
Coordinatively Unsaturated Nickel Nitroxyl Complex: Structure, Physicochemical Properties, and Reactivity toward Dioxygen
 ,
, 
Abstract
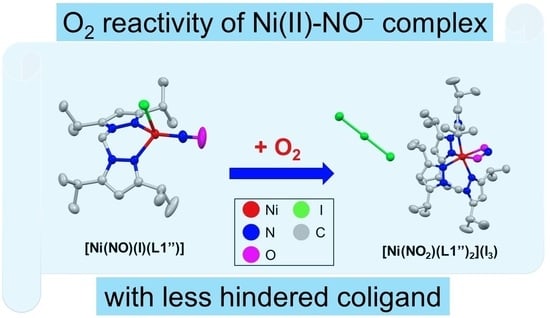
:
1. Introduction
2. Results and Discussion
2.1. Synthesis of Ni–NO Complex
2.2. Structure of Ni-NO Complex
2.3. Characterizations of Ni-NO Complex
2.4. Dioxygen Reactivity of Ni-NO Complex
2.5. Structure of Ni-NO2 Complex
2.6. Characterizations of Ni-NO2 Complex
2.7. Theoretical Considerations
2.7.1. Density Functional Theory (DFT) Calculations for [Ni(NO)(I)(L1″)]
2.7.2. DFT Calculations for [Ni(κ2-O2N)(L1″)2]+
3. Materials and Methods
3.1. Material and General Techniques
3.2. Instrumentation
3.3. Theoretical Calculations
3.4. Preparation of Ligand and Complexes
3.4.1. [Ni(NO)(I)]n
3.4.2. [Ni(NO)(I)(L1″)]
3.4.3. [Ni(κ2-O2N)(L1″)2](I3)
3.5. X-ray Crystal Structure Determination
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Feldman, P.L.; Griffith, O.W.; Stuehr, D.J. The surprising life of nitric oxide. Chem. Eng. News 1993, 71, 26–38. [Google Scholar]
- Beckman, J.S.; Koppenol, W.H. Nitric oxide, superoxide, and peroxynitrite: The good, the bad, and the ugly. Am. J. Physiol. 1996, 271, C1424–C1437. [Google Scholar] [CrossRef] [PubMed]
- Ignarro, L. Nitric Oxide: Biology and Pathobiology; Academic Press: San Diego, CA, USA, 2000. [Google Scholar]
- Li, L.; Li, L. Recent advances in multinuclear metal nitrosyl complexes. Coord. Chem. Rev. 2016, 306, 678–700. [Google Scholar] [CrossRef] [PubMed]
- Maia, L.B.; Moura, J.J.G. How biology handles nitrite. Chem. Rev. 2014, 114, 5273–5357. [Google Scholar] [CrossRef]
- Tsai, M.-L.; Tsou, C.-C.; Liaw, W.-F. Dinitrosyl iron complexes (DNICs): From biomimetic synthesis and spectroscopic characterization toward unveiling the biological and catalytic roles of DNICs. Acc. Chem Res. 2015, 48, 1184–1193. [Google Scholar] [CrossRef]
- Lehnert, N.; Fujisawa, K.; Camarena, S.; Dong, H.T.; White, C.J. Activation of non-heme iron-nitrosyl complexes: Turning up the heat. ACS Catal. 2019, 9, 10499–10518. [Google Scholar] [CrossRef]
- Ferousi, C.; Majer, S.H.; DiMucci, I.M.; Lancaster, K.M. Biological and bioinspired inorganic N–N bond-forming reactions. Chem. Rev. 2020, 120, 5252–5307. [Google Scholar] [CrossRef]
- Pauleta, S.R.; Carepo, M.S.P.; Moura, I. Source and reduction of nitrous oxide. Coord. Chem. Rev. 2019, 387, 436–449. [Google Scholar] [CrossRef]
- Lehnert, N.; Kim, K.; Dong, H.T.; Harland, J.B.; Hunt, A.P.; Manickas, E.C.; Oakley, K.M.; Pham, J.; Reed, G.C.; Alfaro, V.C. The biologically relevant coordination chemistry of iron and nitric oxide: Electronic structure and reactivity. Chem. Rev. 2021, 121, 14682–14905. [Google Scholar] [CrossRef]
- Jørgensen, C.K. Differences between the four halide ligands, and discussion remarks on trigonal-bipyramidal complexes, on oxidation states, and on diagonal elements of one-electron energy. Coord. Chem. Rev. 1966, 1, 164–178. [Google Scholar] [CrossRef]
