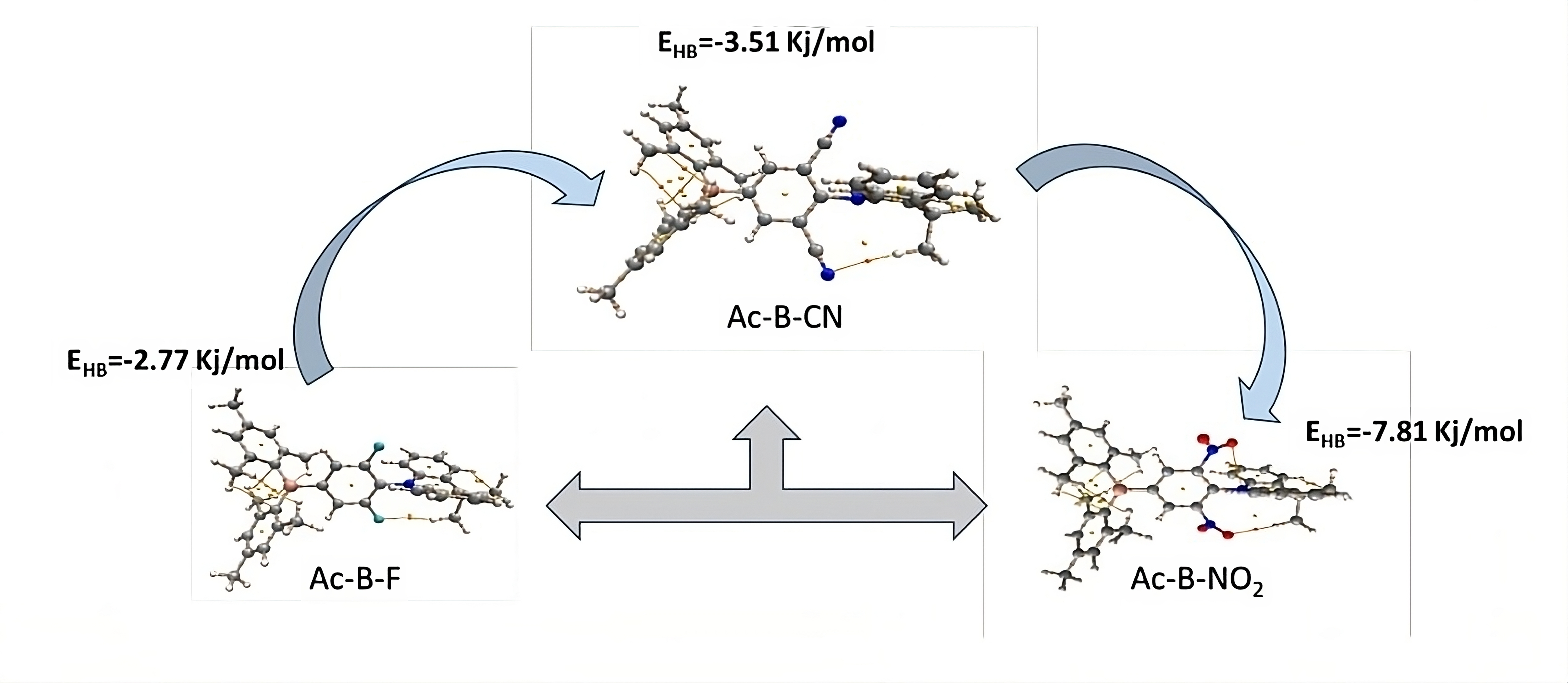
2. Results and Discussion
3. Theoretical Method
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Helfrich, W.; Schneider, W. Recombination Radiation in Anthracene Crystals. Phys. Rev. Lett. 1956, 14, 229. [Google Scholar] [CrossRef]
- Murawski, C.; Gather, M.C. Emerging Biomedical Applications of Organic Light-Emitting Diodes. Adv. Opt. Mater. 2021, 9, 2100269. [Google Scholar] [CrossRef]
- Tang, C.W.; VanSlyke, S.A. Organic electroluminescent diodes. Appl. Phys. Lett. 1987, 51, 913–915. [Google Scholar] [CrossRef]
- Zhao, D.; Qin, Z.; Huang, J.; Yu, J. Progress on material, structure and function for tandem organic light-emitting diodes. Org. Electron. 2017, 51, 220–242. [Google Scholar] [CrossRef]
- Lichtman, J.W.; Conchello, J.-A. Fluorescence microscopy. Nat. Methods 2005, 2, 910–919. [Google Scholar] [CrossRef] [PubMed]
- Baldo, M.A.; O’Brien, D.F.; You, Y.; Shoustikov, A.; Sibley, S.; Thompson, M.E.; Forrest, S.R. Highly efficient phosphorescent emission from organic electroluminescent devices. Nature 1998, 395, 151–154. [Google Scholar] [CrossRef]
- Yersin, H. Triplet Emitters for OLED Applications. Mechanisms of Exciton Trapping and Control of Emission Properties. Transit. Met. Rare Earth Compd. 2004, 241, 1–26. [Google Scholar]
- Chang, C.F.; Cheng, Y.M.; Chi, Y.; Chiu, Y.C.; Lin, C.C.; Lee, G.H.; Chou, P.T.; Chen, C.C.; Chang, C.H.; Wu, C.C. Highly Efficient Blue-Emitting Iridium(III) Carbene Complexes and Phosphorescent OLEDs. Angew. Chem. Int. Ed. 2008, 47, 4542–4545. [Google Scholar] [CrossRef]
- Uoyama, H.; Goushi, K.; Shizu, K.; Nomura, H.; Adachi, C. Highly efficient organic light-emitting diodes from delayed fluorescence. Nature 2012, 492, 234–238. [Google Scholar] [CrossRef]
- Endo, A.; Sato, K.; Yoshimura, K.; Kai, T.; Kawada, A.; Miyazaki, H.; Adachi, C. Efficient up-conversion of triplet excitons into a singlet state and its application for organic light emitting diodes. Appl. Phys. Lett. 2011, 98, 42. [Google Scholar] [CrossRef]
- Zhang, D.-W.; Li, M.; Chen, C.-F. Recent advances in circularly polarized electroluminescence based on organic light-emitting diodes. Chem. Soc. Rev. 2020, 49, 1331–1343. [Google Scholar] [CrossRef] [PubMed]
- Kawasumi, K.; Wu, T.; Zhu, T.; Chae, H.S.; Van Voorhis, T.; Baldo, M.A.; Swager, T.M. Thermally Activated Delayed Fluorescence Materials Based on Homoconjugation Effect of Donor–Acceptor Triptycenes. J. Am. Chem. Soc. 2015, 137, 11908–11911. [Google Scholar] [CrossRef] [PubMed]
- Hirata, S.; Sakai, Y.; Masui, K.; Tanaka, H.; Lee, S.Y.; Nomura, H.; Nakamura, N.; Yasumatsu, M.; Nakanotani, H.; Zhang, Q. Highly efficient blue electroluminescence based on thermally activated delayed fluorescence. Nat. Mater. 2015, 14, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Nakanotani, H.; Higuchi, T.; Furukawa, T.; Masui, K.; Morimoto, K.; Numata, M.; Tanaka, H.; Sagara, Y.; Yasuda, T.; Adachi, C. High-efficiency organic light-emitting diodes with fluorescent emitters. Nat Commun 2014, 5, 4016. [Google Scholar] [CrossRef]
