

A New Diterpenoid of Indonesian Scoparia dulcis Linn: Isolation and Cytotoxic Activity against MCF-7 and T47D Cell Lines
 , , , and
, , , and
Abstract
:1. Introduction
2. Results
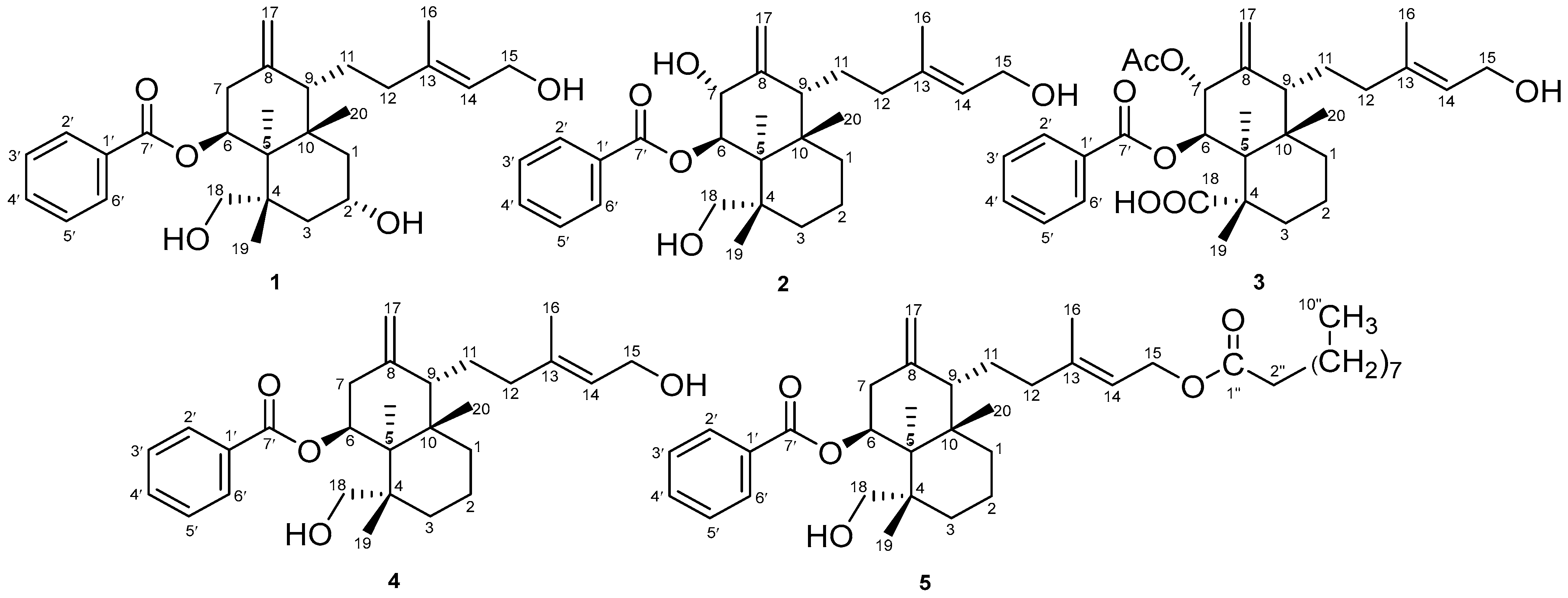
