
Immunomodulatory Gene-Splicing Dysregulation in Tumorigenesis: Unmasking the Complexity

 , ,
, ,  , and
, and
Abstract
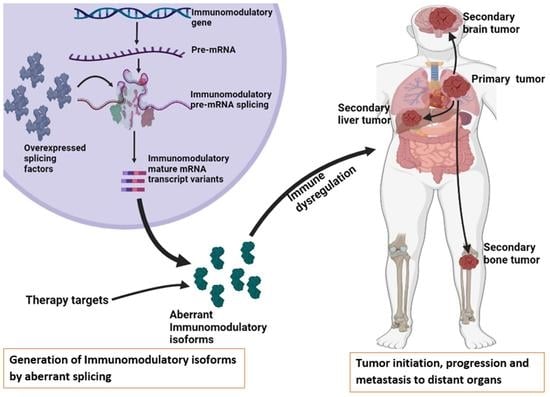
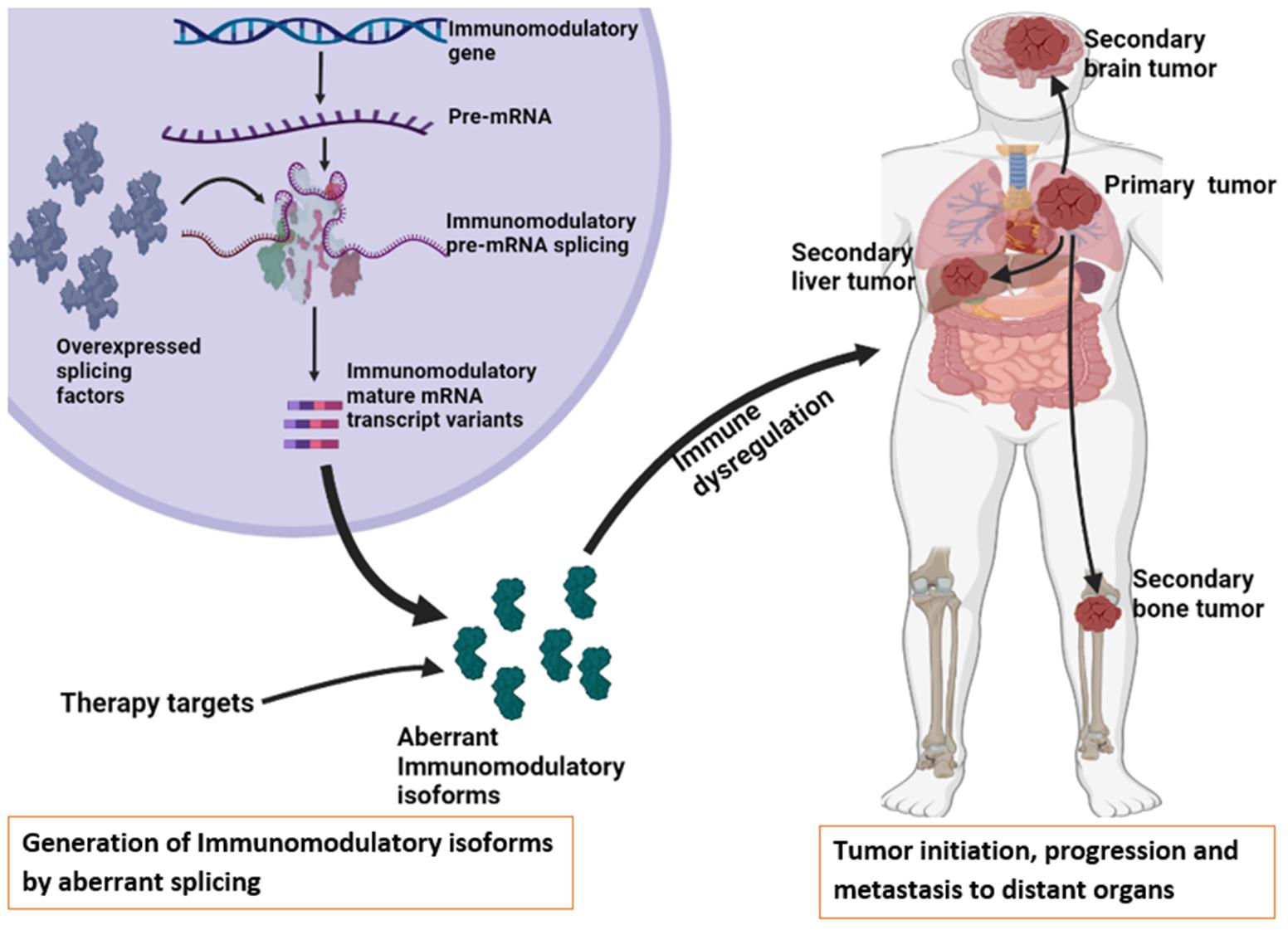
:
1. Introduction
2. Methodology
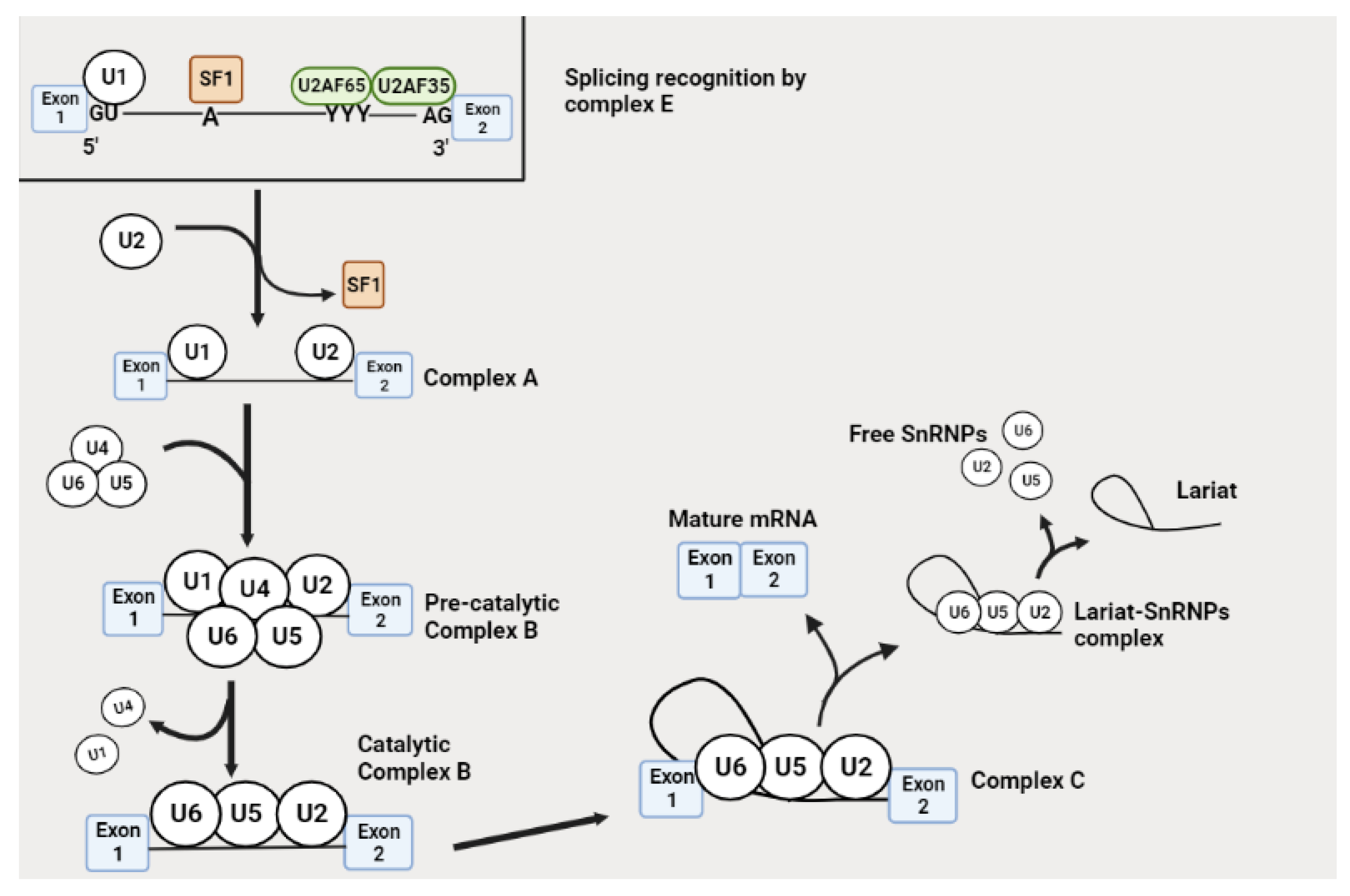
3. Alternative Splicing Mechanism
4. Alternative Splicing in Immunomodulatory Genes
4.1. Cytotoxic T Lymphocyte-Associated Protein 4 (CTLA-4)
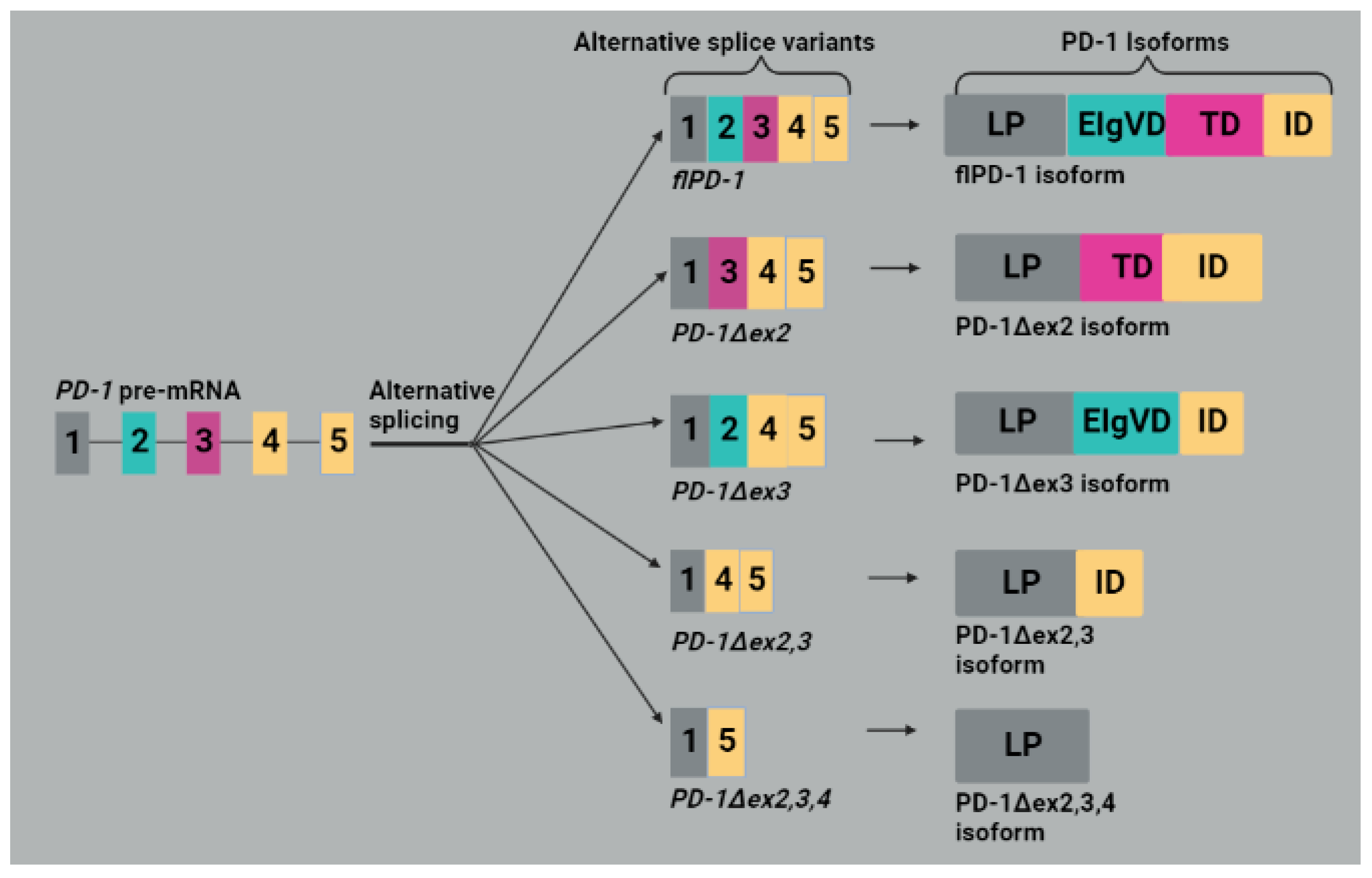
4.2. Programmed Death 1 (PD-1)
4.3. Programmed Death Ligand 1 (PD-L1)
4.4. Human Leukocyte Antigen G (HLA-G)
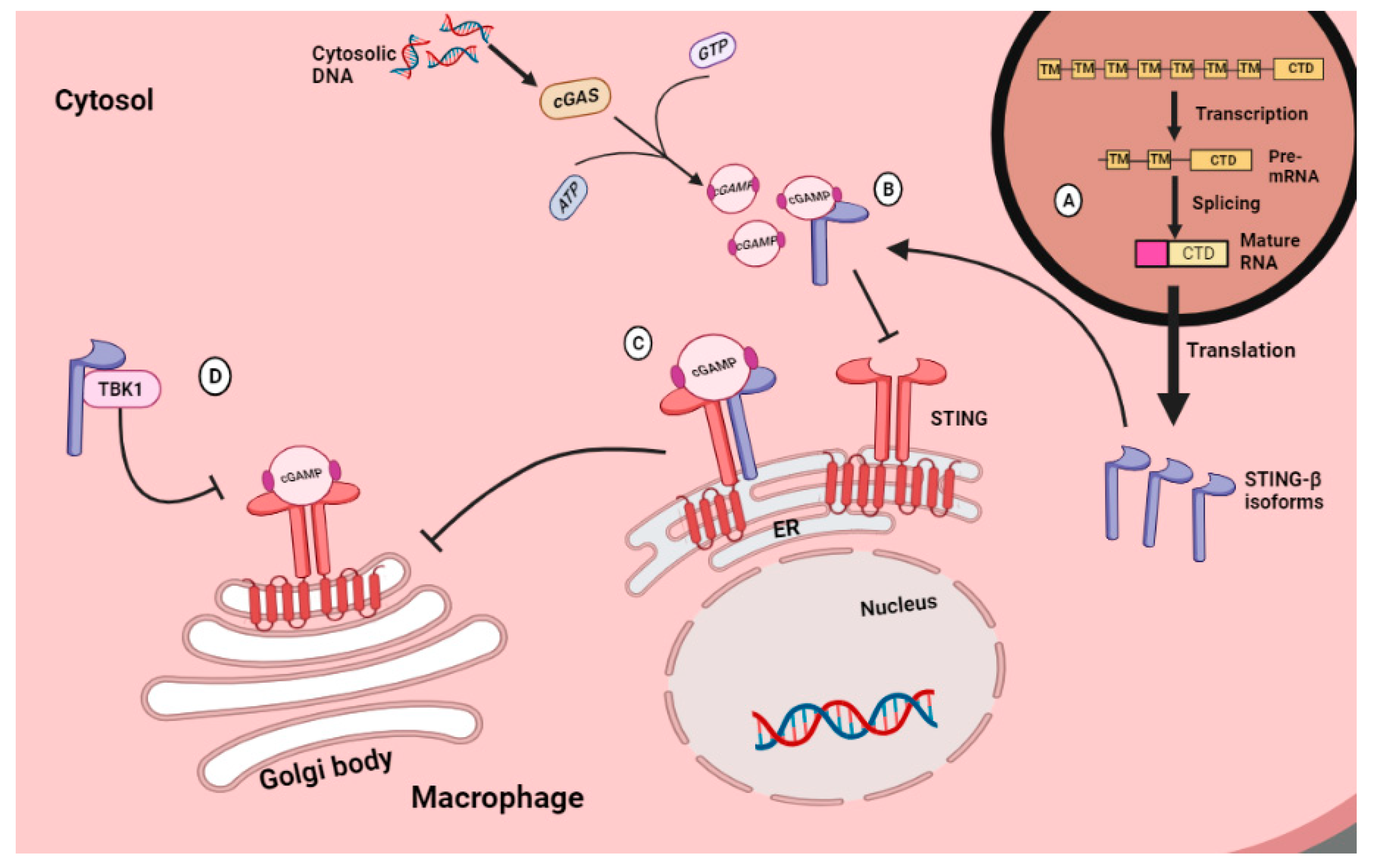
4.5. Simulator of Interferon Genes (STING)
4.6. Toll-like Receptor 4 (TLR-4)
4.7. Myeloid Differentiation Factor 88 (MYD88)
4.8. CD44
4.9. Fibroblast Growth Factor Receptor 2 (FGFR2)
5. Therapeutic Interventions
6. Conclusions and Future Perfectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Fackenthal, J.D. Alternative mRNA Splicing and Promising Therapies in Cancer. Biomolecules 2023, 13, 561. [Google Scholar] [CrossRef] [PubMed]
- Global Burden of Disease 2019 Cancer Collaboration. Cancer Incidence, Mortality, Years of Life Lost, Years Lived with Disability, and Disability-Adjusted Life Years for 29 Cancer Groups from 2010 to 2019: A Systematic Analysis for the Global Burden of Disease Study 2019. JAMA Oncol. 2022, 8, 420–444. [Google Scholar] [CrossRef]
- Öther-Gee Pohl, S.; Myant, K.B. Alternative RNA splicing in tumour heterogeneity, plasticity and therapy. Dis. Models Mech. 2022, 15, dmm049233. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, K.; Nimura, K. Regulation of RNA Splicing: Aberrant Splicing Regulation and Therapeutic Targets in Cancer. Cells 2021, 10, 923. [Google Scholar] [CrossRef] [PubMed]
- Murphy, A.J.; Li, A.H.; Li, P.; Sun, H. Therapeutic Targeting of Alternative Splicing: A New Frontier in Cancer Treatment. Front. Oncol. 2022, 12, 868664. [Google Scholar] [CrossRef] [PubMed]
- Bernard, A.; Boidot, R.; Végran, F. Alternative Splicing in Cancer and Immune Cells. Cancers 2022, 14, 1726. [Google Scholar] [CrossRef]
- Marin, J.J.G.; Reviejo, M.; Soto, M.; Lozano, E.; Asensio, M.; Ortiz-Rivero, S.; Berasain, C.; Avila, M.A.; Herraez, E. Impact of Alternative Splicing Variants on Liver Cancer Biology. Cancers 2021, 14, 18. [Google Scholar] [CrossRef]
- Dai, H.; Fan, Q.; Wang, C. Recent applications of immunomodulatory biomaterials for disease immunotherapy. Exploration 2022, 2, 20210157. [Google Scholar] [CrossRef]
- Disis, M.L. Immune regulation of cancer. J. Clin. Oncol. 2010, 28, 4531–4538. [Google Scholar] [CrossRef]
- Kim, S.K.; Cho, S.W. The Evasion Mechanisms of Cancer Immunity and Drug Intervention in the Tumor Microenvironment. Front. Pharmacol. 2022, 13, 868695. [Google Scholar] [CrossRef]
- Viganò, S.; Perreau, M.; Pantaleo, G.; Harari, A. Positive and negative regulation of cellular immune responses in physiologic conditions and diseases. J. Immunol. Res. 2012, 2012, 485781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de la Fuente, H.; Cibrián, D.; Sánchez-Madrid, F. Immunoregulatory molecules are master regulators of inflammation during the immune response. FEBS Lett. 2012, 586, 2897–2905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, H.; Niimi, A.; Yasuhara, T.; Permata, T.B.M.; Hagiwara, Y.; Isono, M.; Nuryadi, E.; Sekine, R.; Oike, T.; Kakoti, S.; et al. DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nat. Commun. 2017, 8, 1751. [Google Scholar] [CrossRef] [Green Version]
- Russell, B.M.; Avigan, D.E. Immune dysregulation in multiple myeloma: The current and future role of cell-based immunotherapy. Int. J. Hematol. 2023, 117, 652–659. [Google Scholar] [CrossRef] [PubMed]
- Hao, Z.; Lin, M.; Du, F.; Xin, Z.; Wu, D.; Yu, Q.; Wu, Y.; Zhu, Z.; Li, W.; Chen, Y.; et al. Systemic Immune Dysregulation Correlates With Clinical Features of Early Non-Small Cell Lung Cancer. Front. Immunol. 2021, 12, 754138. [Google Scholar] [CrossRef]