- Chen, X.-K.; Zhang, S.-F.; Fan, J.-X.; Ren, A.-M. Nature of Highly Efficient Thermally Activated Delayed Fluorescence in Organic Light-Emitting Diode Emitters: Nonadiabatic Effect between Excited States. J. Phys. Chem. C 2015, 119, 9728–9733. [Google Scholar] [CrossRef]
- Huang, S.; Zhang, Q.; Shiota, Y.; Nakagawa, T.; Kuwabara, K.; Yoshizawa, K.; Adachi, C. Computational Prediction for Singlet- and Triplet-Transition Energies of Charge-Transfer Compounds. J. Chem. Theory Comput. 2013, 9, 3872–3877. [Google Scholar] [CrossRef]
- Sun, H.; Zhong, C.; Bredas, J.-L. Reliable Prediction with Tuned Range-Separated Functionals of the Singlet–Triplet Gap in Organic Emitters for Thermally Activated Delayed Fluorescence. J. Chem. Theory Comput. 2015, 11, 3851–3858. [Google Scholar] [CrossRef] [PubMed]
- Naveen, K.R.; Lee, H.; Braveenth, R.; Karthik, D.; Yang, K.J.; Hwang, S.J.; Kwon, J.H. Achieving High Efficiency and Pure Blue Color in Hyperfluorescence Organic Light Emitting Diodes using Organo-Boron Based Emitters. Adv. Funct. Mater. 2022, 32, 2110356–2110366. [Google Scholar] [CrossRef]
- Dias, F.B.; Bourdakos, K.N.; Jankus, V.; Moss, K.C.; Kamtekar, K.T.; Bhalla Santos, J.; Bryce, M.R.; Monkman, A.P. Triplet Harvesting with 100% Efficiency by Way of Thermally Activated Delayed Fluorescence in Charge Transfer OLED Emitters. Adv. Mater. 2013, 25, 3707–3714. [Google Scholar] [CrossRef]
- Kim, J.U.; Park, I.S.; Chan, C.-Y.; Tanaka, M.; Tsuchiya, Y.; Nakanotani, H.; Adachi, C. Nanosecond-time-scale delayed fluorescence molecule for deep-blue OLEDs with small efficiency rolloff. Nat. Commun. 2020, 11, 1765. [Google Scholar] [CrossRef]
- Etherington, M.K.; Gibson, J.; Higginbotham, H.F.; Penfold, T.J.; Monkman, A.P. Revealing the spin–vibronic coupling mechanism of thermally activated delayed fluorescence. Nat. Commun. 2016, 7, 13680. [Google Scholar] [CrossRef]
- Monkman, A.P. Vibrational coupling in TADF and how molecular structure can control this complex triplet harvesting process (Conference Presentation). In Organic Light Emitting Materials and Devices XXI; SPIE: Paris, France, 2017; p. 1036204. [Google Scholar]
- Fan, J.; Cai, L.; Lin, L.; Wang, C. Understanding the light-emitting mechanism of an X-shape organic thermally activated delayed fluorescence molecule: First-principles study. Chem. Phys. Lett. 2016, 664, 33–38. [Google Scholar] [CrossRef]
- Sagara, Y.; Shizu, K.; Tanaka, H.; Miyazaki, H.; Goushi, K.; Kaji, H.; Adachi, C. Highly Efficient Thermally Activated Delayed Fluorescence Emitters with a Small Singlet Triplet Energy Gap and Large Oscillator Strength. Chem. Lett. 2015, 44, 360–362. [Google Scholar] [CrossRef]
