
Access to Enantiomerically Pure P-Chiral 1-Phosphanorbornane Silyl Ethers
 , , , , , and
, , , , , and
Abstract
: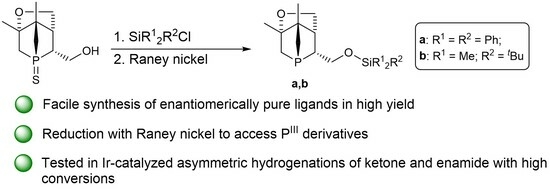
1. Introduction
2. Results and Discussion
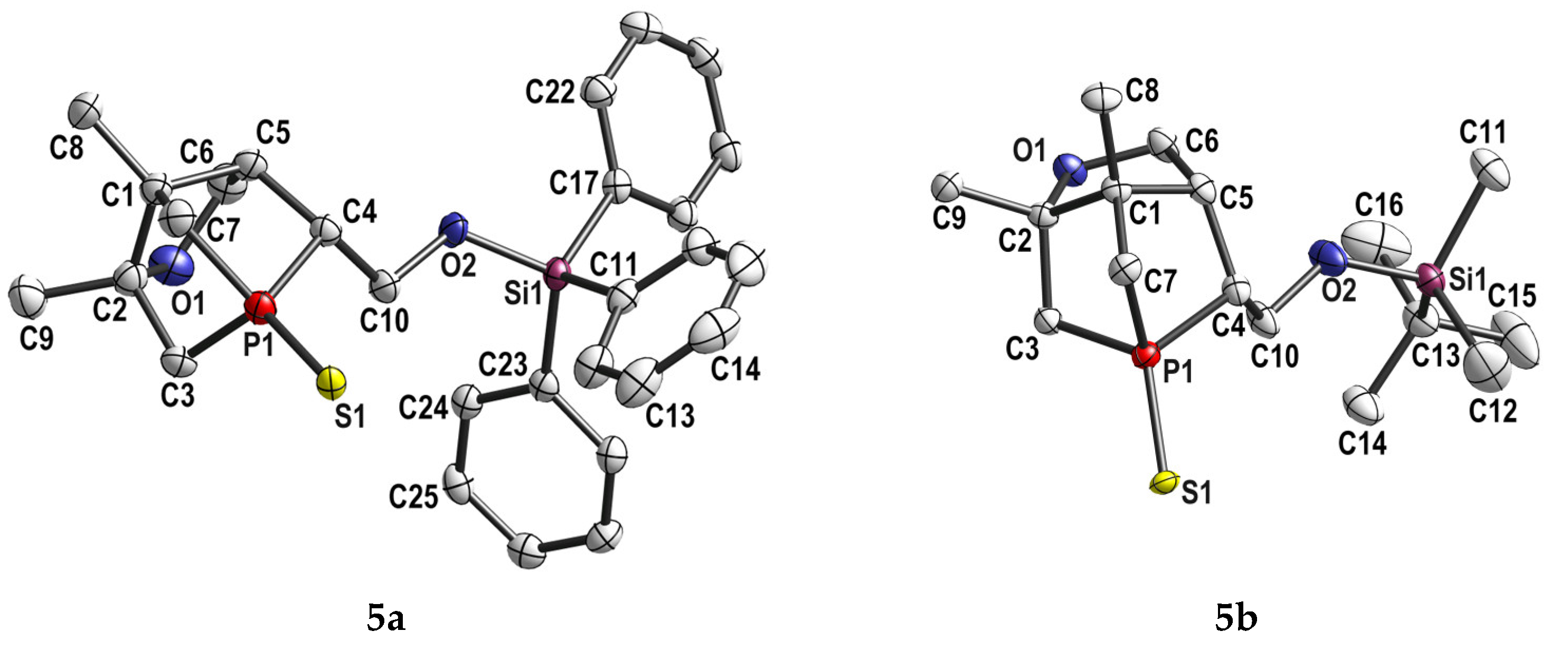
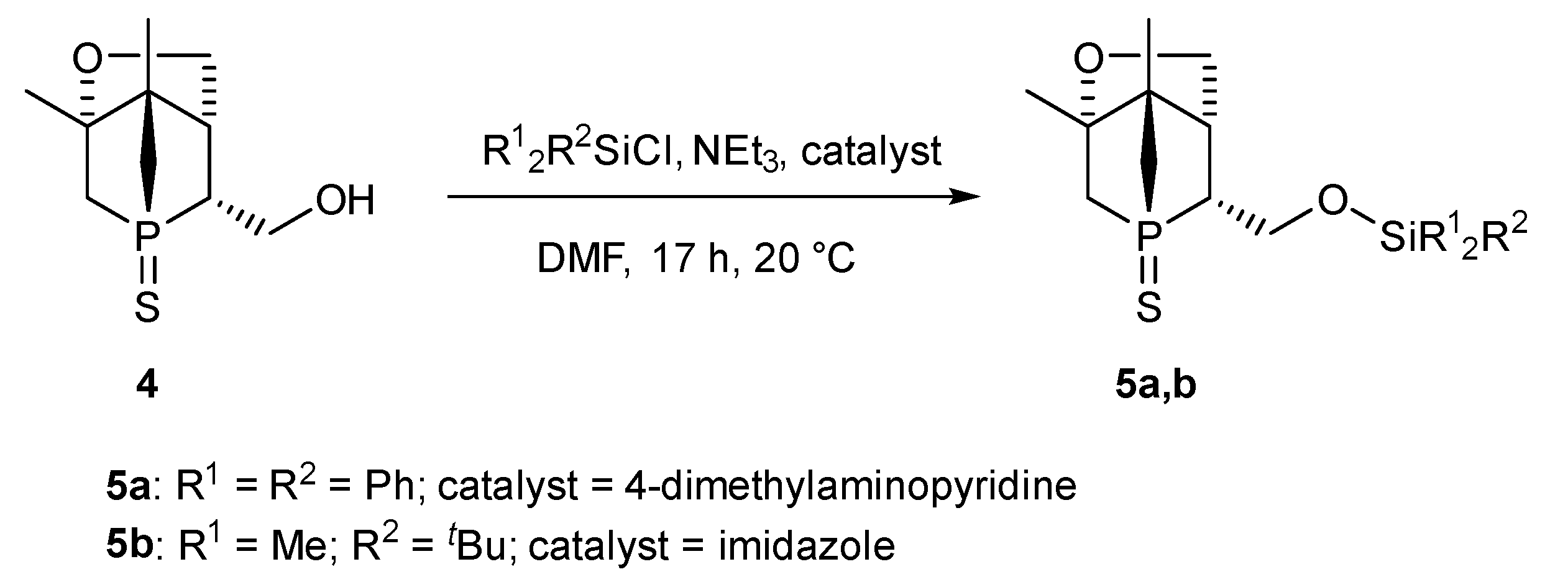
2.1. Synthesis and Characterization of 5a,b
2.2. Desulfurization of Compounds 5a,b
3. Catalysis
4. Conclusions
5. Materials
5.1. General Information
5.2. Synthesis
5.2.1. Synthesis of 5a
- 1H NMR (400 MHz, CDCl3): δ 7.61 (m, 5H), 7.50–7.25 (m, 10H), 4.33 (m, 1H, H-5 or H-6a), 4.16–4.01 (m, 2H), 3.94 (dd, J = 9.9, 5.7 Hz, 1H, H-5 or H-6a), 2.67–2.45 (m, 2H), 2.25 (m, 1H, H-6 or H-7 or H-2), 2.00 (m, 1H), 1.92–1.78 (m, 2H), 1.24 (s, 3H, H-3a or H-4a), 1.20 (s, 3H, H-3a or H-4a) ppm; 13C{1H} NMR (101 MHz, CDCl3): δ 135.4 (s, C-aryl), 135.2 (s, C-aryl), 133.4 (s, C-aryl quart.), 130.3 (s, C-aryl), 129.8 (s, C-aryl), 128.0 (s, C-aryl), 127.7 (s, C-aryl), 86.3 (s, C-quart.), 66.3 (s), 59.0 (d, JC,P = 6.1 Hz), 51.3 (d, 2JC,P = 19.5 Hz, C-quart.), 47.3 (d, 2JC,P = 2 Hz, C-5), 44.8 (d, 1JC,P = 46.7 Hz, C-6), 41.4 (d, 1JC,P = 44.8 Hz, C-2 or C-7), 40.3 (d, 1JC,P = 51.8 Hz, C-2 or C-7), 23.9 (d, 3JC,P = 7.2 Hz, C-3a or C-4a), 18.3 (d, 3JC,P = 15.9 Hz, C-3a or C-4a) ppm; 31P{1H} NMR (162 MHz, CDCl3): δ 43.4 (s) ppm; HRMS (ESI, MeCN), m/z: found: 491.1617, calculated for [M + H]+: 491.1624; found: 508.1878, calc. for [M + NH4]+: 508.1890; found: 513.1447, calc. for [M + Na]+: 513.1444; found: 998.3474, calc. for [2M + NH4]+: 998.3441; found: 1003.3032, calc. for [2M + Na]+: 1003.2996; IR (KBr, /cm−1): 3067 (w), 2975 (w), 2881 (w), 1588 (w), 1485 (w), 1427 (m), 1381 (w), 1369 (w), 1306 (w), 1250 (w), 1189 (w), 1114 (s), 1077 (s), 1053 (m), 1042 (m), 1012 (m), 996 (m), 958 (m), 928 (w), 881 (m), 862 (w), 841 (w), 800 (m), 775 (m), 738 (m), 709 (s), 697 (s), 675 (m), 619 (m), 609 (w), 582 (w), 506 (s), 481 (s), 448 (m), 435 (m).