- Su, Z.; Huang, D. Alternative Splicing of Pre-mRNA in the Control of Immune Activity. Genes 2021, 12, 574. [Google Scholar] [CrossRef]
- Deng, Y.; Zhao, H.; Ye, L.; Hu, Z.; Fang, K.; Wang, J. Correlations Between the Characteristics of Alternative Splicing Events, Prognosis, and the Immune Microenvironment in Breast Cancer. Front. Genet. 2021, 12, 686298. [Google Scholar] [CrossRef]
- Peng, Q.; Zhou, Y.; Oyang, L.; Wu, N.; Tang, Y.; Su, M.; Luo, X.; Wang, Y.; Sheng, X.; Ma, J.; et al. Impacts and mechanisms of alternative mRNA splicing in cancer metabolism, immune response, and therapeutics. Mol. Ther. 2022, 30, 1018–1035. [Google Scholar] [CrossRef]
- Khurana, L.; ElGindi, M.; Tilstam, P.V.; Pantouris, G. Elucidating the role of an immunomodulatory protein in cancer: From protein expression to functional characterization. Methods Enzym. 2019, 629, 307–360. [Google Scholar] [CrossRef]
- Cerasuolo, A.; Buonaguro, L.; Buonaguro, F.M.; Tornesello, M.L. The Role of RNA Splicing Factors in Cancer: Regulation of Viral and Human Gene Expression in Human Papillomavirus-Related Cervical Cancer. Front. Cell Dev. Biol. 2020, 8, 474. [Google Scholar] [CrossRef]
- Ren, P.; Lu, L.; Cai, S.; Chen, J.; Lin, W.; Han, F. Alternative Splicing: A New Cause and Potential Therapeutic Target in Autoimmune Disease. Front. Immunol. 2021, 12, 713540. [Google Scholar] [CrossRef] [PubMed]
- Wahl, M.C.; Will, C.L.; Lührmann, R. The Spliceosome: Design Principles of a Dynamic RNP Machine. Cell 2009, 136, 701–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, M.; Kakoulidou, M.; Giscombe, R.; Pirskanen, R.; Lefvert, A.K.; Klareskog, L.; Wang, X. Identification of CTLA-4 isoforms produced by alternative splicing and their association with myasthenia gravis. Clin. Immunol. 2008, 128, 374–381. [Google Scholar] [CrossRef]
- Sobhani, N.; Tardiel-Cyril, D.R.; Davtyan, A.; Generali, D.; Roudi, R.; Li, Y. CTLA-4 in Regulatory T Cells for Cancer Immunotherapy. Cancers 2021, 13, 1440. [Google Scholar] [CrossRef] [PubMed]
- Contardi, E.; Palmisano, G.L.; Tazzari, P.L.; Martelli, A.M.; Falà, F.; Fabbi, M.; Kato, T.; Lucarelli, E.; Donati, D.; Polito, L.; et al. CTLA-4 is constitutively expressed on tumor cells and can trigger apoptosis upon ligand interaction. Int. J. Cancer 2005, 117, 538–550. [Google Scholar] [CrossRef]
- Oyewole-Said, D.; Konduri, V.; Vazquez-Perez, J.; Weldon, S.A.; Levitt, J.M.; Decker, W.K. Beyond T-Cells: Functional Characterization of CTLA-4 Expression in Immune and Non-Immune Cell Types. Front. Immunol. 2020, 11, 608024. [Google Scholar] [CrossRef]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef] [Green Version]
- Ueda, H.; Howson, J.M.M.; Esposito, L.; Heward, J.; Snook; Chamberlain, G.; Rainbow, D.B.; Hunter, K.M.D.; Smith, A.N.; Di Genova, G.; et al. Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature 2003, 423, 506–511. [Google Scholar] [CrossRef]
- Deng, K.; Yao, J.; Huang, J.; Ding, Y.; Zuo, J. Abnormal alternative splicing promotes tumor resistance in targeted therapy and immunotherapy. Transl. Oncol. 2021, 14, 101077. [Google Scholar] [CrossRef]
- Ouyang, J.; Zhang, Y.; Xiong, F.; Zhang, S.; Gong, Z.; Yan, Q.; He, Y.; Wei, F.; Zhang, W.; Zhou, M.; et al. The role of alternative splicing in human cancer progression. Am. J. Cancer Res. 2021, 11, 4642–4667. [Google Scholar]
- Leung, A.M.; Lee, A.F.; Ozao-Choy, J.; Ramos, R.I.; Hamid, O.; O’Day, S.J.; Shin-Sim, M.; Morton, D.L.; Faries, M.B.; Sieling, P.A.; et al. Clinical Benefit from Ipilimumab Therapy in Melanoma Patients may be Associated with Serum CTLA4 Levels. Front. Oncol. 2014, 4, 110. [Google Scholar] [CrossRef]
- Teng, W.; Jeng, W.J.; Chen, W.T.; Lin, C.C.; Lin, C.Y.; Lin, S.M.; Sheen, I.S. Soluble form of CTLA-4 is a good predictor for tumor recurrence after radiofrequency ablation in hepatocellular carcinoma patients. Cancer Med. 2022, 11, 3786–3795. [Google Scholar] [CrossRef] [PubMed]