- Ansari, R.; Shao, W.; Yoon, S.-J.; Kim, J.; Kieffer, J. Charge Transfer as the Key Parameter Affecting the Color Purity of Thermally Activated Delayed Fluorescence Emitters. ACS Appl. Mater. Interfaces 2021, 13, 28529–28537. [Google Scholar] [CrossRef] [PubMed]
- Pan, K.C.; Li, S.W.; Ho, Y.Y.; Shiu, Y.J.; Tsai, W.L.; Jiao, M.; Lee, W.K.; Wu, C.C.; Chung, C.L.; Chatterjee, T. Efficient and Tunable Thermally Activated Delayed Fluorescence Emitters Having Orientation-Adjustable CN-Substituted Pyridine and Pyrimidine Acceptor Units. Adv. Funct. Mater. 2016, 26, 7560–7571. [Google Scholar] [CrossRef]
- Zhao, B.; Wang, H.; Han, C.; Ma, P.; Li, Z.; Chang, P.; Xu, H. Highly Efficient Deep-Red Non-Doped Diodes Based on a T-Shape Thermally Activated Delayed Fluorescence Emitter. Angew. Chem. Int. Ed. 2020, 59, 19042–19047. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.S.; Nobuyasu, R.S.; Fox, M.A.; Aguilar, J.A.; Hall, D.; Batsanov, A.S.; Ren, Z.; Dias, F.B.; Bryce, M.R. Impact of Methoxy Substituents on Thermally Activated Delayed Fluorescence and Room-Temperature Phosphorescence in All-Organic Donor–Acceptor Systems. J. Org. Chem. 2019, 84, 3801–3816. [Google Scholar] [CrossRef]
- Kitamoto, Y.; Namikawa, T.; Suzuki, T.; Miyata, Y.; Kita, H.; Sato, T.; Oi, S. Dimesitylarylborane-based luminescent emitters exhibiting highly-efficient thermally activated delayed fluorescence for organic light-emitting diodes. Org. Electron. 2016, 34, 208–217. [Google Scholar] [CrossRef]
- Shizu, K.; Tanaka, H.; Uejima, M.; Sato, T.; Tanaka, K.; Kaji, H.; Adachi, C. Strategy for Designing Electron Donors for Thermally Activated Delayed Fluorescence Emitters. J. Phys. Chem. 2015, 119, 1291–1297. [Google Scholar] [CrossRef]
- Ganesan, P.; Ranganathan, R.; Chi, Y.; Liu, X.K.; Lee, C.S.; Liu, S.H.; Lee, G.H.; Lin, T.C.; Chen, Y.T.; Chou, P.T. Functional Pyrimidine-Based Thermally Activated Delay Fluorescence Emitters: Photophysics, Mechanochromism, and Fabrication of Organic Light-Emitting Diodes. Chem. A Eur. J. 2017, 23, 2858–2866. [Google Scholar] [CrossRef]
- Cho, E.; Liu, L.; Coropceanu, V.; Brédas, J.-L. Impact of secondary donor units on the excited-state properties and thermally activated delayed fluorescence (TADF) efficiency of pentacarbazole-benzonitrile emitters. J. Chem. Phys. 2020, 153, 144708. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, W.-C.; Liu, H.; Chen, Z.; Chai, D.; Lee, C.-S.; Yang, C. Double-twist pyridine–carbonitrile derivatives yielding excellent thermally activated delayed fluorescence emitters for high-performance OLEDs. J. Mater. Chem. 2020, 8, 602–606. [Google Scholar] [CrossRef]