5.2.2. Synthesis of 5b
- 1H NMR (400 MHz, CDCl3): δ 4.20–4.07 (m, 2H, H-5a/6a), 3.94 (m, 2H, H-5a/6a), 2.62–2.41 (m, 2H, H-5 or H-6), 2.31 (m, 1H, H-5 or H-6), 2.02 (m, 1H, H-2 or H-7), 1.96–1.83 (m, 2H, H-7 or H-2), 1.26 (s, 3H, H-3a or H-4a), 1.20 (s, 3H, H-3a or H-4a), 0.88 (s, 9H, H-9a), 0.09 (s, 6H, H-8) ppm; 13C{1H} NMR (101 MHz, CDCl3): δ 86.3 (s, C-quart.), 66.2 (s, C-5a), 58.1 (d, 2JC,P = 5.6 Hz, C-6a), 51.3 (d, 2JC,P = 19.4 Hz, C-quart.), 47.3 (d, 2JC,P = 2.3 Hz, C-5), 44.8 (d, 1JC,P = 47.0 Hz, C-6), 41.6 (d, 1JC,P = 44.8 Hz, C-2 or C-7), 40.3 (d, 1JC,P = 51.8 Hz, C-2 or C-7), 25.8 (s, C-9a), 23.9 (d, 3JC,P = 7.3 Hz, C-3a or C-4a), 18.3 (d, 3JC,P = 16.0 Hz, C-3a or C-4a), 18.1 (s, C-9), −5.5 (d, J = 6.1 Hz, C-8) ppm; 31P{1H} NMR (162 MHz, CDCl3) δ 43.6 (s) ppm; HRMS (ESI, MeCN), m/z: found: 347.1631, calc. for [M + H]+: 347.1624; found: 369.1451, calc. for [M + Na]+: 369.1444; IR (KBr, /cm−1): 2948 (m), 2925 (m), 2877 (m), 2853 (m), 1497 (w), 1468 (w), 1426 (w), 1383 (w), 1360 (w), 1311 (w), 1258 (m), 1245 (m), 1198 (w), 1162 (w), 1122 (m), 1077 (s), 1041 (m), 1029 (m), 1011 (m), 959 (m), 930 (w), 881 (s), 867 (s), 829 (m), 815 (m), 783 (s), 768 (s), 749 (m), 721 (s), 675 (s), 658 (s), 586 (w), 563 (w), 511 (m), 481 (w), 447 (m).
5.2.3. Synthesis of 6a
- 1H NMR (400 MHz, THF-d8): δ 7.54–7.44 (m, 3H), 7.39–7.33 (m, 3H), 7.33–7.19 (m, 6H), 7.13 (m, 3H), 3.86 (m, 2H, H-2 or H-7), 3.61–3.51 (m, 2H, H-5a or H-6a), 2.38 (m, 1H, H-6 or H-5), 2.13 (m, 1H, H-5 or H-6), 1.44–1.16 (m, 4H, H-2 or H-7, H-5a or H-6a), 0.99 (s, 3H, H-3a or H-4a), 0.97 (s, 3H, H-3a or H-4a) ppm; 13C{1H} NMR (101 MHz, THF-d8): δ 135.1 (s, C-aryl), 134.9 (s, C-aryl), 134.2 (s, C-aryl quart.), 129.6 (s, C-aryl), 129.5 (s, C-aryl), 127.5 (s, C-aryl), 127.4 (s, C-aryl), 86.9 (s, C-quart.), 63.7 (s), 62 (d, 1JC,P = 14.9 Hz, C-2 or C-7), 47.5 (d, 2JC,P = 3.3 Hz, C-5), 45.1 (d, 1JC,P = 13.5 Hz, C-6), 38 (d, 1JC,P = 16.0 Hz, C-2 or C-7), 36.7 (d, J = 6.6 Hz, C-6a or C-5a), 23.7 (s, C-3a or C-4a), 17.5 (s, C-3a or C-4a) ppm; 31P{1H} NMR (162 MHz, C6D6): δ −45.6 (s) ppm; HRMS (ESI, MeCN), m/z: found: 475.1795, calc. for [M + O + H]+: 475.1863; found: 497.1644, calc for [M + O + Na]+: 497.1672.