- Ward, F.J.; Dahal, L.N.; Wijesekera, S.K.; Abdul-Jawad, S.K.; Kaewarpai, T.; Xu, H.; Vickers, M.A.; Barker, R.N. The soluble isoform of CTLA-4 as a regulator of T-cell responses. Eur. J. Immunol. 2013, 43, 1274–1285. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Tian, X.; Wang, Y.; Kang, X.; Song, W. Soluble cytotoxic T-lymphocyte–associated antigen 4 (sCTLA-4) as a potential biomarker for diagnosis and evaluation of the prognosis in Glioma. BMC Immunol. 2021, 22, 33. [Google Scholar] [CrossRef] [PubMed]
- Simone, R.; Tenca, C.; Fais, F.; Luciani, M.; De Rossi, G.; Pesce, G.; Bagnasco, M.; Saverino, D. A soluble form of CTLA-4 is present in paediatric patients with acute lymphoblastic leukaemia and correlates with CD1d+ expression. PLoS ONE 2012, 7, e44654. [Google Scholar] [CrossRef]
- Li, Y.; Cui, X.; Yang, Y.J.; Chen, Q.Q.; Zhong, L.; Zhang, T.; Cai, R.L.; Miao, J.Y.; Yu, S.C.; Zhang, F. Serum sPD-1 and sPD-L1 as Biomarkers for Evaluating the Efficacy of Neoadjuvant Chemotherapy in Triple-Negative Breast Cancer Patients. Clin. Breast Cancer 2019, 19, 326–332.e321. [Google Scholar] [CrossRef]
- Zhou, J.; Mahoney, K.M.; Giobbie-Hurder, A.; Zhao, F.; Lee, S.; Liao, X.; Rodig, S.; Li, J.; Wu, X.; Butterfield, L.H.; et al. Soluble PD-L1 as a Biomarker in Malignant Melanoma Treated with Checkpoint Blockade. Cancer Immunol. Res. 2017, 5, 480–492. [Google Scholar] [CrossRef] [Green Version]
- Gong, B.; Kiyotani, K.; Sakata, S.; Nagano, S.; Kumehara, S.; Baba, S.; Besse, B.; Yanagitani, N.; Friboulet, L.; Nishio, M.; et al. Secreted PD-L1 variants mediate resistance to PD-L1 blockade therapy in non-small cell lung cancer. J. Exp. Med. 2019, 216, 982–1000. [Google Scholar] [CrossRef]
- Ugurel, S.; Rebmann, V.; Ferrone, S.; Tilgen, W.; Grosse-Wilde, H.; Reinhold, U. Soluble human leukocyte antigen–G serum level is elevated in melanoma patients and is further increased by interferon-α immunotherapy. Cancer 2001, 92, 369–376. [Google Scholar] [CrossRef]
- Wei, F.; Yang, F.; Li, J.; Zheng, Y.; Yu, W.; Yang, L.; Ren, X. Soluble Toll-like receptor 4 is a potential serum biomarker in non-small cell lung cancer. Oncotarget 2016, 7, 40106–40114. [Google Scholar] [CrossRef] [Green Version]
- Cardona Gloria, Y.; Bernhart, S.H.; Fillinger, S.; Wolz, O.O.; Dickhöfer, S.; Admard, J.; Ossowski, S.; Nahnsen, S.; Siebert, R.; Weber, A.N.R. Absence of Non-Canonical, Inhibitory MYD88 Splice Variants in B Cell Lymphomas Correlates with Sustained NF-κB Signaling. Front. Immunol. 2021, 12, 616451. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Zhao, W.; Shao, W. Prognostic value of CD44 and CD44v6 expression in patients with non-small cell lung cancer: Meta-analysis. Tumor Biol. 2014, 35, 7383–7389. [Google Scholar] [CrossRef] [PubMed]
- Olsson, E.; Honeth, G.; Bendahl, P.O.; Saal, L.H.; Gruvberger-Saal, S.; Ringnér, M.; Vallon-Christersson, J.; Jönsson, G.; Holm, K.; Lövgren, K.; et al. CD44 isoforms are heterogeneously expressed in breast cancer and correlate with tumor subtypes and cancer stem cell markers. BMC Cancer 2011, 11, 418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, W.M.; Teng, E.; Chong, H.S.; Lopez, K.A.P.; Tay, A.Y.L.; Salto-Tellez, M.; Shabbir, A.; So, J.B.Y.; Chan, S.L. CD44v8-10 Is a Cancer-Specific Marker for Gastric Cancer Stem Cells. Cancer Res. 2014, 74, 2630–2641. [Google Scholar] [CrossRef] [PubMed]
- Teles, S.P.; Oliveira, P.; Ferreira, M.; Carvalho, J.; Ferreira, P.; Oliveira, C. Integrated Analysis of Structural Variation and RNA Expression of FGFR2 and Its Splicing Modulator ESRP1 Highlight the ESRP1(amp)-FGFR2(norm)-FGFR2-IIIc(high) Axis in Diffuse Gastric Cancer. Cancers 2019, 12, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, C.; Ohm-Laursen, L.; Barington, T.; Husby, S.; Lillevang, S.T. Alternative splice variants of the human PD-1 gene. Cell. Immunol. 2005, 235, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, C.; Luong, G.; Sun, Y. A snapshot of the PD-1/PD-L1 pathway. J. Cancer 2021, 12, 2735–2746. [Google Scholar] [CrossRef]