- Lv, X.; Huang, R.; Sun, S.; Zhang, Q.; Xiang, S.; Ye, S.; Leng, P.; Dias, F.B.; Wang, L. Blue TADF Emitters Based on Indenocarbazole Derivatives with High Photoluminescence and Electroluminescence Efficiencies. ACS Appl. Mater. Interfaces 2019, 11, 10758–10767. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Lee, W.; Lee, T.; Jung, J.; Yoo, S.; Lee, M.H. Triarylboron-based TADF emitters with perfluoro substituents: High-efficiency OLEDs with a power efficiency over 100 lm W−1. J. Mater. Chem. 2020, 8, 4253–4263. [Google Scholar] [CrossRef]
- Marcus, R.A. Electron Transfer Reactions in Chemistry: Theory and Experiment (Nobel Lecture). Angew. Chem. Int. Ed. Engl. 1993, 32, 1111–1121. [Google Scholar] [CrossRef]
- Hilborn, R.C. Einstein coefficients, cross sections, f values, dipole moments, and all that. Am. J. Phys. 1982, 50, 982–986. [Google Scholar] [CrossRef]
- Thangaraji, V.; Rajamalli, P.; Jayakumar, J.; Huang, M.-J.; Chen, Y.-W.; Cheng, C.-H. Quinolinylmethanone-Based Thermally Activated Delayed Fluorescence Emitters and the Application in OLEDs: Effect of Intramolecular H-Bonding. ACS Appl. Mater. Interfaces 2019, 11, 17128–17133. [Google Scholar] [CrossRef]
- Olivier, Y.; Sancho-Garcia, J.-C.; Muccioli, L.; D’Avino, G.; Beljonne, D. Computational Design of Thermally Activated Delayed Fluorescence Materials: The Challenges Ahead. J. Phys. Chem. Lett. 2018, 9, 6149–6163. [Google Scholar] [CrossRef]
- De Silva, P.; Kim, C.A.; Zhu, T.; Van Voorhis, T. Extracting Design Principles for Efficient Thermally Activated Delayed Fluorescence (TADF) from a Simple Four-State Model. Chem. Mater. 2019, 31, 6995–7006. [Google Scholar] [CrossRef]
- Samanta, P.K.; Kim, D.; Coropceanu, V.; Brédas, J.-L. Up-Conversion Intersystem Crossing Rates in Organic Emitters for Thermally Activated Delayed Fluorescence: Impact of the Nature of Singlet vs Triplet Excited States. J. Am. Chem. Soc. 2017, 139, 4042–4051. [Google Scholar] [CrossRef]
- Yang, Z.; Mao, Z.; Xie, Z.; Zhang, Y.; Liu, S.; Zhao, J.; Xu, J.; Chi, Z.; Aldred, M.P. Recent advances in organic thermally activated delayed fluorescence materials. Chem. Soc. Rev. 2017, 46, 915–1016. [Google Scholar] [CrossRef]
- Yang, C.Y.; Lee, K.H.; Lee, J.Y. Zig-Zag Type Molecular Design Strategy of N-Type Hosts for Sky-Blue Thermally-Activated Delayed Fluorescence Organic Light-Emitting Diodes. Chem. A Eur. J. 2020, 26, 2429–2435. [Google Scholar] [CrossRef]
- Hussain, A.; Yuan, H.; Li, W.; Zhang, J. Theoretical investigations of the realization of sky-blue to blue TADF materials via CH/N and H/CN substitution at the diphenylsulphone acceptor. J. Mater. Chem. 2019, 7, 6685–6691. [Google Scholar] [CrossRef]
- Chen, X.K.; Tsuchiya, Y.; Ishikawa, Y.; Zhong, C.; Adachi, C.; Brédas, J.-L. A New Design Strategy for Efficient Thermally Activated Delayed Fluorescence Organic Emitters: From Twisted to Planar Structures. Adv. Mater. 2017, 29, 1702767. [Google Scholar] [CrossRef] [PubMed]