5.2.4. Synthesis of 6b
- 1H NMR (400 MHz, C6D6): δ 3.89–3.82 (m, 2H), 3.77–3.67 (m, 2H), 2.34 (m, 1H), 1.95–1.90 (m, 1H), 1.75 (dt, J = 15.4, 3.1 Hz, 1H, H-2 or H-7), 1.43–1.33 (m, 1H), 1.21–1.15 (m, 1H), 1.1 (s, 3H, H-3a or H-4a), 0.91 (s, 9H, H-8a), 0.89 (s, 3H, H-3a or H-4a), 0.29 (s, 6H, H-7) ppm; 13C{1H} NMR (101 MHz, C6D6): δ 64.1(s, C-5a), 61.3 (d, J = 15.0 Hz, C-6a), 47.6 (d, 2JC,P = 3.6 Hz, C-5), 45.2 (d, 1JC,P = 12.9 Hz, C-6), 38.52 (d, 1JC,P = 15.7 Hz, C-2 or C-7), 37.03 (d, 1JC,P = 6.4 Hz, C-2 or C-7), 25.7 (s, C-9a), 24.5 (s, C-3a or C-4a), 18.1 (s, C-3a or C-4a), −5.7 (d, J = 11.6 Hz, C-8) ppm; 31P NMR (162 MHz, C6D6): δ −45.6 (s) ppm; HRMS (ESI, MeCN), m/z: found: 337.1722, calc. for [M + Na]+: 337.1723; found: 353.1668, calc. for [M + O + Na]+: 353.1672; found: 369.1420, calc. for [M + O + K]+: 369.1412.
5.3. Catalysis
General Procedure for Hydrogenations
5.4. X-ray Crystallography Data
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Dutartre, M.; Bayardon, J.; Jugé, S. Applications and stereoselective syntheses of P-chirogenic phosphorus compounds. Chem. Soc. Rev. 2016, 45, 5771–5794. [Google Scholar] [CrossRef] [PubMed]
- Imamoto, T. Synthesis and applications of high-performance P-chiral phosphine ligands. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2021, 97, 520–542. [Google Scholar] [CrossRef] [PubMed]
- Cabré, A.; Verdaguer, X.; Riera, A. Recent Advances in the Enantioselective Synthesis of Chiral Amines via Transition Metal-Catalyzed Asymmetric Hydrogenation. Chem. Rev. 2022, 122, 269–339. [Google Scholar] [CrossRef] [PubMed]
- Kamer, P.C.J.; van Leeuwen, P.W.N.M. Phosphorus(III) Ligands in Homogeneous Catalysis; John Wiley & Sons: Hoboken, NJ, USA, 2012; ISBN 9780470666272. [Google Scholar]
- van Leeuwen, P.W.N.M.; Kamer, P.C.J.; Claver, C.; Pàmies, O.; Diéguez, M. Phosphite-containing ligands for asymmetric catalysis. Chem. Rev. 2011, 111, 2077–2118. [Google Scholar] [CrossRef]
- Tang, W.; Zhang, X. New chiral phosphorus ligands for enantioselective hydrogenation. Chem. Rev. 2003, 103, 3029–3070. [Google Scholar] [CrossRef]
- Xu, G.; Senanayake, C.H.; Tang, W. P-Chiral Phosphorus Ligands Based on a 2,3-Dihydrobenzo d1,3oxaphosphole Motif for Asymmetric Catalysis. Acc. Chem. Res. 2019, 52, 1101–1112. [Google Scholar] [CrossRef]
- Fu, W.; Tang, W. Chiral Monophosphorus Ligands for Asymmetric Catalytic Reactions. ACS Catal. 2016, 6, 4814–4858. [Google Scholar] [CrossRef]
- Horner, L.; Siegel, H.; Büthe, H. Asymmetric Catalytic Hydrogenation with an Optically Active Phosphinerhodium Complex in Homogeneous Solution. Angew. Chem. Int. Ed. 1968, 7, 942. [Google Scholar] [CrossRef]
- Knowles, W.S.; Sabacky, M.J. Catalytic asymmetric hydrogenation employing a soluble, optically active, rhodium complex. Chem. Commun. 1968, 1445–1446. [Google Scholar] [CrossRef]