- Kaim, W.; Schwederski, B. Non-innocent ligands in bioinorganic chemistry—An overview. Coord. Chem. Rev. 2010, 254, 1580–1588. [Google Scholar] [CrossRef]
- Mingos, D.M.P. A review of complexes of ambivalent and ambiphilic Lewis acid/bases with symmetry signatures and an alternative notation for these noninnocent ligands. J. Organomet. Chem. 2014, 751, 153–173. [Google Scholar] [CrossRef]
- Enemark, J.H.; Feltham, R.D. Principles of structure, bonding, and reactivity for metal nitrosyl complexes. Coord. Chem. Rev. 1974, 13, 339–406. [Google Scholar] [CrossRef]
- Matsunaga, Y.; Fujisawa, K.; Ibi, N.; Miyashita, Y.; Okamoto, K. Structural and spectroscopic characterization of first-row transition metal(II) substituted blue copper model complexes with hydrotris(pyrazolyl)borate. Inorg. Chem. 2005, 44, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Gorelsky, S.I.; Basumallick, L.; Vura-Weis, J.; Sarangi, R.; Hodgson, K.O.; Hedman, B.; Fujisawa, K.; Solomon, E.I. Spectroscopic and DFT investigation of [M{HB(3,5-iPr2pz)3}(SC6F5)] (M = Mn, Fe, Co, Ni, Cu, and Zn) model complexes: Periodic trends in metal–thiolate bonding. Inorg. Chem. 2005, 44, 4947–4960. [Google Scholar] [CrossRef] [PubMed]
- Kitajima, N.; Fujisawa, K.; Fujimoto, C.; Moro-oka, Y.; Hashimoto, S.; Kitagawa, T.; Toriumi, T.; Tatsumi, K.; Nakamura, A. A new model for dioxygen binding in hemocyanin. Synthesis, characterization, and molecular structure of the μ-η2:η2 peroxo dinuclear copper(II) complexes, [Cu(HB(3,5-R2pz)3)]2(O2) (R = i-Pr and Ph). J. Am. Chem. Soc. 1992, 114, 1277–1291. [Google Scholar] [CrossRef]
- Irving, H.; Williams, R.J.P. Order of stability of metal complexes. Nature 1948, 162, 746–747. [Google Scholar] [CrossRef]
- Imai, S.; Fujisawa, K.; Kobayashi, T.; Shirasawa, N.; Fujii, H.; Yoshimura, T.; Kitajima, N.; Moro-oka, Y. 63Cu NMR study of copper(I) carbonyl complexes with various hydrotris(pyrazolyl)borates: Correlation between 63Cu chemical shifts and CO stretching vibrations. Inorg. Chem. 1998, 37, 3066–3070. [Google Scholar] [CrossRef]
- Fujisawa, K.; Soma, S.; Kurihara, H.; Ohta, A.; Dong, H.T.; Minakawa, Y.; Zhao, J.; Alp, E.E.; Hu, M.Y.; Lehnert, N. Stable ferrous mononitroxyl {FeNO}8 complex with a hindered hydrotris(pyrazolyl)borate coligand: Structure, spectroscopic characterization, and reactivity toward NO and O2. Inorg. Chem. 2019, 58, 4059–4062. [Google Scholar] [CrossRef]
- Fujisawa, K.; Soma, S.; Kurihara, K.; Dong, H.T.; Bilodeau, M.; Lehnert, N. A cobalt–nitrosyl complex with a hindered hydrotris(pyrazolyl)borate coligand: Detailed electronic structure, and reactivity towards dioxygen. Dalton Trans. 2017, 46, 13273–13289. [Google Scholar] [CrossRef]
- Soma, S.; Stappen, C.V.; Kiss, M.; Szilagyi, R.K.; Lehnert, N.; Fujisawa, K. Distorted tetrahedral nickel-nitrosyl complexes: Spectroscopic characterization and electronic structure. J. Biol. Inorg. Chem. 2016, 21, 757–775. [Google Scholar] [CrossRef] [PubMed]