- Meng, G.; Chen, X.; Wang, X.; Wang, N.; Peng, T.; Wang, S. Isomeric Bright Sky-Blue TADF Emitters Based on Bisacridine Decorated DBNA: Impact of Donor Locations on Luminescent and Electroluminescent Properties. Adv. Opt. Mater. 2019, 7, 1900130. [Google Scholar] [CrossRef]
- ADF 2013.01, SCM, Theoretical Chemistry. Vrije Universiteit, Amsterdam, The Netherlands. Available online: http://www.scm.com (accessed on 10 September 2023).
- Zhang, Q.; Li, J.; Shizu, K.; Huang, S.; Hirata, S.; Miyazaki, H.; Adachi, C. Design of Efficient Thermally Activated Delayed Fluorescence Materials for Pure Blue Organic Light Emitting Diodes. J. Am. Chem. Soc. 2012, 134, 14706–14709. [Google Scholar] [CrossRef]
- Chen, M.C.; Chen, D.G.; Chou, P.T. Fluorescent Chromophores Containing the Nitro Group: Relatively Unexplored Emissive Properties. ChemPlusChem 2021, 86, 11–27. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Zhang, J.; Liang, Q.; Wei, Y.; Duan, C.; Han, C.; Xu, H. Charge-Transfer Exciton Manipulation Based on Hydrogen Bond for Efficient White Thermally Activated Delayed Fluorescence. Adv. Funct. Mater. 2020, 30, 1908568. [Google Scholar] [CrossRef]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chem. Phys. Lett. 1998, 285, 170. [Google Scholar] [CrossRef]
- Gupta, A.K.; Zhang, Z.; Spuling, E.; Kaczmarek, M.; Wang, Y.; Hassan, Z.; Samuel, I.D.; Bräse, S.; Zysman-Colman, E. Electron-withdrawing group modified carbazolophane donors for deep blue thermally activated delayed fluorescence OLEDs. Mater. Adv. 2021, 2, 6684–6693. [Google Scholar] [CrossRef]
- Yi, C.-L.; Ko, C.-L.; Yeh, T.-C.; Chen, C.-Y.; Chen, Y.-S.; Chen, D.-G.; Chou, P.-T.; Hung, W.-Y.; Wong, K.-T. Harnessing a New Co-Host System and Low Concentration of New TADF Emitters Equipped with Trifluoromethyl- and Cyano-Substituted Benzene as Core for High-Efficiency Blue OLEDs. ACS Appl. Mater. Interfaces 2019, 12, 2724–2732. [Google Scholar] [CrossRef] [PubMed]
- Woon, K.L.; Nadiah, Z.N.; Hasan, Z.A.; Ariffin, A.; Chen, S.-A. Tuning the singlet-triplet energy splitting by fluorination at 3,6 positions of the 1,4-biscarbazoylbenzene. Dyes Pigments 2016, 132, 1–6. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed]
- Ditchfield, R.; Hehre, W.J.; Pople, J.A. Self-Consistent Molecular Orbital Methods. 9. Extended Gaussian-type basis for molecular-orbital studies of organic molecules. J. Chem. Phys. 1971, 54, 724. [Google Scholar] [CrossRef]
- Bauernschmitt, R.; Ahlrichs, R. Treatment of electronic excitations within the adiabatic approximation of time dependent density functional theory. Chem. Phys. Lett. 1996, 256, 454–464. [Google Scholar] [CrossRef]
- Martin, R.L. Natural transition orbitals. J. Chem. Phys. 2003, 118, 4775–4777. [Google Scholar] [CrossRef]