- Wang, J.; Okazaki, I.-M.; Yoshida, T.; Chikuma, S.; Kato, Y.; Nakaki, F.; Hiai, H.; Honjo, T.; Okazaki, T. PD-1 deficiency results in the development of fatal myocarditis in MRL mice. Int. Immunol. 2010, 22, 443–452. [Google Scholar] [CrossRef]
- Kuai, W.; Xu, X.; Yan, J.; Zhao, W.; Li, Y.; Wang, B.; Yuan, N.; Li, Z.; Jia, Y. Prognostic Impact of PD-1 and Tim-3 Expression in Tumor Tissue in Stage I-III Colorectal Cancer. BioMed Res. Int. 2020, 2020, 5294043. [Google Scholar] [CrossRef]
- Ishikawa, M.; Nakayama, K.; Nakamura, K.; Yamashita, H.; Ishibashi, T.; Minamoto, T.; Iida, K.; Razia, S.; Ishikawa, N.; Nakayama, S.; et al. High PD-1 expression level is associated with an unfavorable prognosis in patients with cervical adenocarcinoma. Arch. Gynecol. Obstet. 2020, 302, 209–218. [Google Scholar] [CrossRef]
- Elhag, O.A.; Hu, X.J.; Wen-Ying, Z.; Li, X.; Yuan, Y.Z.; Deng, L.F.; Liu, D.L.; Liu, Y.L.; Hui, G. Reconstructed adeno-associated virus with the extracellular domain of murine PD-1 induces antitumor immunity. Asian Pac. J. Cancer Prev. 2012, 13, 4031–4036. [Google Scholar] [CrossRef] [Green Version]
- Hudson, K.; Cross, N.; Jordan-Mahy, N.; Leyland, R. The Extrinsic and Intrinsic Roles of PD-L1 and Its Receptor PD-1: Implications for Immunotherapy Treatment. Front. Immunol. 2020, 11, 568931. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 pathway: Current researches in cancer. Am. J. Cancer Res. 2020, 10, 727–742. [Google Scholar]
- Chen, J.; Jiang, C.C.; Jin, L.; Zhang, X.D. Regulation of PD-L1: A novel role of pro-survival signalling in cancer. Ann. Oncol. 2016, 27, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Weng, M.; Xia, S.; Zhang, M.; Chen, C.; Tang, J.; Huang, D.; Yu, H.; Sun, W.; Zhang, H.; et al. Distinct roles of programmed death ligand 1 alternative splicing isoforms in colorectal cancer. Cancer Sci. 2021, 112, 178–193. [Google Scholar] [CrossRef]
- Hassounah, N.B.; Malladi, V.S.; Huang, Y.; Freeman, S.S.; Beauchamp, E.M.; Koyama, S.; Souders, N.; Martin, S.; Dranoff, G.; Wong, K.K.; et al. Identification and characterization of an alternative cancer-derived PD-L1 splice variant. Cancer Immunol. Immunother. 2019, 68, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Tronik-Le Roux, D.; Renard, J.; Vérine, J.; Renault, V.; Tubacher, E.; LeMaoult, J.; Rouas-Freiss, N.; Deleuze, J.F.; Desgrandschamps, F.; Carosella, E.D. Novel landscape of HLA-G isoforms expressed in clear cell renal cell carcinoma patients. Mol. Oncol. 2017, 11, 1561–1578. [Google Scholar] [CrossRef] [Green Version]
- Naji, A.; Le Rond, S.; Durrbach, A.; Krawice-Radanne, I.; Creput, C.; Daouya, M.; Caumartin, J.; LeMaoult, J.; Carosella, E.D.; Rouas-Freiss, N. CD3+CD4low and CD3+CD8low are induced by HLA-G: Novel human peripheral blood suppressor T-cell subsets involved in transplant acceptance. Blood 2007, 110, 3936–3948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, A.; Yan, W.H. Human Leukocyte Antigen-G (HLA-G) Expression in Cancers: Roles in Immune Evasion, Metastasis and Target for Therapy. Mol. Med. 2015, 21, 782–791. [Google Scholar] [CrossRef]
- Amiot, L.; Ferrone, S.; Grosse-Wilde, H.; Seliger, B. Biology of HLA-G in cancer: A candidate molecule for therapeutic intervention? Cell Mol. Life Sci. 2011, 68, 417–431. [Google Scholar] [CrossRef] [Green Version]
- Wuerfel, F.M.; Huebner, H.; Häberle, L.; Gass, P.; Hein, A.; Jud, S.M.; Hack, C.C.; Wunderle, M.; Schulz-Wendtland, R.; Erber, R.; et al. HLA-G and HLA-F protein isoform expression in breast cancer patients receiving neoadjuvant treatment. Sci. Rep. 2020, 10, 15750. [Google Scholar] [CrossRef]
- Attia, J.V.D.; Dessens, C.E.; van de Water, R.; Houvast, R.D.; Kuppen, P.J.K.; Krijgsman, D. The Molecular and Functional Characteristics of HLA-G and the Interaction with Its Receptors: Where to Intervene for Cancer Immunotherapy? Int. J. Mol. Sci. 2020, 21, 8678. [Google Scholar] [CrossRef] [PubMed]
