
Theoretical Investigation on the “ON-OFF” Mechanism of a Fluorescent Probe for Thiophenols: Photoinduced Electron Transfer and Intramolecular Charge Transfer


Abstract
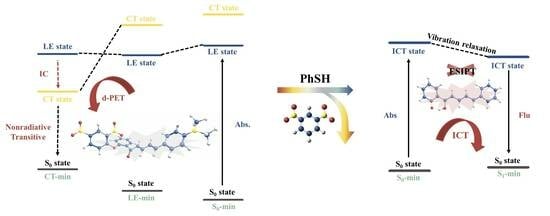
:
1. Introduction
2. Results and Discussion
2.1. DAPH-DNP
2.2. DAPH
3. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Ji, H.-F.; Shen, L.; Zhang, H.-Y. Theoretical Reinvestigation of Opposite Electronic Effects on Bond Lengths in Thiophenols and Thiophenolic Radicals. J. Struct. Chem. 2005, 46, 347–351. [Google Scholar] [CrossRef]
- dos Santos, D.J.V.A.; Newton, A.S.; Bernardino, R.; Guedes, R.C. Substituent Effects on O–H and S–H Bond Dissociation Enthalpies of Disubstituted Phenols and Thiophenols. Int. J. Quantum Chem. 2008, 108, 754–761. [Google Scholar] [CrossRef]
- Wang, H.; Wu, X.; Yang, S.; Tian, H.; Liu, Y.; Sun, B. A Rapid and Visible Colorimetric Fluorescent Probe for Benzenethiol Flavor Detection. Food Chem. 2019, 286, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Hao, Y.; Ma, X.; Chen, S.; Xu, M. A Dicyanoisophorone-Based Highly Sensitive and Selective near-Infrared Fluorescent Probe for Sensing Thiophenol in Water Samples and Living Cells. Environ. Pollut. 2020, 265, 114958. [Google Scholar] [CrossRef]
- Xiao, M.-M.; Ren, H.; Liu, T.-Z.; Li, Z.-Y.; Wang, J.-Z.; Miao, J.-Y.; Zhao, B.-X. Two Fluorescent Turn-on Probes for Detecting Thiophenols in Environmental Water and in Living Cell Imaging. Microchem. J. 2022, 175, 107220. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Yang, Y.; Zhu, H.-L. Selective and Rapid Detection of Thiophenol by a Novel Fluorescent Probe with Cellular Imaging. Anal. Lett. 2022, 55, 2727–2737. [Google Scholar] [CrossRef]
- Wu, F.; Wang, H.; Xu, J.; Yuan, H.-Q.; Zeng, L.; Bao, G.-M. A New Fluorescent Chemodosimeter for Ultra-Sensitive Determination of Toxic Thiophenols in Environmental Water Samples and Cancer Cells. Sens. Actuators B Chem. 2018, 254, 21–29. [Google Scholar] [CrossRef]
- Nádudvari, Á.; Kozielska, B.; Abramowicz, A.; Fabiańska, M.; Ciesielczuk, J.; Cabała, J.; Krzykawski, T. Heavy Metal- and Organic-Matter Pollution Due to Self-Heating Coal-Waste Dumps in the Upper Silesian Coal Basin (Poland). J. Hazard. Mater. 2021, 412, 125244. [Google Scholar] [CrossRef]
- He, J.-X.; Akao, T.; Tani, T. Development of a Simple HPLC Method for Determination of Paeoniflorin-Metabolizing Activity of Intestinal Bacteria in Rat Feces. Chem. Pharm. Bull. 2002, 50, 1233–1237. [Google Scholar] [CrossRef]
- Hu, G.; Wang, Z.; Yang, W.; Shen, W.; Sun, W.; Xu, H.; Hu, Y. Dicyanisophorone-Based near-Infrared Fluorescent Probe for the Detection of Thiophenol and Its Application in Living Cells and Actual Water Samples. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2022, 272, 120984. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Xia, Q.; Feng, W.; Feng, G. A Dicyanoisophorone-Based near-Infrared Fluorescent Probe and Its Application for Detecting Thiophenols in Water and Living Cells. Dye. Pigment. 2018, 159, 604–609. [Google Scholar] [CrossRef]
- Wu, Y.; Shi, A.; Liu, H.; Li, Y.; Lun, W.; Zeng, H.; Fan, X. A Novel Near-Infrared Xanthene-Based Fluorescent Probe for Detection of Thiophenol in Vitro and in Vivo. New J. Chem. 2020, 44, 17360–17367. [Google Scholar] [CrossRef]
