
Synthesis and Characterization of New Triazole-Bispidinone Scaffolds and Their Metal Complexes for Catalytic Applications
 , , , and
, , , and 
Abstract
:
1. Introduction
2. Results and Discussion
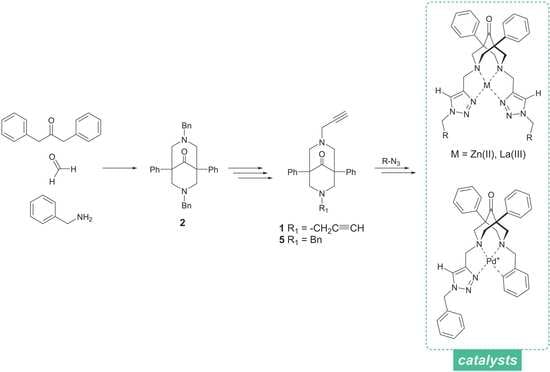
2.1. Synthesis of the Bispidine-Triazole Ligands
2.2. NMR Studies on the Coordination Chemistry
2.3. ESI-MS Studies on the Coordination Chemistry
2.4. Single-Crystal X-ray Characterization of the Free Ligand 8a
2.5. Molecular Modeling
2.6. Catalysis Applications of the Complexes
3. Conclusions
4. Materials and Methods
4.1. General Remarks
4.2. Synthetic Procedures
4.3. Crystallographic Data
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Comba, P.; Haaf, C.; Wadepohl, H. Novel Bispidine Ligands and Their First-Row Transition Metal Complexes: Trigonal Bipyramidal and Trigonal Prismatic Geometries. Inorg. Chem. 2009, 48, 6604–6614. [Google Scholar] [CrossRef]
- Sacchetti, A.; Rossetti, A. Synthesis of Natural Compounds Based on the [3,7]-Diazabicyclo[3.3.1]nonane (Bispidine) Core. Eur. J. Org. Chem. 2021, 1491–1507. [Google Scholar] [CrossRef]
- Black, D.S.C.; Deacon, G.B.; Rose, M. Synthesis and metal complexes of symmetrically N-substituted bispidinones. Tetrahedron 1995, 51, 2055–2076. [Google Scholar] [CrossRef]
- Comba, P.; Hunoldt, S.; Morgen, M.; Pietzsch, J.; Stephan, H.; Wadepohl, H. Optimization of pentadentate bispidines as bifunctional chelators for 64Cu positron emission tomography (PET). Inorg. Chem. 2013, 52, 8131–8143. [Google Scholar] [CrossRef] [PubMed]
- Comba, P.; Jermilova, U.; Orvig, C.; Patrick, B.O.; Ramogida, C.F.; Rück, K.; Schneider, C.; Starke, M. Octadentate Picolinic Acid-Based Bispidine Ligand for Radiometal Ions. Chem. Eur. J. 2017, 23, 15945–15956. [Google Scholar] [CrossRef]
- Rossetti, A.; Landoni, S.; Meneghetti, F.; Castellano, C.; Mori, M.; Colombo Dugoni, G.; Sacchetti, A. Application of chiral bi- and tetra-dentate bispidine-derived ligands in the copper(ii)-catalyzed asymmetric Henry reaction. New J. Chem. 2018, 42, 12072–12081. [Google Scholar] [CrossRef]
- Lippi, M.; Wadepohl, H.; Comba, P.; Cametti, M. A Bispidine Based CuII/ZnII Heterobimetallic Coordination Polymer. Eur. J. Inorg. Chem. 2022, 2022, e202200221. [Google Scholar] [CrossRef]
- Mori, M.; Fumagalli, E.; Castellano, C.; Tresoldi, A.; Sacchetti, A.; Meneghetti, F. Synthesis and characterization of a tetradentate bispidine-based ligand and its zinc(II) complex. Inorganica Chim. Acta 2022, 538, 120968. [Google Scholar] [CrossRef]
- Gao, F.; Sihver, W.; Bergmann, R.; Walther, M.; Stephan, H.; Belter, B.; Neuber, C.; Haase-Kohn, C.; Bolzati, C.; Pietzsch, J.; et al. Radiochemical and radiopharmacological characterization of a 64Cu-labeled α-MSH analog conjugated with different chelators. J. Label. Compd. Radiopharm. 2019, 62, 495–509. [Google Scholar] [CrossRef]
- Bleher, K.; Comba, P.; Kass, D.; Ray, K.; Wadepohl, H. Reactivities of iron(IV)-oxido compounds with pentadentate bispidine ligands. J. Inorg. Biochem. 2023, 241, 112123. [Google Scholar] [CrossRef]
- Rossetti, A.; Lippi, M.; Martí-Rujas, J.; Sacchetti, A.; Cametti, M. Highly Dynamic and Tunable Behavior of 1D Coordination Polymers Based on the Bispidine Ligand. Chem. Eur. J. 2018, 24, 19368–19372. [Google Scholar] [CrossRef]