- Pistoia, V.; Morandi, F.; Wang, X.; Ferrone, S. Soluble HLA-G: Are they clinically relevant? Semin. Cancer Biol. 2007, 17, 469–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Yu, S.; Han, Y.; Wang, Y.; Sun, Y. Human leukocyte antigen-G expression and polymorphisms promote cancer development and guide cancer diagnosis/treatment. Oncol. Lett. 2018, 15, 699–709. [Google Scholar] [CrossRef] [Green Version]
- Loumagne, L.; Baudhuin, J.; Favier, B.; Montespan, F.; Carosella, E.D.; Rouas-Freiss, N. In vivo evidence that secretion of HLA-G by immunogenic tumor cells allows their evasion from immunosurveillance. Int. J. Cancer 2014, 135, 2107–2117. [Google Scholar] [CrossRef]
- Shang, G.; Zhang, C.; Chen, Z.J.; Bai, X.C.; Zhang, X. Cryo-EM structures of STING reveal its mechanism of activation by cyclic GMP-AMP. Nature 2019, 567, 389–393. [Google Scholar] [CrossRef]
- Decout, A.; Katz, J.D.; Venkatraman, S.; Ablasser, A. The cGAS–STING pathway as a therapeutic target in inflammatory diseases. Nat. Rev. Immunol. 2021, 21, 548–569. [Google Scholar] [CrossRef] [PubMed]
- Motwani, M.; Pesiridis, S.; Fitzgerald, K.A. DNA sensing by the cGAS–STING pathway in health and disease. Nat. Rev. Genet. 2019, 20, 657–674. [Google Scholar] [CrossRef]
- Zhu, Y.; An, X.; Zhang, X.; Qiao, Y.; Zheng, T.; Li, X. STING: A master regulator in the cancer-immunity cycle. Mol. Cancer 2019, 18, 152. [Google Scholar] [CrossRef] [Green Version]
- Ho, S.S.; Zhang, W.Y.; Tan, N.Y.; Khatoo, M.; Suter, M.A.; Tripathi, S.; Cheung, F.S.; Lim, W.K.; Tan, P.H.; Ngeow, J.; et al. The DNA Structure-Specific Endonuclease MUS81 Mediates DNA Sensor STING-Dependent Host Rejection of Prostate Cancer Cells. Immunity 2016, 44, 1177–1189. [Google Scholar] [CrossRef] [Green Version]
- Takashima, K.; Takeda, Y.; Oshiumi, H.; Shime, H.; Okabe, M.; Ikawa, M.; Matsumoto, M.; Seya, T. STING in tumor and host cells cooperatively work for NK cell-mediated tumor growth retardation. Biochem. Biophys. Res. Commun. 2016, 478, 1764–1771. [Google Scholar] [CrossRef]
- Xia, T.; Konno, H.; Ahn, J.; Barber, G.N. Deregulation of STING Signaling in Colorectal Carcinoma Constrains DNA Damage Responses and Correlates With Tumorigenesis. Cell Rep. 2016, 14, 282–297. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Zhu, Y.; Zhang, X.; An, X.; Weng, M.; Shi, J.; Wang, S.; Liu, C.; Luo, S.; Zheng, T. An alternatively spliced STING isoform localizes in the cytoplasmic membrane and directly senses extracellular cGAMP. J. Clin. Investig. 2022, 132, e144339. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Hong, Z.; Sun, B.; Guo, Z.; Wang, C.; Zhu, J. The Alternatively Spliced Isoforms of Key Molecules in the cGAS-STING Signaling Pathway. Front. Immunol. 2021, 12, 771744. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Yu, D.; Peng, L.; Wu, Y.; Fan, Y.; Gu, T.; Yao, Y.-L.; Zhong, J.; Chen, X.; Yao, Y.-G. An Alternative Splicing of Tupaia STING Modulated Anti-RNA Virus Responses by Targeting MDA5-LGP2 and IRF3. J. Immunol. 2020, 204, 3191–3204. [Google Scholar] [CrossRef] [PubMed]
- Flood, B.A.; Higgs, E.F.; Li, S.; Luke, J.J.; Gajewski, T.F. STING pathway agonism as a cancer therapeutic. Immunol. Rev. 2019, 290, 24–38. [Google Scholar] [CrossRef]
- Chen, H.; Pei, R.; Zhu, W.; Zeng, R.; Wang, Y.; Wang, Y.; Lu, M.; Chen, X. An Alternative Splicing Isoform of MITA Antagonizes MITA-Mediated Induction of Type I IFNs. J. Immunol. 2014, 192, 1162–1170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.H.; Fung, S.Y.; Gao, W.W.; Deng, J.J.; Cheng, Y.; Chaudhary, V.; Yuen, K.S.; Ho, T.H.; Chan, C.P.; Zhang, Y.; et al. A novel transcript isoform of STING that sequesters cGAMP and dominantly inhibits innate nucleic acid sensing. Nucleic Acids Res. 2018, 46, 4054–4071. [Google Scholar] [CrossRef]