- Fujisawa, K.; Tateda, A.; Miyashita, Y.; Okamoto, K.; Paulat, F.; Praneeth, V.N.N.; Merkle, A.; Lehnert, N. Structural and spectroscopic characterization of mononuclear copper(I) nitrosyl complexes: End-on versus side-on coordination of NO to copper(I). J. Am. Chem. Soc. 2008, 130, 1205–1213. [Google Scholar] [CrossRef] [PubMed]
- Lehnert, N.; Cornelissen, U.; Neese, F.; Ono, T.; Noguchi, Y.; Okamoto, K.; Fujisawa, K. Synthesis and spectroscopic characterization of copper(II)-nitrito complexes with hydrotris(pyrazolyl)borate and related coligands. Inorg. Chem. 2007, 46, 3916–3933. [Google Scholar] [CrossRef] [PubMed]
- Fujisawa, K.; Tada, N.; Ishikawa, Y.; Higashimura, H.; Miyashita, Y.; Okamoto, K. The most hindered hydrotris(pyrazolyl)borate ligand, X-ray structure of chlorocopper(II) complex: [Cu(Cl){HB(3-Ad-5-Pripz)3}] as compared with [Cu(Cl){HB(3-But-5-Pripz)3}]. Inorg. Chem. Commun. 2004, 7, 209–212. [Google Scholar] [CrossRef]
- Fujisawa, K.; Noguchi, Y.; Miyashita, Y.; Okamoto, O.; Lehnert, N. Mononuclear and binuclear copper(I) complexes ligated by bis(3,5-diisopropyl-1-pyrazolyl)methane: Insight into the fundamental coordination chemistry of three-coordinate copper(I) complexes with a neutral coligand. Inorg. Chem. 2007, 46, 10607–10623. [Google Scholar] [CrossRef]
- Fujisawa, K.; Kataoka, T.; Terashima, K.; Tiekink, E.R.T. The crystal structure of nitroxyl-κN-{hydridotris(3-trifluoromethyl-5-methylpyrazolyl-1-yl-κN3)borato}nickel(II), C15H13BF9N7NiO. Z. Kristallogr.-N. Cryst. Struct. 2022, 237, 1075–1078. [Google Scholar]
- Landry, V.K.; Pang, K.; Quan, S.M.; Parkin, G. Tetrahedral nickel nitrosyl complexes with tripodal [N3] and [Se3] donor ancillary ligands: Structural and computational evidence that a linear nitrosyl is a trivalent ligand. Dalton Trans. 2007, 8, 820–824. [Google Scholar] [CrossRef]
- Tomson, N.C.; Crimmin, M.R.; Petrenko, T.; Rosebrugh, L.E.; Sproules, S.; Boyd, W.R.; Bergman, R.G.; DeBeer, S.; Toste, F.D.; Wieghardt, K. A Step beyond the Feltham–Enemark notation: Spectroscopic and correlated ab initio computational support for an antiferromagnetically coupled M(II)–(NO)– description of Tp*M(NO) (M = Co, Ni). J. Am. Chem. Soc. 2011, 133, 18785–18801. [Google Scholar] [CrossRef]
- Griego, L.; Woods, T.J.; Mirica, L.M. A five-coordinate Ni(I) complex supported by 1,4,7-triisopropyl-1,4,7-triazacyclononane. Chem. Commun. 2022, 58, 7360–7363. [Google Scholar] [CrossRef]
- Maffett, L.S.; Gunter, K.L.; Kreisel, K.A.; Yap, G.P.A.; Rabinovich, D. Nickel nitrosyl complexes in a sulfur-rich environment: The first poly(mercaptoimidazolyl)borate derivatives. Polyhedron 2007, 26, 4758–4764. [Google Scholar] [CrossRef]
- Nieto, I.; Bontchev, R.P.; Ozarowski, A.; Smirnov, D.; Krzystek, J.; Telser, J.; Smith, J.M. Synthesis and spectroscopic investigations of four-coordinate nickel complexes supported by a strongly donating scorpionate ligand. Inorg. Chim. Acta 2009, 362, 4449–4460. [Google Scholar] [CrossRef]
- Muñoz, S.B., III; Foster, W.K.; Lin, H.-J.; Margarit, C.G.; Dickie, D.A.; Smith, J.M. Tris(carbene)borate ligands featuring imidazole-2-ylidene, benzimidazol-2-ylidene and 1,3,4-triazol-2-ylidene donors. evaluation of donor properties in four-coordinate {NiNO}10 complexes. Inorg. Chem. 2012, 51, 12660–12668. [Google Scholar] [CrossRef] [PubMed]