- Bader, R.F. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Silvi, B.; Savin, A. Classification of chemical bonds based on topological analysis of electron localization functions. Nature 1994, 371, 683–686. [Google Scholar] [CrossRef]
- Lasorne, B.; Worth, G.A.; Robb, M.A. Excited-state dynamics. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 460–475. [Google Scholar] [CrossRef]
- Mignolet, B.; Curchod, B.F.E. Excited-State Molecular Dynamics Triggered by Light Pulses—Ab Initio Multiple Spawning vs Trajectory Surface Hopping. J. Phys. Chem. A 2019, 123, 3582–3591. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Emitter | S1 | S2 | S3 | T1 | T2 | T3 | ∆EST (V) | ∆EST | KRISC | Kr |
|---|---|---|---|---|---|---|---|---|---|---|
| Ac-B | 3.48 | 3.91 | 4.12 | 3.43 | 3.46 | 3.51 | 0.05 | (0.04) # | 3.12 × 104 | 2.4 × 106 |
| Ac-B-F | 3.36 | 3.83 | 4.04 | 3.34 | 3.36 | 3.45 | 0.02 | - | 2.09 × 106 | 4.0 × 106 |
| Ac-B-CN | 2.71 | 3.10 | 3.67 | 2.69 | 3.07 | 3.26 | 0.02 | - | 3.50 × 106 | 1.0 × 105 |
| Ac-B-NO2 | 2.33 | 2.59 | 3.41 | 2.29 | 2.55 | 3.24 | 0.04 | 1.26 × 107 | 7.2 × 106 |
| Emitter | f (S1/S2/S3) | λ (nm) (S1/S2/S3) |
|---|---|---|
| Ac-B | 0.0015/0.1255/0.1331 | 355/317/301 (295/360) ** |
| Ac-B-F | 0.0027/0.1370/0.1023 | 369/324/307 |
| Ac-B-CN | 0.0001/0.0004/0.1293 | 457/400/337 |
| Ac-B-NO2 | 0.0101/0.0018/0.0385 | 531/478/363 |
| Emitters | Adiabatic Energy Gap (S1−T1) (eV) | Kr (s−1) | H-Bond Energy (kJ/mol) |
|---|---|---|---|
| Ac-B | 0.02 | 3.2 × 105 | - |
| Ac-B-F | 0.06 | 5.8 × 105 | - |
| Ac-B-CN | 0.01 | 3.5 × 104 | −0.20 |
| Ac-B-NO2 | −0.04 | 1.8 × 105 | −1.66 |
| Substrate | S1 | T1 | ||
|---|---|---|---|---|
| LE% | CT% | LE% | CT% | |
| Ac-B | 19.21 | 80.79 | 77.08 | 22.92 |
| Ac-B-F | 05.07 | 94.93 | 07.37 | 92.63 |
| Ac-B-CN | 06.53 | 93.47 | 06.70 | 93.30 |
| Ac-B-NO2 | 07.37 | 92.63 | 08.66 | 91.34 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Asiri, J.A.; Hasan, W.M.I.; Jedidi, A.; Elroby, S.A.; Aziz, S.G.; Osman, O.I. Organoboron Complexes as Thermally Activated Delayed Fluorescence (TADF) Materials for Organic Light-Emitting Diodes (OLEDs): A Computational Study. Molecules 2023, 28, 6952. https://doi.org/10.3390/molecules28196952
Asiri JA, Hasan WMI, Jedidi A, Elroby SA, Aziz SG, Osman OI. Organoboron Complexes as Thermally Activated Delayed Fluorescence (TADF) Materials for Organic Light-Emitting Diodes (OLEDs): A Computational Study. Molecules. 2023; 28(19):6952. https://doi.org/10.3390/molecules28196952
Chicago/Turabian StyleAsiri, Jamilah A., Walid M. I. Hasan, Abdesslem Jedidi, Shaaban A. Elroby, Saadullah G. Aziz, and Osman I. Osman. 2023. "Organoboron Complexes as Thermally Activated Delayed Fluorescence (TADF) Materials for Organic Light-Emitting Diodes (OLEDs): A Computational Study" Molecules 28, no. 19: 6952. https://doi.org/10.3390/molecules28196952