- Hu, J.; Xu, J.; Feng, X.; Li, Y.; Hua, F.; Xu, G. Differential Expression of the TLR4 Gene in Pan-Cancer and Its Related Mechanism. Front. Cell Dev. Biol. 2021, 9, 700661. [Google Scholar] [CrossRef]
- Kawasaki, T.; Kawai, T. Toll-Like Receptor Signaling Pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Yang, F.; Wei, F.; Ren, X. The role of toll-like receptor 4 in tumor microenvironment. Oncotarget 2017, 8, 66656–66667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaure, C.; Liu, Y. A Comparative Review of Toll-Like Receptor 4 Expression and Functionality in Different Animal Species. Front. Immunol. 2014, 5, 316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, F.F.Y.; Alper, S. Alternative pre-mRNA splicing as a mechanism for terminating Toll-like Receptor signaling. Front. Immunol. 2022, 13, 1023567. [Google Scholar] [CrossRef] [PubMed]
- Iwami, K.-I.; Matsuguchi, T.; Masuda, A.; Kikuchi, T.; Musikacharoen, T.; Yoshikai, Y. Cutting Edge: Naturally Occurring Soluble Form of Mouse Toll-Like Receptor 4 Inhibits Lipopolysaccharide Signaling1. J. Immunol. 2000, 165, 6682–6686. [Google Scholar] [CrossRef] [Green Version]
- Zunt, S.L.; Burton, L.V.; Goldblatt, L.I.; Dobbins, E.E.; Srinivasan, M. Soluble forms of Toll-like receptor 4 are present in human saliva and modulate tumour necrosis factor-alpha secretion by macrophage-like cells. Clin. Exp. Immunol. 2009, 156, 285–293. [Google Scholar] [CrossRef]
- El-Kharashy, G.; Gowily, A.; Okda, T.; Houssen, M. Association between serum soluble Toll-like receptor 2 and 4 and the risk of breast cancer. Mol. Clin. Oncol. 2021, 14, 38. [Google Scholar] [CrossRef]
- Zheng, C.; Chen, J.; Chu, F.; Zhu, J.; Jin, T. Inflammatory Role of TLR-MyD88 Signaling in Multiple Sclerosis. Front. Mol. Neurosci. 2020, 12, 314. [Google Scholar] [CrossRef]
- Deguine, J.; Barton, G.M. MyD88: A central player in innate immune signaling. F1000Prime Rep. 2014, 6, 97. [Google Scholar] [CrossRef] [Green Version]
- Zhu, G.; Cheng, Z.; Huang, Y.; Zheng, W.; Yang, S.; Lin, C.; Ye, J. MyD88 mediates colorectal cancer cell proliferation, migration and invasion via NF-κB/AP-1 signaling pathway. Int. J. Mol. Med. 2020, 45, 131–140. [Google Scholar] [CrossRef] [Green Version]
- De Arras, L.; Alper, S. Limiting of the innate immune response by SF3A-dependent control of MyD88 alternative mRNA splicing. PLoS Genet. 2013, 9, e1003855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Z.; Li, Q.; Meng, R.; Yi, B.; Xu, Q. METTL3 regulates alternative splicing of MyD88 upon the lipopolysaccharide-induced inflammatory response in human dental pulp cells. J. Cell Mol. Med. 2018, 22, 2558–2568. [Google Scholar] [CrossRef]
- Chen, H.; Gao, S.; Liu, W.; Wong, C.-C.; Wu, J.; Wu, J.; Liu, D.; Gou, H.; Kang, W.; Zhai, J.; et al. RNA N6-Methyladenosine Methyltransferase METTL3 Facilitates Colorectal Cancer by Activating the m6A-GLUT1-mTORC1 Axis and Is a Therapeutic Target. Gastroenterology 2021, 160, 1284–1300.e1216. [Google Scholar] [CrossRef] [PubMed]
- Ishqi, H.M.; Husain, M.A.; Rehman, S.U.; Sarwar, T.; Tabish, M. Identification and expression of alternatively spliced novel isoforms of cancer associated MYD88 lacking death domain in mouse. Mol. Biol. Rep. 2018, 45, 699–711. [Google Scholar] [CrossRef] [PubMed]
- Senbanjo, L.T.; Chellaiah, M.A. CD44: A Multifunctional Cell Surface Adhesion Receptor Is a Regulator of Progression and Metastasis of Cancer Cells. Front. Cell Dev. Biol. 2017, 5, 18. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Zhao, S.; Karnad, A.; Freeman, J.W. The biology and role of CD44 in cancer progression: Therapeutic implications. J. Hematol. Oncol. 2018, 11, 64. [Google Scholar] [CrossRef] [Green Version]
- Mackay, C.R.; Terpe, H.J.; Stauder, R.; Marston, W.L.; Stark, H.; Günthert, U. Expression and modulation of CD44 variant isoforms in humans. J. Cell Biol. 1994, 124, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Prochazka, L.; Tesarik, R.; Turanek, J. Regulation of alternative splicing of CD44 in cancer. Cell. Signal. 2014, 26, 2234–2239. [Google Scholar] [CrossRef]