- Enemark, J.H. Four-coordinate metal nitrosyls. I. the structure of azidonitrosylbis(triphenylphosphine)nickel, Ni(N3)(NO)(P(C6H5)3). Inorg. Chem. 1971, 10, 1952–1957. [Google Scholar] [CrossRef]
- Haymore, B.; Feltham, R.D. Nirosyliron, -cobalt, and -nickel iodides. Inorg. Synth. 1973, 14, 81–89. [Google Scholar]
- Kurihara, H.; Ohta, A.; Fujisawa, K. Structures and properties of dinitrosyl iron and cobalt complexes ligated by bis(3,5-diisopropyl-1-pyrazolyl)methane. Inorganics 2019, 7, 116. [Google Scholar] [CrossRef]
- Yang, L.; Powell, D.R.; Houser, R.P. Structural variation in copper(I) complexes with pyridylmethylamide ligands: Structural analysis with a new four-coordinate geometry index, τ4. Dalton Trans. 2007, 7, 955–964. [Google Scholar] [CrossRef]
- Tasman, H.A.; Boswijk, K.H. Re-investigation of the crystal structure of CsI3. Acta Cryst. 1955, 8, 59–60. [Google Scholar] [CrossRef]
- Maki, A.G.; Forneris, R. Infrared and spectra of some trihalide ions: ICl2−,IBr2−, I3−, I2Br−, and BrICl−. Spectrochim. Acta Part A 1967, 23, 867–880. [Google Scholar] [CrossRef]
- Thyagarajan, S.; Incarvito, C.D.; Rheingold, A.L.; Theopold, K.H. In pursuit of a stable peroxynitrite complex–NOx (x = 1−3) derivatives of Tpt-Bu,MeCo. Inorg. Chim. Acta 2003, 345, 333–339. [Google Scholar] [CrossRef]
- Kiefer, W.; Bernstein, H.J. The UV-laser excited resonance Raman spectrum of the I3– ion. Chem. Phys. Lett. 1972, 16, 5–9. [Google Scholar] [CrossRef]
- Andrews, L.; Prochaska, E.S.; Loewenschuss, A. Resonance Raman and ultraviolet absorption spectra of the triiodide ion produced by alkali iodide-iodine argon matrix reactions. Inorg. Chem. 1980, 19, 463–465. [Google Scholar] [CrossRef]
- Kaya, K.; Mikami, N.; Udagawa, Y.; Ito, M. Resonance Raman effect of I3− ion by ultraviolet laser excitation. Chem. Phys. Lett. 1972, 16, 151–153. [Google Scholar] [CrossRef]
- Gabes, W.; Stufkens, D.J. Electronic absorption spectra of symmetrical and asymmetrical trihalide ions. Spectrochim. Acta Part A 1974, 30, 1835–1841. [Google Scholar] [CrossRef]
- Stappen, C.V.; Lehnert, N. Mechanism of N-N bond formation by transition metal-nitrosyl complexes: Modeling flavodiiron nitric oxide reductases. Inorg. Chem. 2018, 57, 4252–4269. [Google Scholar] [CrossRef]
- Armarego, W.L.F.; Chai, C.L.L. Purification of Laboratory Chemicals, 7th ed.; Butterworth-Heinemann: Oxford, UK, 2012. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Schäfer, A.; Horn, H.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets for atoms Li to Kr. J. Chem. Phys. 1992, 97, 2571–2577. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Perdew, J.P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 33, 8822–8824. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. the role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- CrystalClear, Data Collection and Processing Software; Rigaku Corporation: Tokyo, Japan, 2001.