- Grosshauser, M.; Comba, P.; Kim, J.Y.; Ohto, K.; Thuéry, P.; Lee, Y.H.; Kim, Y.; Harrowfield, J. Ferro- and antiferromagnetic coupling in a chlorido-bridged, tetranuclear Cu(ii) complex. Dalt. Trans. 2014, 43, 5662–5666. [Google Scholar] [CrossRef]
- Nonat, A.M.; Roux, A.; Sy, M.; Charbonnière, L.J. 2,4-Substituted bispidines as rigid hosts for versatile applications: From κ-opioid receptor to metal coordination. Dalt. Trans. 2019, 48, 16476–16492. [Google Scholar] [CrossRef]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Eremina, O.E.; Kapitanova, O.O.; Medved’ko, A.V.; Zelenetskaya, A.S.; Egorova, B.V.; Shekhovtsova, T.N.; Vatsadze, S.Z.; Veselova, I.A. Plier Ligands for Trapping Neurotransmitters into Complexes for Sensitive Analysis by SERS Spectroscopy. Biosensors 2023, 13, 124. [Google Scholar] [CrossRef]
- Dalinger, A.I.; Medved’ko, A.V.; Balalaeva, A.I.; Vatsadze, I.; Dalinger, I.L.; Vatsadze, S.Z. Synthesis of Novel Azides and Triazoles on the Basis of 1H-Pyrazole-3(5)-Carboxylic Acids. Chem. Heterocycl. Compd. 2020, 56, 180–191. [Google Scholar] [CrossRef]
- Vatsadze, S.Z.; Medved’ko, A.V.; Bodunov, A.A.; Lyssenko, K.A. Bispidine-based bis-azoles as a new family of supramolecular receptors: The theoretical approach. Mendeleev Commun. 2020, 30, 344–346. [Google Scholar] [CrossRef]
- Elliott, P.I.P. Chapter 1 Organometallic complexes with 1,2,3-triazole-derived ligands. In Organometallic Chemistry; The Royal Society of Chemistry: London, UK, 2014; Volume 39, pp. 1–25. ISBN 978-1-84973-583-4. [Google Scholar]
- Creary, X.; Chormanski, K.; Peirats, G.; Renneburg, C. Electronic Properties of Triazoles. Experimental and Computational Determination of Carbocation and Radical-Stabilizing Properties. J. Org. Chem. 2017, 82, 5720–5730. [Google Scholar] [CrossRef] [PubMed]
- Bulygina, L.A.; Kagramanov, N.D.; Khrushcheva, N.S.; Lyssenko, K.A.; Peregudov, A.S.; Sokolov, V.I. Unsymmetrical pincer CNN palladium complex of 7-ferrocenylmethyl-3-methyl-3,7-diazabicyclo[3.3.1]nonane. J. Organomet. Chem. 2017, 846, 169–175. [Google Scholar] [CrossRef]
- Bulygina, L.A.; Khrushcheva, N.S.; Peregudov, A.S.; Sokolov, V.I. Cyclopalladate complex of 3-benzyl-7-methyl-3,7-diazabicyclo[3.3.1]nonane. Russ. Chem. Bull. 2016, 65, 2479–2484. [Google Scholar] [CrossRef]
- Spek, A.L. Single-crystal structure validation with the program platon. J. Appl. Crystallogr. 2003, 36, 7–13. [Google Scholar] [CrossRef]
- MacRae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Cremer, D.; Pople, J.A. General definition of ring puckering coordinates. J. Am. Chem. Soc. 1975, 97, 1354–1358. [Google Scholar] [CrossRef]
- Luzzio, F.A. The Henry reaction: Recent examples. Tetrahedron 2001, 57, 915–945. [Google Scholar] [CrossRef]
- Noboru, O. The Nitro Group in Organic Synthesis; Wiley Online Books: Hoboken, NJ, USA, 2001; ISBN 9780471224488. [Google Scholar]
- Ballini, R. Synthesis of natural products via aliphatic nitroderivatives. In Structure and Chemistry (Part E); Atta-ur-Rahman, B.T.-S.N.P.C., Ed.; Elsevier: Amsterdam, The Netherlands, 1996; Volume 19, pp. 117–184. ISBN 1572-5995. [Google Scholar]