- Wielenga, V.J.M.; Heider, K.H.; Offerhaus, G.J.A.; Adolf, G.R.; van den Berg, F.M.; Ponta, H.; Herrlich, P.; Pals, S.T. Expression of CD44 Variant Proteins in Human Colorectal Cancer Is Related to Tumor Progression. Cancer Res. 1993, 53, 4754–4756. [Google Scholar] [PubMed]
- Situ, D.; Long, H.; Lin, P.; Zhu, Z.; Wang, J.; Zhang, X.; Xie, Z.; Rong, T. Expression and prognostic relevance of CD44v6 in stage I non-small cell lung carcinoma. J. Cancer Res. Clin. Oncol. 2010, 136, 1213–1219. [Google Scholar] [CrossRef]
- Ejima, R.; Suzuki, H.; Tanaka, T.; Asano, T.; Kaneko, M.K.; Kato, Y. Development of a Novel Anti-CD44 Variant 6 Monoclonal Antibody C44Mab-9 for Multiple Applications against Colorectal Carcinomas. Int. J. Mol. Sci. 2023, 24, 4007. [Google Scholar] [CrossRef]
- Yang, Y.; Meng, W.-J.; Wang, Z.-Q. Cancer Stem Cells and the Tumor Microenvironment in Gastric Cancer. Front. Oncol. 2022, 11, 803974. [Google Scholar] [CrossRef]
- Hyung, S.; Han, B.; Jung, J.; Kim, S.T.; Hong, J.Y.; Park, S.H.; Zang, D.Y.; Park, J.O.; Park, Y.S.; Kim, K.-M.; et al. Incidence of FGFR2 Amplification and FGFR2 Fusion in Patients with Metastatic Cancer Using Clinical Sequencing. J. Oncol. 2022, 2022, 9714570. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Liu, W.; Serra, S.; Asa, S.L.; Ezzat, S. FGFR2 isoforms support epithelial-stromal interactions in thyroid cancer progression. Cancer Res. 2012, 72, 2017–2027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zugman, M.; Botrus, G.; Pestana, R.C.; Uson Junior, P.L.S. Precision Medicine Targeting FGFR2 Genomic Alterations in Advanced Cholangiocarcinoma: Current State and Future Perspectives. Front. Oncol. 2022, 12, 860453. [Google Scholar] [CrossRef]
- Gong, S.G. The Fgfr2W290Rmouse model of Crouzon syndrome. Childs Nerv. Syst. 2012, 28, 1495–1503. [Google Scholar] [CrossRef]
- Yashiro, M.; Kuroda, K.; Masuda, G.; Okuno, T.; Miki, Y.; Yamamoto, Y.; Sera, T.; Sugimoto, A.; Kushiyama, S.; Nishimura, S.; et al. Clinical difference between fibroblast growth factor receptor 2 subclass, type IIIb and type IIIc, in gastric cancer. Sci. Rep. 2021, 11, 4698. [Google Scholar] [CrossRef]
- Oltean, S.; Febbo, P.G.; Garcia-Blanco, M.A. Dunning rat prostate adenocarcinomas and alternative splicing reporters: Powerful tools to study epithelial plasticity in prostate tumors in vivo. Clin. Exp. Metastasis 2008, 25, 611–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Q.; Caballero, O.L.; Davis, I.D.; Jonasch, E.; Tamboli, P.; Yung, W.K.; Weinstein, J.N.; Strausberg, R.L.; Yao, J. Tumor-specific isoform switch of the fibroblast growth factor receptor 2 underlies the mesenchymal and malignant phenotypes of clear cell renal cell carcinomas. Clin. Cancer Res. 2013, 19, 2460–2472. [Google Scholar] [CrossRef] [Green Version]
- Rosato, B.; Ranieri, D.; Nanni, M.; Torrisi, M.R.; Belleudi, F. Role of FGFR2b expression and signaling in keratinocyte differentiation: Sequential involvement of PKCδ and PKCα. Cell Death Dis. 2018, 9, 565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranieri, D.; Nanni, M.; Persechino, F.; Torrisi, M.R.; Belleudi, F. Role of PKCε in the epithelial-mesenchymal transition induced by FGFR2 isoform switch. Cell Commun. Signal. 2020, 18, 76. [Google Scholar] [CrossRef]
- Ueda, J.; Matsuda, Y.; Yamahatsu, K.; Uchida, E.; Naito, Z.; Korc, M.; Ishiwata, T. Epithelial splicing regulatory protein 1 is a favorable prognostic factor in pancreatic cancer that attenuates pancreatic metastases. Oncogene 2014, 33, 4485–4495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassn Mesrati, M.; Syafruddin, S.E.; Mohtar, M.A.; Syahir, A. CD44: A Multifunctional Mediator of Cancer Progression. Biomolecules 2021, 11, 1850. [Google Scholar] [CrossRef]