- CrysAlisPro, Data Collection and Processing Software; Rigaku Corporation: Tokyo, Japan, 2015.
- Burla, M.C.; Caliandro, R.; Camalli, M.; Carrozzini, B.; Cascarano, G.L.; De Caro, L.; Giacovazzo, C.; Polidori, G.; Siliqi, D.; Spagna, R. IL MILIONE: A suite of computer programs for crystal structure solution of proteins. J. Appl. Crystallogr. 2007, 40, 609–613. [Google Scholar] [CrossRef]
- Crystal Structure, 4.3. Crystal Structure Analysis Package. Rigaku Corporation: Tokyo, Japan, 2003.
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. 2015, C71, 3–8. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complexes a | Ligand Type | d (Ni–N)/Å b | d (N–O)/ Å b | ∠ (Ni–N–O)/ ° b | ν(N–O)/ cm−1 b | {MNO}n | Ref. |
|---|---|---|---|---|---|---|---|
| [Ni(NO)(I)(L1″)] | (NO)N2I | 1.6467 (16) | 1.136 (3) | 176.68 (19) | 1777 (KBr) | {NiNO}10 | tw c |
| [Ni(NO)(L3)] | (NO)N3 | 1.651 (6) | 1.160 (10) | 179.1 (7) | 1780 (KBr) | {NiNO}10 | [22] |
| [Ni(NO)(L0f)] | (NO)N3 | 1.6361 (16) | 1.160 (2) | 172.42 (17) | 1823 (KBr) | {NiNO}10 | [27] |
| [Ni(NO)(L0)] | (NO)N3 | 1.619 (6), 1.617 (6) | 1.170 (7), 1.158 (7) | 178.5 (6), 175.3 (7) | 1786 (KBr) | na d | [28] |
| {NiNO}10 | [29] | ||||||
| [Ni(NO)(iPr3tacn)](PF6) | (NO)N3 | 1.677 (4) 1.646 (3) | 1.123 (5) 1.162 (4) | 180.0 170.9 (2) | 1770 (ATR) | {NiNO}10 | [30] |
| [Ni(NO)(Tmp-tol)] | (NO)S3 | 1.665 (3) | 1.131 (4) | 173.9 (4) | 1752 (KBr) | {NiNO}10 | [31] |
| [Ni(NO)(TseMes)] | (NO)Se3 | 1.633 (4) | 1.156 (5) | 180.0 | 1763, 1752 (KBr) | na d | [28] |
| [Ni(NO){HB(tBuIm)3}] | (NO)C3 | 1.620 (5) | 1.184 (7) | 178.5 (4) | 1703 (toluene) | {NiNO}10 | [32] |
| [Ni(NO){PhB(CyCH2Im)3}] | (NO)C3 | 1.633 (9)–1.668 (1) | 1.174 (1)–1.197 (1) | 172.7 (1)–177.8 (1) | 1693 (THF) | {NiNO}10 | [33] |
| [Ni(NO){HB(MeBz)3}] | (NO)C3 | 1.643 (2)–1.646 (2) | 1.183 (3)–1.191 (3) | 169.3 (2)–174.8 (2) | 1714 (THF) | {NiNO}10 | [33] |
| [Ni(NO){HB(p-tBuPhTz)3}] | (NO)C3 | 1.640 (2) | 1.163 (3) | 176.3 (3) | 1746 (THF) | {NiNO}10 | [33] |
| Method a | d (Ni–N) /Å | d (N–O) /Å | ∠ (Ni–N–O) /° | ν(N–O) /cm−1 | ν(Ni–NO) /cm−1 | δ(Ni–N–O) /cm−1 | SD b (Ni) | SD b (NO) |
|---|---|---|---|---|---|---|---|---|
| Exp. | 1.647 | 1.135 | 176.7 | 1777 | 572 | 410 | ||
| BP86 (CS) | 1.646 | 1.172 | 165.1 | 1799 | 618 | 410 | 0 | 0 |
| B3LYP (CS) | 1.630 | 1.156 | 163.8 | 1883 | 651 | 412 | 0 | 0 |