- Vineyard, B.D.; Knowles, W.S.; Sabacky, M.J.; Bachman, G.L.; Weinkauff, D.J. Asymmetric hydrogenation. Rhodium chiral bisphosphine catalyst. J. Am. Chem. Soc. 1977, 99, 5946–5952. [Google Scholar] [CrossRef]
- Knowles, W.S. Asymmetric hydrogenations (Nobel lecture). Angew. Chem. Int. Ed. 2002, 41, 1999–2007. [Google Scholar] [PubMed]
- Noyori, R. Asymmetric Catalysis: Science and Opportunities (Nobel Lecture). Angew. Chem. Int. Ed. 2002, 41, 2008–2022. [Google Scholar] [CrossRef]
- Sandoval, C.A.; Ohkuma, T.; Muñiz, K.; Noyori, R. Mechanism of asymmetric hydrogenation of ketones catalyzed by BINAP/1,2-diamine-rutheniumII complexes. J. Am. Chem. Soc. 2003, 125, 13490–13503. [Google Scholar] [CrossRef] [PubMed]
- Akutagawa, S. Asymmetric synthesis by metal BINAP catalysts. Appl. Catal. A Gen. 1995, 128, 171–207. [Google Scholar] [CrossRef]
- Li, J.J. Noyori Asymmetric Hydrogenation. In Name Reactions; Li, J.J., Ed.; Springer International Publishing: Berlin/Heidelberg, Germany, 2021; pp. 399–402. ISBN 978-3-030-50864-7. [Google Scholar]
- Seo, C.S.G.; Morris, R.H. Catalytic Homogeneous Asymmetric Hydrogenation: Successes and Opportunities. Organometallics 2019, 38, 47–65. [Google Scholar] [CrossRef]
- Biosca, M.; Salomó, E.; de La Cruz-Sánchez, P.; Riera, A.; Verdaguer, X.; Pàmies, O.; Diéguez, M. Extending the Substrate Scope in the Hydrogenation of Unfunctionalized Tetrasubstituted Olefins with Ir-P Stereogenic Aminophosphine-Oxazoline Catalysts. Org. Lett. 2019, 21, 807–811. [Google Scholar] [CrossRef]
- Andersson, P.G. Iridium Catalysis; Springer: Berlin/Heidelberg, Germany, 2011; ISBN 9783642153341. [Google Scholar]
- Hu, X.-P.; Wang, D.-S.; Yu, C.-B.; Zhou, Y.-G.; Zheng, Z. Adventure in Asymmetric Hydrogenation: Synthesis of Chiral Phosphorus Ligands and Asymmetric Hydrogenation of Heteroaromatics. In Asymmetric Catalysis from a Chinese Perspective; Ma, S., Ed.; Scholars Portal: Berlin/Heidelberg, Germany, 2011; pp. 313–354. ISBN 978-3-642-19471-9. [Google Scholar]
- Akiyama, T.; Ojima, I. Catalytic Asymmetric Synthesis; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2022. [Google Scholar]
- Mazuela, J.; Verendel, J.J.; Coll, M.; Schäffner, B.; Börner, A.; Andersson, P.G.; Pàmies, O.; Diéguez, M. Iridium phosphite-oxazoline catalysts for the highly enantioselective hydrogenation of terminal alkenes. J. Am. Chem. Soc. 2009, 131, 12344–12353. [Google Scholar] [CrossRef]
- Crabtree, R. Iridium compounds in catalysis. Acc. Chem. Res. 1979, 12, 331–337. [Google Scholar] [CrossRef]
- Roseblade, S.J.; Pfaltz, A. Iridium-catalyzed asymmetric hydrogenation of olefins. Acc. Chem. Res. 2007, 40, 1402–1411. [Google Scholar] [CrossRef]
- Helmchen, G.; Pfaltz, A. Phosphinooxazolines—A new class of versatile, modular P,N-ligands for asymmetric catalysis. Acc. Chem. Res. 2000, 33, 336–345. [Google Scholar] [CrossRef]
- von Matt, P.; Pfaltz, A. Chiral Phosphinoaryldihydrooxazoles as Ligands in Asymmetric Catalysis: Pd-Catalyzed Allylic Substitution. Angew. Chem. Int. Ed. 1993, 32, 566–568. [Google Scholar] [CrossRef]