- Murugavel, G.; Sadhu, P.; Punniyamurthy, T. Copper(II)-Catalyzed Nitroaldol (Henry) Reactions: Recent Developments. Chem. Rec. 2016, 16, 1906–1917. [Google Scholar] [CrossRef]
- Saranya, S.; Harry, N.A.; Ujwaldev, S.M.; Anilkumar, G. Recent Advances and Perspectives on the Zinc-Catalyzed Nitroaldol (Henry) Reaction. Asian J. Org. Chem. 2017, 6, 1349–1360. [Google Scholar] [CrossRef]
- Burla, M.C.; Caliandro, R.; Carrozzini, B.; Cascarano, G.L.; Cuocci, C.; Giacovazzo, C.; Mallamo, M.; Mazzone, A.; Polidori, G. Crystal structure determination and refinement via SIR2014. J. Appl. Crystallogr. 2015, 48, 306–309. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | R | Yield (%) |
|---|---|---|
| 7a | benzyl | 95 |
| 7b | p-Cl-benzyl | 75 |
| 7c | p-NO2-benzyl | 78 |
| 7d | 2-(azidomethyl)pyridine | 84 |
| 7e | 4-(azidomethyl)pyridine | 83 |
| 7f | n-octyl | 86 |
| 7g | n-undecyl | 73 |
| 7h | 3-hydroxypropyl | 85 |
| 7i | -CH2PhCH2- | traces |
| 7j | -CH2(CH2)4CH2- | traces |
| Entry | Compound | ∆δ for H2,4,6,8 eq (ppm) | ∆δ for H2,4,6,8 ax (ppm) | ∆δ for HA (ppm) | ∆δ for HB (ppm) | ∆δ for HC (ppm) |
|---|---|---|---|---|---|---|
| 1 | 7b·Zn(II) | 0.46 | 0.44 | 0.18 | 0.50 | 0.15 |
| 2 | 7d·Zn(II) | 0.50 | 0.61 | 0.27 | 0.24 | 0.15 |
| 3 | 7d·La(III) | 0.45 | 0.70 | 0.43 | 0.23 | 0.03 |
| 4 | 7a·La(III) | 0.46 | 0.71 | 0.42 | 0.24 | 0.04 |
| 5 | 7h·La(III) | 0.45 | 0.70 | 0.40 | 0.40 | 0.05 |
| ∆δ for H2,4,6,8 eq (ppm) | ∆δ for H2,4,6,8 ax (ppm) | ∆δ for HA-A (ppm) | ∆δ for HB (ppm) | ∆δ for HC (ppm) | ∆δ for HD (ppm) |
|---|---|---|---|---|---|
| 0.82–0.84 | 0.80 | 0.66 | 0.28 | 0.18 | 0.68 |
 | |||
|---|---|---|---|
| Entry | Aldehyde | Catalyst Loading | Yield (%) |
| 1 a | p-NO2-PhCHO | Zn (2% mol) | 7 |
| 2 a | p-NO2-PhCHO | 7a·Zn (2% mol) | 79 |
| 3 a | p-NO2-PhCHO | 7a·Zn (15% mol) | 99 |
| 4 a | p-NO2-PhCHO | 7a·Zn (15% mol) + TEA (2% mol) | 81 |
| 5 a | p-NO2-PhCHO | 7a·Cu (2% mol) | 22 |
| 6 a | p-NO2-PhCHO | 7a·Cu (15% mol) | 56 |
| 7 | p-Br-PhCHO | 7a·Zn (15% mol) | 92 |
| 8 | p-F-PhCHO | 7a·Zn (15% mol) | 91 |
| 9 | p-CF3-PhCHO | 7a·Zn (15% mol) | 94 |
| 10 | o-NO2-PhCHO | 7a·Zn (15% mol) | 81 |
| 11 | 2-naphthaldehyde | 7a·Zn (15% mol) | 88 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rossetti, A.; Sacchetti, A.; Meneghetti, F.; Colombo Dugoni, G.; Mori, M.; Castellano, C. Synthesis and Characterization of New Triazole-Bispidinone Scaffolds and Their Metal Complexes for Catalytic Applications. Molecules 2023, 28, 6351. https://doi.org/10.3390/molecules28176351
Rossetti A, Sacchetti A, Meneghetti F, Colombo Dugoni G, Mori M, Castellano C. Synthesis and Characterization of New Triazole-Bispidinone Scaffolds and Their Metal Complexes for Catalytic Applications. Molecules. 2023; 28(17):6351. https://doi.org/10.3390/molecules28176351
Chicago/Turabian StyleRossetti, Arianna, Alessandro Sacchetti, Fiorella Meneghetti, Greta Colombo Dugoni, Matteo Mori, and Carlo Castellano. 2023. "Synthesis and Characterization of New Triazole-Bispidinone Scaffolds and Their Metal Complexes for Catalytic Applications" Molecules 28, no. 17: 6351. https://doi.org/10.3390/molecules28176351