- Wang, K.; Zhao, C.-X.; Leng, T.-H.; Wang, C.-Y.; Lu, Y.-X.; Shen, Y.-J.; Zhu, W.-H. Dual Quenching Strategy for Sensitive Detection of Toxic Thiolphenols Based on a NIR-Illuminant Platform with a Large Stokes Shift. Dye. Pigment. 2018, 151, 194–201. [Google Scholar] [CrossRef]
- Lin, L.; Zeng, X.; Shen, Y.; Zhu, H.; Qian, Y. An Ultrasensitive Fluorescent Probe for Rapid Determination of Thiophenols. Talanta 2017, 165, 321–325. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhang, K.; Liu, J.; Wang, Y.; Yu, F. The Sensing Mechanism of Fluorescent Probe for PhSH and the Process of ESIPT. Photochem. Photobiol. Sci. 2022, 21, 1055–1065. [Google Scholar] [CrossRef]
- Cao, Y.; Yu, X.; Sun, C.; Cui, J. Theoretical Investigation on the ESIPT Process and Detection Mechanism for Dual-Proton Type Fluorescent Probe. Int. J. Mol. Sci. 2022, 23, 2132. [Google Scholar] [CrossRef]
- Ni, H.; Wang, Q.; Jin, L.; Wang, W.; Dai, L.; Zhao, C. High Selectivity and Reversibility/Reusability Red Emitting Fluorescent Probe for Copper Ions Detection and Imaging in Living Cells. J. Lumin. 2019, 206, 125–131. [Google Scholar] [CrossRef]
- Donadio, G.; Martino, R.D.; Oliva, R.; Petraccone, L.; Vecchio, P.D.; Luccia, B.D.; Ricca, E.; Isticato, R.; Donato, A.D.; Notomista, E. A New Peptide-Based Fluorescent Probe Selective for Zinc(II) and Copper(II). J. Mater. Chem. B 2016, 4, 6979–6988. [Google Scholar] [CrossRef]
- Choi, Y.; Shin, S.-H.; Jung, H.; Kwon, O.; Seo, J.-K.; Kee, J.-M. Specific Fluorescent Probe for Protein Histidine Phosphatase Activity. ACS Sens. 2019, 4, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Li, M.; Liu, Y. Detection Sensitivity Enhancement of Naphthalimide PET Fluorescent Probes by 4-Methoxy-Substitution. Molecules 2020, 25, 4465. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Wu, M.; Wang, S.; Qin, J.; Li, P. An AIE/PET-Based Fluorescent Probe for Zn2+/Al3+ Detection and Its Application in Fluorescence-Assisted Diagnosis for Prostate Cancer. Dye. Pigment. 2022, 203, 110372. [Google Scholar] [CrossRef]
- Mittapalli, R.R.; Namashivaya, S.S.R.; Oshchepkov, A.S.; Kuczyńska, E.; Kataev, E.A. Design of Anion-Selective PET Probes Based on Azacryptands: The Effect of pH on Binding and Fluorescence Properties. Chem. Commun. 2017, 53, 4822–4825. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Yang, B.-Z.; Ren, A.-M. Theoretical Investigation on Ratiometric Two-Photon Fluorescent Probe for Zn2+ Detection Based on ICT Mechanism. J. Mol. Struct. 2016, 1114, 65–77. [Google Scholar] [CrossRef]
- Li, P.-Y.; Han, C.-Z.; Gong, B.; Liu, D.; Wang, J.-P. TDDFT Study on the ESPT and ICT Mechanism of a Bifunctional Fluorescent Probe for Detecting Fluoride and Sulphite. Chem. Phys. Lett. 2022, 802, 139782. [Google Scholar] [CrossRef]
- Lin, Y.; Yu, A.; Wang, J.; Kong, D.; Liu, H.; Li, J.; Jia, C. A Curcumin-Based AIEE-Active Fluorescent Probe for Cu2+ Detection in Aqueous Solution. RSC Adv. 2022, 12, 16772–16778. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Hu, X.; Yang, B.; Liu, B. Dual Sites Fluorescence Probe for Hydrogen Sulfide: AIEE Activity and Supramolecular Assembly with β-Cyclodextrin. Sens. Actuators B Chem. 2019, 282, 743–749. [Google Scholar] [CrossRef]