- Busacca, C.A.; Grossbach, D.; So, R.C.; O’Brien, E.M.; Spinelli, E.M. Probing electronic effects in the asymmetric Heck reaction with the BIPI ligands. Org. Lett. 2003, 5, 595–598. [Google Scholar] [CrossRef] [PubMed]
- Carroll, M.P.; Guiry, P.J. P,N ligands in asymmetric catalysis. Chem. Soc. Rev. 2014, 43, 819–833. [Google Scholar] [CrossRef]
- Elsevier, C.J.; de Vries, J.G. (Eds.) The Handbook of Homogeneous Hydrogenation; Wiley-VCH: Weinheim, Germany, 2007; ISBN 3527311610. [Google Scholar]
- Helmchen, G.; Kudis, S.; Sennhenn, P.; Steinhagen, H. Enantioselective catalysis with complexes of asymmetric P,N-chelate ligands. Pure Appl. Chem. 1997, 69, 513–518. [Google Scholar] [CrossRef]
- Peters, B.B.C.; Zheng, J.; Birke, N.; Singh, T.; Andersson, P.G. Iridium-catalyzed enantioconvergent hydrogenation of trisubstituted olefins. Nat. Commun. 2022, 13, 361. [Google Scholar] [CrossRef] [PubMed]
- Frank, D.J.; Franzke, A.; Pfaltz, A. Asymmetric hydrogenation using rhodium complexes generated from mixtures of monodentate neutral and anionic phosphorus ligands. Chem. Eur. J. 2013, 19, 2405–2415. [Google Scholar] [CrossRef]
- Minnaard, A.J.; Feringa, B.L.; Lefort, L.; de Vries, J.G. Asymmetric hydrogenation using monodentate phosphoramidite ligands. Acc. Chem. Res. 2007, 40, 1267–1277. [Google Scholar] [CrossRef] [PubMed]
- Giacomina, F.; Meetsma, A.; Panella, L.; Lefort, L.; de Vries, A.H.M.; de Vries, J.G. High enantioselectivity is induced by a single monodentate phosphoramidite ligand in iridium-catalyzed asymmetric hydrogenation. Angew. Chem. Int. Ed. 2007, 46, 1497–1500. [Google Scholar] [CrossRef]
- Komarov, I.V.; Börner, A. Highly Enantioselective or Not?—Chiral Monodentate Monophosphorus Ligands in the Asymmetric Hydrogenation. Angew. Chem. Int. Ed. 2001, 40, 1197–1200. [Google Scholar] [CrossRef]
- Cabré, A.; Riera, A.; Verdaguer, X. P-Stereogenic Amino-Phosphines as Chiral Ligands: From Privileged Intermediates to Asymmetric Catalysis. Acc. Chem. Res. 2020, 53, 676–689. [Google Scholar] [CrossRef]
- Xie, X.; Li, S.; Chen, Q.; Guo, H.; Yang, J.; Zhang, J. Synthesis and application of novel P-chiral monophosphorus ligands. Org. Chem. Front. 2022, 9, 1589–1592. [Google Scholar] [CrossRef]
- Murai, T. Axis-to-Center Chirality Transfer Reactions of Phosphates with a Binaphthyl Group and their Congeners: New Synthetic Routes to P-Chirogenic Organophosphorus Compounds. Chem. Lett. 2023, advance publication. [Google Scholar] [CrossRef]
- Mathey, F. The organic chemistry of phospholes. Chem. Rev. 1988, 88, 429–453. [Google Scholar] [CrossRef]
- Wonneberger, P.; König, N.; Kraft, F.B.; Sárosi, M.B.; Hey-Hawkins, E. Access to 1-Phospha-2-azanorbornenes by Phospha-aza-Diels-Alder Reactions. Angew. Chem. Int. Ed. 2019, 58, 3208–3211. [Google Scholar] [CrossRef] [PubMed]