| B3LYP (BS) | 1.720 | 1.169 | 167.1 | 1820 | 439 | 356 | 1.05 | −1.19 |
| Method | d (Ni–O1) /Å | d (Ni–O2) /Å | d (N1–O1) /Å | d (N1–O2) /Å | ∠ (O1–N1–O2) /° | νas(O–N–O) /cm−1 |
|---|---|---|---|---|---|---|
| Exp. | 2.088 | 2.120 | 1.256 | 1.267 | 111.5 | 1201 |
| BP86 | 2.178 | 2.107 | 1.268 | 1.274 | 111.8 | 1166 |
| Complex | [Ni(NO)(I)(L1″)](thf) | [Ni(κ2-NO2)(L1″)2](I3) |
|---|---|---|
| CCDC number | 2285305 | 2285306 |
| Empirical formula | C23H40IN5NiO2 | C38H64I3N9NiO2 |
| Formula weight | 604.21 | 1118.40 |
| Crystal system | Triclinic | Monoclinic |
| Space group | P (#2) | P21/n (#14) |
| a/Å | 9.02661 (11) | 9.3444 (2) |
| b/Å | 9.61413 (11) | 17.2498 (3) |
| c/Å | 16.4456 (2) | 30.0610 (5) |
| α/° | 77.6870 (10) | 90 |
| β/° | 89.2370 (10) | 86.7699 (17) |
| γ/° | 87.2540 (10) | 90 |
| V/Å3 | 1392.76 (3) | 4837.81 (16) |
| Z | 2 | 4 |
| Dcalc/g cm−3 | 1.441 | 1.535 |
| μ(MoKα)/cm−1 | 18.318 | 23.555 |
| 2θ range, ° | 6–55 | 6–55 |
| Reflections collected | 45,524 | 115,535 |
| Unique reflections | 6385 | 10,976 |
| Rint | 0.0335 | 0.0280 |
| Number of variables | 289 | 478 |
| Refls./Para. ratio | 22.09 | 22.96 |
| Residuals: R1 (I > 2 σ (I)) a | 0.0233 | 0.0747 |
| Residuals: R (All refl.) | 0.0247 | 0.0843 |
| Residuals: wR2 (All refl.) a | 0.0653 | 0.1898 |
| Goodness of fit ind. | 1.064 | 1.103 |
| Max/min peak, /e Å−3 | 0.76/−0.30 | 2.46/−1.05 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fujisawa, K.; Kataoka, T.; Terashima, K.; Kurihara, H.; de Santis Gonçalves, F.; Lehnert, N. Coordinatively Unsaturated Nickel Nitroxyl Complex: Structure, Physicochemical Properties, and Reactivity toward Dioxygen. Molecules 2023, 28, 6206. https://doi.org/10.3390/molecules28176206
Fujisawa K, Kataoka T, Terashima K, Kurihara H, de Santis Gonçalves F, Lehnert N. Coordinatively Unsaturated Nickel Nitroxyl Complex: Structure, Physicochemical Properties, and Reactivity toward Dioxygen. Molecules. 2023; 28(17):6206. https://doi.org/10.3390/molecules28176206
Chicago/Turabian StyleFujisawa, Kiyoshi, Taisei Kataoka, Kohei Terashima, Haruka Kurihara, Felipe de Santis Gonçalves, and Nicolai Lehnert. 2023. "Coordinatively Unsaturated Nickel Nitroxyl Complex: Structure, Physicochemical Properties, and Reactivity toward Dioxygen" Molecules 28, no. 17: 6206. https://doi.org/10.3390/molecules28176206