- Gordon, A.; Johnston, E.; Lau, D.K.; Starling, N. Targeting FGFR2 Positive Gastroesophageal Cancer: Current and Clinical Developments. OncoTargets Ther. 2022, 15, 1183–1196. [Google Scholar] [CrossRef] [PubMed]
- Tsimafeyeu, I.; Statsenko, G.; Vladimirova, L.; Besova, N.; Artamonova, E.; Raskin, G.; Rykov, I.; Mochalova, A.; Utyashev, I.; Gorbacheva, S.; et al. A phase 1b study of the allosteric extracellular FGFR2 inhibitor alofanib in patients with pretreated advanced gastric cancer. Investig. New Drugs 2023, 41, 324–332. [Google Scholar] [CrossRef]
- Kennedy, P.T.; Saulters, E.L.; Duckworth, A.D.; Lim, Y.J.; Woolley, J.F.; Slupsky, J.R.; Cragg, M.S.; Ward, F.J.; Dahal, L.N. Soluble CTLA-4 raises the threshold for T-cell activation and modulates anti-tumour immunity. bioRxiv 2023. [Google Scholar] [CrossRef]
- Zhou, X.; Li, C.; Chen, T.; Li, W.; Wang, X.; Yang, Q. Targeting RNA N6-methyladenosine to synergize with immune checkpoint therapy. Mol. Cancer 2023, 22, 36. [Google Scholar] [CrossRef]
- Gu, D.; Ao, X.; Yang, Y.; Chen, Z.; Xu, X. Soluble immune checkpoints in cancer: Production, function and biological significance. J. Immunother. Cancer 2018, 6, 132. [Google Scholar] [CrossRef] [Green Version]
- Bonner, E.A.; Lee, S.C. Therapeutic Targeting of RNA Splicing in Cancer. Genes 2023, 14, 1378. [Google Scholar] [CrossRef]
- Urbanski, L.M.; Leclair, N.; Anczuków, O. Alternative-splicing defects in cancer: Splicing regulators and their downstream targets, guiding the way to novel cancer therapeutics. Wiley Interdiscip. Rev. RNA 2018, 9, e1476. [Google Scholar] [CrossRef]
- Yamauchi, H.; Nishimura, K.; Yoshimi, A. Aberrant RNA splicing and therapeutic opportunities in cancers. Cancer Sci. 2022, 113, 373–381. [Google Scholar] [CrossRef]
- Bashari, A.; Siegfried, Z.; Karni, R. Targeting splicing factors for cancer therapy. RNA 2023, 29, 506–515. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isoform | Tumor Type | Expression Levels in Tumor Tissue | Expression Levels in Normal Tissue | Clinical Results | References |
|---|---|---|---|---|---|
| sCTLA-4 | Melanoma | High | Unknown | Associated with drug response and better patient survival | [31] |
| Glioma | High | Low | Shorter survival | [34] | |
| PD-1Δex3 | Breast (TNBC) | High | Low | Associated with high tumor stage | [36] |
| PD-L1-1 | Melanoma | High | Low | Melanoma progression | [37] |
| PD-L1-3 | |||||
| PD-L1-9 | |||||
| PD-L1-242 | NSCLC | High | Low | Drug resistance | [38] |
| PD-L1-229 | |||||
| sHLA-G | Melanoma | High | Low | Advanced disease stage and tumor load | [39] |
| sTLR-4 | NSCLC | High | Low | Tumor metastasis and poor survival | [40] |
| MyD88s | B cell lymphoma | Comparable | Comparable | Unknown | [41] |
| MyD88L | High | Low | |||
| CD44V6 | NSCLC | High | Unknown | Poor patient survival | [42] |
| CD44s | NSCLC | High | Unknown | Poor patient survival | |
| CD44V8-10 | Breast cancer (basal-like subtype) | Unknown | Unknown | Better overall 10-year patient survival | [43] |
| GC | High | Low | Unknown | [44] | |
| FGFR2IIIc | GC | Low | High | Low expression correlates with better patient survival | [45] |
| FGFR2IIIb | GC | High | Low | Unknown | [45] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maebele, L.T.; Mulaudzi, T.V.; Yasasve, M.; Dlamini, Z.; Damane, B.P. Immunomodulatory Gene-Splicing Dysregulation in Tumorigenesis: Unmasking the Complexity. Molecules 2023, 28, 5984. https://doi.org/10.3390/molecules28165984
Maebele LT, Mulaudzi TV, Yasasve M, Dlamini Z, Damane BP. Immunomodulatory Gene-Splicing Dysregulation in Tumorigenesis: Unmasking the Complexity. Molecules. 2023; 28(16):5984. https://doi.org/10.3390/molecules28165984
Chicago/Turabian StyleMaebele, Lorraine Tshegofatso, Thanyani Victor Mulaudzi, Madhavan Yasasve, Zodwa Dlamini, and Botle Precious Damane. 2023. "Immunomodulatory Gene-Splicing Dysregulation in Tumorigenesis: Unmasking the Complexity" Molecules 28, no. 16: 5984. https://doi.org/10.3390/molecules28165984