- Ramazanova, K.; Lönnecke, P.; Hey-Hawkins, E. Facile Synthesis of Enantiomerically Pure P-Chiral 1-Alkoxy-2,3-dihydrophospholes via Nucleophilic P-N Bond Cleavage of a 1-Phospha-2-azanorbornene. Chem. Eur. J. 2023, 29, e202300790. [Google Scholar] [CrossRef]
- Wonneberger, P.; König, N.; Sárosi, M.B.; Hey-Hawkins, E. Reductive Rearrangement of a 1-Phospha-2-azanorbornene. Chem. Eur. J. 2021, 27, 7847–7852. [Google Scholar] [CrossRef]
- Möller, T.; Sárosi, M.B.; Hey-Hawkins, E. Asymmetric phospha-Diels-Alder reaction: A stereoselective approach towards P-chiral phosphanes through diastereotopic face differentiation. Chem. Eur. J. 2012, 18, 16604–16607. [Google Scholar] [CrossRef] [PubMed]
- Möller, T.; Wonneberger, P.; Kretzschmar, N.; Hey-Hawkins, E. P-chiral phosphorus heterocycles: A straightforward synthesis. Chem. Commun. 2014, 50, 5826–5828. [Google Scholar] [CrossRef]
- Möller, T.; Wonneberger, P.; Sárosi, M.B.; Coburger, P.; Hey-Hawkins, E. P-chiral 1-phosphanorbornenes: From asymmetric phospha-Diels-Alder reactions towards ligand design and functionalisation. Dalton Trans. 2016, 45, 1904–1917. [Google Scholar] [CrossRef]
- Denmark, S.E.; Hammer, R.P.; Weber, E.J.; Habermas, K.L. Diphenylmethylsilyl ether (DPMS): A protecting group for alcohols. J. Org. Chem. 1987, 52, 165–168. [Google Scholar] [CrossRef]
- Bols, M.; Pedersen, C.M. Silyl-protective groups influencing the reactivity and selectivity in glycosylations. Beilstein J. Org. Chem. 2017, 13, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Crouch, R.D. Recent Advances in Silyl Protection of Alcohols. Synth. Commun. 2013, 43, 2265–2279. [Google Scholar] [CrossRef]
- Wuts, P.G.; Green, T.W. Greene’s Protective Groups in Organic Synthesis, 4th ed.; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2007; ISBN 0471697540. [Google Scholar]
- Davies, J.S.; Higginbotham, C.L.; Tremeer, E.J.; Brown, C.; Treadgold, R.C. Protection of hydroxy groups by silylation: Use in peptide synthesis and as lipophilicity modifiers for peptides. J. Chem. Soc. Perkin Trans. 1 1992, 3043–3048. [Google Scholar] [CrossRef]
- Patschinski, P.; Zhang, C.; Zipse, H. The Lewis base-catalyzed silylation of alcohols—A mechanistic analysis. J. Org. Chem. 2014, 79, 8348–8357. [Google Scholar] [CrossRef] [PubMed]
- Corey, E.J.; Venkateswarlu, A. Protection of hydroxyl groups as tert-butyldimethylsilyl derivatives. J. Am. Chem. Soc. 1972, 94, 6190–6191. [Google Scholar] [CrossRef]
- Weinhold, F.; West, R. The Nature of the Silicon–Oxygen Bond. Organometallics 2011, 30, 5815–5824. [Google Scholar] [CrossRef]
- Kaftory, M.; Kapon, M.; Botoshansky, M. The Structural Chemistry of Organosilicon Compounds. In The Chemistry of Organic Silicon Compounds; Patai, S., Rappoport, Z., Eds.; Wiley: Chichester, UK, 1998; pp. 181–265. ISBN 9780471967576. [Google Scholar]
- Wender, P.A.; Bi, F.C.; Brodney, M.A.; Gosselin, F. Asymmetric synthesis of the tricyclic core of NGF-inducing cyathane diterpenes via a transition-metal-catalyzed 5 + 2 cycloaddition. Org. Lett. 2001, 3, 2105–2108. [Google Scholar] [CrossRef]