- Catalán, J.; Ignacio Fernández-Alonso, J. A Theoretical Study of the Stereochemistry of the Intramolecular Hydrogen Bond of Salicylic Acid. J. Mol. Struct. 1975, 27, 59–65. [Google Scholar] [CrossRef]
- Peters, K.; Applebury, M.L.; Rentzepis, P.M. Primary Photochemical Event in Vision: Proton Translocation. Proc. Natl. Acad. Sci. USA 1977, 74, 3119–3123. [Google Scholar] [CrossRef]
- Goodman, J.; Brus, L.E. Proton Transfer and Tautomerism in an Excited State of Methyl Salicylate. J. Am. Chem. Soc. 1978, 100, 7472–7474. [Google Scholar] [CrossRef]
- Li, Z.; Wu, Y.; Shen, Y.; Gu, B. Simple NIR-Emitting ESIPT Fluorescent Probe for Thiophenol with a Remarkable Stokes Shift and Its Application. ACS Omega 2020, 5, 10808–10814. [Google Scholar] [CrossRef]
- Liu, Z.; Lu, T.; Chen, Q. An Sp-Hybridized All-Carboatomic Ring, Cyclo[18]Carbon: Electronic Structure, Electronic Spectrum, and Optical Nonlinearity. Carbon 2020, 165, 461–467. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- CanceS, E.; Mennucci, B.; Tomasi, J. A New Integral Equation Formalism for the Polarizable Continuum Model: Theoretical Background and Applications to Isotropic and Anisotropic Dielectrics. J. Chem. Phys. 1997, 107, 3032–3041. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Guido, C.A.; Chrayteh, A.; Scalmani, G.; Mennucci, B.; Jacquemin, D. Simple Protocol for Capturing Both Linear-Response and State-Specific Effects in Excited-State Calculations with Continuum Solvation Models. J. Chem. Theory Comput. 2021, 17, 5155–5164. [Google Scholar] [CrossRef]
- Jiang, G.; Ma, Y.; Ding, J.; Liu, J.; Liu, R.; Zhou, P. N-Protonation as a Switch of the Twisted Excited States with ππ* or nπ* Character and Correlation with the π-electrons Characteristic of Rotatable Bonds. Chem.–Eur. J. 2023, 29, e202300625. [Google Scholar] [CrossRef]
- Tang, Z.; Bai, T.; Zhou, P. Sensing Mechanism of a Fluorescent Probe for Cysteine: Photoinduced Electron Transfer and Invalidity of Excited-State Intramolecular Proton Transfer. J. Phys. Chem. A 2020, 124, 6920–6927. [Google Scholar] [CrossRef]
- Chi, W.; Chen, J.; Liu, W.; Wang, C.; Qi, Q.; Qiao, Q.; Tan, T.M.; Xiong, K.; Liu, X.; Kang, K.; et al. A General Descriptor ΔE Enables the Quantitative Development of Luminescent Materials Based on Photoinduced Electron Transfer. J. Am. Chem. Soc. 2020, 142, 6777–6785. [Google Scholar] [CrossRef]
- del Valle, J.C.; Catalán, J. Kasha’s Rule: A Reappraisal. Phys. Chem. Chem. Phys. 2019, 21, 10061–10069. [Google Scholar] [CrossRef]
- Loudet, A.; Burgess, K. BODIPY Dyes and Their Derivatives: Syntheses and Spectroscopic Properties. Chem. Rev. 2007, 107, 4891–4932. [Google Scholar] [CrossRef]
- Zhang, X.; Chi, L.; Ji, S.; Wu, Y.; Song, P.; Han, K.; Guo, H.; James, T.D.; Zhao, J. Rational Design of D-PeT Phenylethynylated-Carbazole Monoboronic Acid Fluorescent Sensors for the Selective Detection of α-Hydroxyl Carboxylic Acids and Monosaccharides. J. Am. Chem. Soc. 2009, 131, 17452–17463. [Google Scholar] [CrossRef] [PubMed]
- TURBOMOLE V7.6 2022, a Development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, Since 2007. Available online: https://www.turbomole.org (accessed on 30 September 2023).