- de Vries, E.F.J.; Brussee, J.; van der Gen, A. Intramolecular Reductive Cleavage of tert-Butyldimethylsilyl Ethers. Selective Mono-Deprotection of Bis-Silyl-Protected Diols. J. Org. Chem. 1994, 59, 7133–7137. [Google Scholar] [CrossRef]
- Crouch, R.D. Selective deprotection of silyl ethers. Tetrahedron 2013, 69, 2383–2417. [Google Scholar] [CrossRef]
- Pfaltz, A.; Blankenstein, J.; Hilgraf, R.; Hörmann, E.; McIntyre, S.; Menges, F.; Schönleber, M.; Smidt, S.P.; Wüstenberg, B.; Zimmermann, N. Iridium-Catalyzed Enantioselective Hydrogenation of Olefins. Adv. Synth. Catal. 2003, 345, 33–43. [Google Scholar] [CrossRef]
- Peters, B.B.C.; Andersson, P.G. The Implications of the Brønsted Acidic Properties of Crabtree-Type Catalysts in the Asymmetric Hydrogenation of Olefins. J. Am. Chem. Soc. 2022, 144, 16252–16261. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.K.; Becker, E.D.; Cabral de Menezes, S.M.; Goodfellow, R.; Granger, P. NMR Nomenclature: Nuclear Spin Properties and Conventions for Chemical Shifts. IUPAC Recommendations 2001. Solid State Nucl. Magn. Reson. 2002, 22, 458–483. [Google Scholar] [CrossRef] [PubMed]
- Rigaku Corporation. CrysAlisPro Software System; Rigaku Oxford Diffraction: Wroclaw, Poland, 1995–2023. [Google Scholar]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta. Cryst. Sect. A Found. Adv. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Brandenburg, K. Crystal Impact GbR, version 4.6.8; DIAMOND 4: Bonn, Germany.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
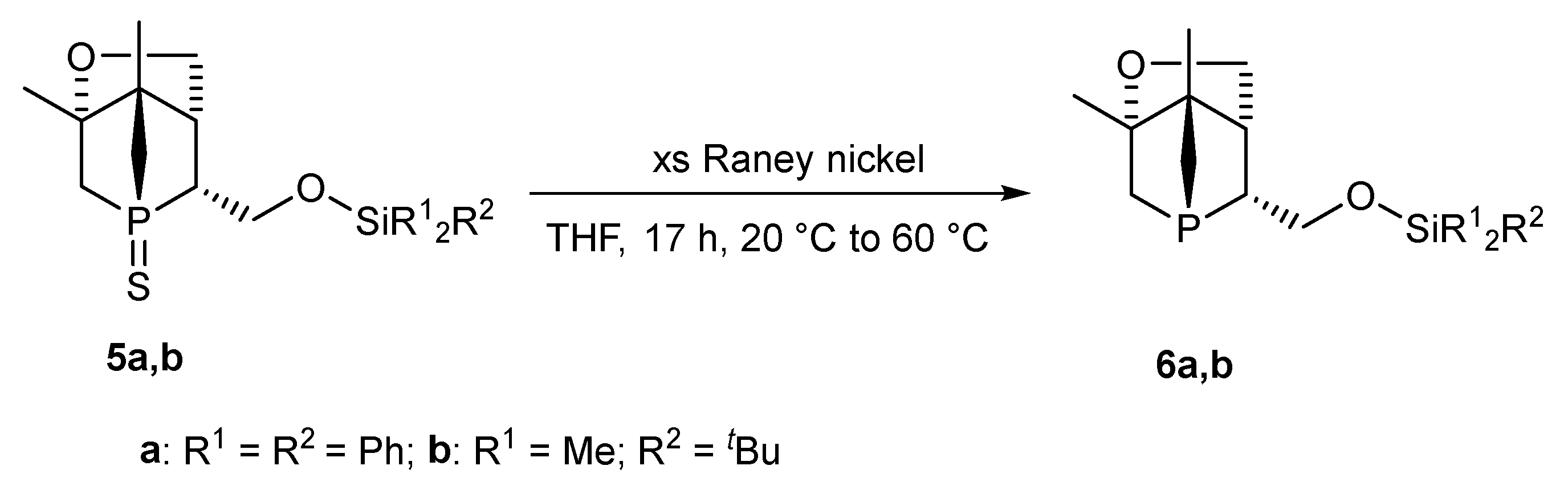{kind=link}
{kind=link}
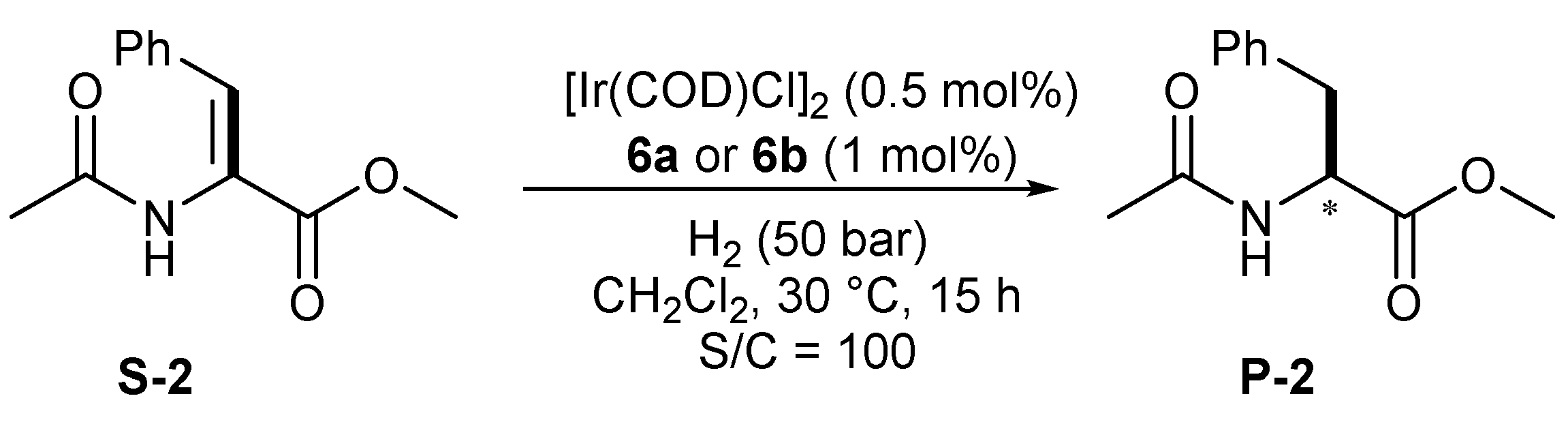{kind=link}
| Entry | M/L | Conversion | ee |
|---|---|---|---|
| 1 | 1:3 | - | - |
| 2 a | 1:2 | - | - |
| 3 a | 1:3 | 6% | racemate |
| 4 b | 1:3 | 20% | racemate |
| Entry | Solvent | Conversion | ee |
|---|---|---|---|
| 1 a | CH2Cl2 | 98% | 8% |
| 2 b | CH2Cl2 | >99% | 9% |
| 3 b | MeOH | 90% | 8% |
| 4 b | THF | 50% | - |
| 5 c | CH2Cl2 | 95% | 8% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramazanova, K.; Chakrabortty, S.; Kallmeier, F.; Kretzschmar, N.; Tin, S.; Lönnecke, P.; de Vries, J.G.; Hey-Hawkins, E. Access to Enantiomerically Pure P-Chiral 1-Phosphanorbornane Silyl Ethers. Molecules 2023, 28, 6210. https://doi.org/10.3390/molecules28176210
Ramazanova K, Chakrabortty S, Kallmeier F, Kretzschmar N, Tin S, Lönnecke P, de Vries JG, Hey-Hawkins E. Access to Enantiomerically Pure P-Chiral 1-Phosphanorbornane Silyl Ethers. Molecules. 2023; 28(17):6210. https://doi.org/10.3390/molecules28176210
Chicago/Turabian StyleRamazanova, Kyzgaldak, Soumyadeep Chakrabortty, Fabian Kallmeier, Nadja Kretzschmar, Sergey Tin, Peter Lönnecke, Johannes G. de Vries, and Evamarie Hey-Hawkins. 2023. "Access to Enantiomerically Pure P-Chiral 1-Phosphanorbornane Silyl Ethers" Molecules 28, no. 17: 6210. https://doi.org/10.3390/molecules28176210