- Mewes, J.-M.; You, Z.-Q.; Wormit, M.; Kriesche, T.; Herbert, J.M.; Dreuw, A. Experimental Benchmark Data and Systematic Evaluation of Two a Posteriori, Polarizable-Continuum Corrections for Vertical Excitation Energies in Solution. J. Phys. Chem. A 2015, 119, 5446–5464. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Revision A.03 2016. Available online: https://gaussian.com/relnotes_a03/ (accessed on 30 September 2023).
- Gauss, J. Calculation of NMR Chemical Shifts at Second-Order Many-Body Perturbation Theory Using Gauge-Including Atomic Orbitals. Chem. Phys. Lett. 1992, 191, 614–620. [Google Scholar] [CrossRef]
- Schafer, A.; Horn, H.; Ahlrichs, R. Fully Optimized Contracted Gaussian Basis Sets for Atoms Li to Kr. J. Chem. Phys. 1992, 97, 2571. [Google Scholar] [CrossRef]
- Coitiño, E.L.; Tomasi, J.; Cammi, R. On the Evaluation of the Solvent Polarization Apparent Charges in the Polarizable Continuum Model: A New Formulation. J. Comput. Chem. 1995, 16, 20–30. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Electronic Transition | Energy (nm/eV) | f | Contrib | CI | Exp (nm/eV) | ||
|---|---|---|---|---|---|---|---|
| LR | cLR | ||||||
| DAPH- DNP | |||||||
| Absorption | S0 → S1 | 395/3.14 | 395/3.14 | 1.8031 | H → L + 2 | 81.3% | 447/2.77 |
| Absorption | S0 → S2 | 335/3.70 | 339/3.66 | 0.0007 | H-4 → L + 2 | 48.1% | / |
| Emission | S1 → S0 | 800/1.55 | 800/1.55 | 0.4340 | H → L | 63.8% | / |
| Emission | S2 → S0 | 477/2.60 | 458/2.71 | 1.8673 | H → L + 2 | 70.1% | / |
| DAPH | |||||||
| Absorption | S0 → S1 | 419/2.96 | 429/2.89 | 1.8406 | H → L | 86.1% | 462/2.68 |
| Emission | S1 → S0 | 453/2.74 | 475/2.61 | 1.8491 | H → L | 88.1% | 654/1.90 |
| D (Å) | Sr | H (Å) | t (Å) | HDI | EDI | |
|---|---|---|---|---|---|---|
| DAPH-DNP | ||||||
| S0 → S1 | 2.979 | 0.62145 | 3.658 | −0.122 | 6.88 | 7.21 |
| S0 → S2 | 2.225 | 0.38421 | 2.693 | 0.201 | 17.99 | 6.99 |
| S1 → S0 | 3.671 | 0.57021 | 3.971 | 0.192 | 6.84 | 5.99 |
| DAPH | ||||||
| S0 → S1 | 2.159 | 0.73058 | 3.814 | −1.399 | 6.97 | 7.30 |
| S1 → S0 | 1.991 | 0.73102 | 3.868 | −1.599 | 6.91 | 7.30 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Zhang, M.; Li, W.; Wang, Y.; Zhou, P. Theoretical Investigation on the “ON-OFF” Mechanism of a Fluorescent Probe for Thiophenols: Photoinduced Electron Transfer and Intramolecular Charge Transfer. Molecules 2023, 28, 6921. https://doi.org/10.3390/molecules28196921
Wang Y, Zhang M, Li W, Wang Y, Zhou P. Theoretical Investigation on the “ON-OFF” Mechanism of a Fluorescent Probe for Thiophenols: Photoinduced Electron Transfer and Intramolecular Charge Transfer. Molecules. 2023; 28(19):6921. https://doi.org/10.3390/molecules28196921
Chicago/Turabian StyleWang, Yuxi, Meng Zhang, Wenzhi Li, Yi Wang, and Panwang Zhou. 2023. "Theoretical Investigation on the “ON-OFF” Mechanism of a Fluorescent Probe for Thiophenols: Photoinduced Electron Transfer and Intramolecular Charge Transfer" Molecules 28, no. 19: 6921. https://doi.org/10.3390/molecules28196921