2.1. Properties of 2α-Hydroxyscopadiol (Compound 1)
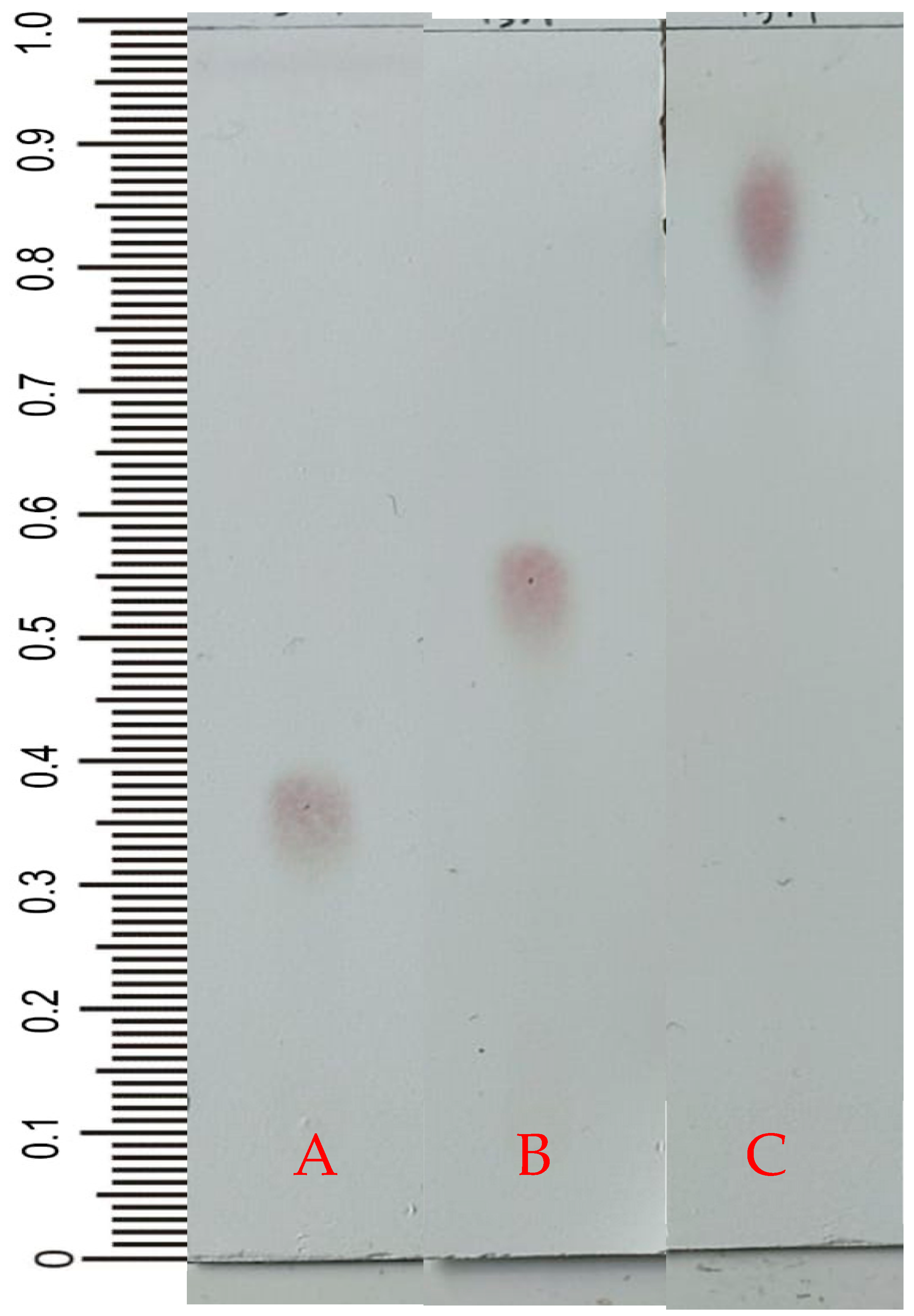
2.2. Thin-Layer Chromatography
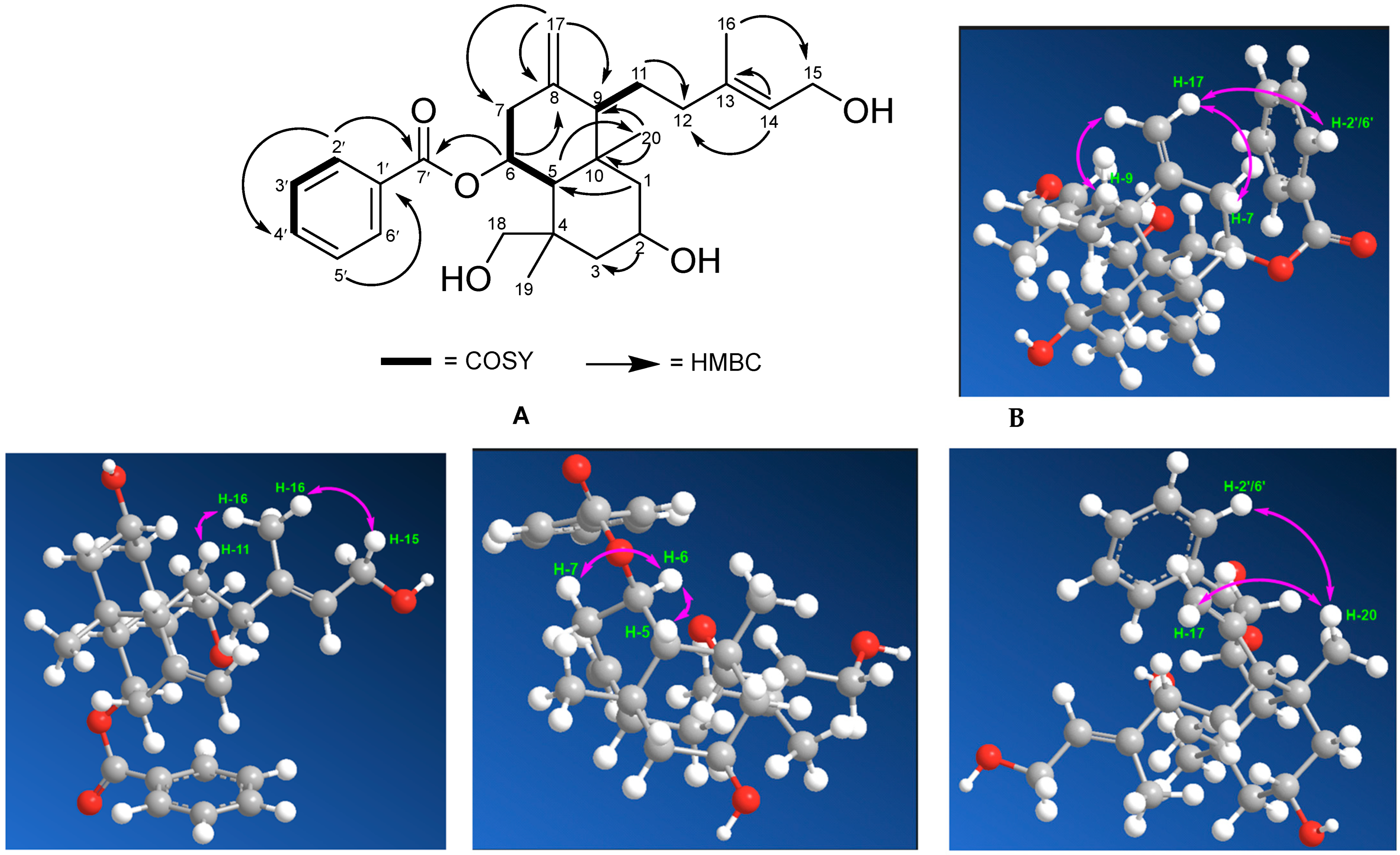
2.3. Spectroscopic Analysis
COSY and HMBC Correlations of Compound (1)
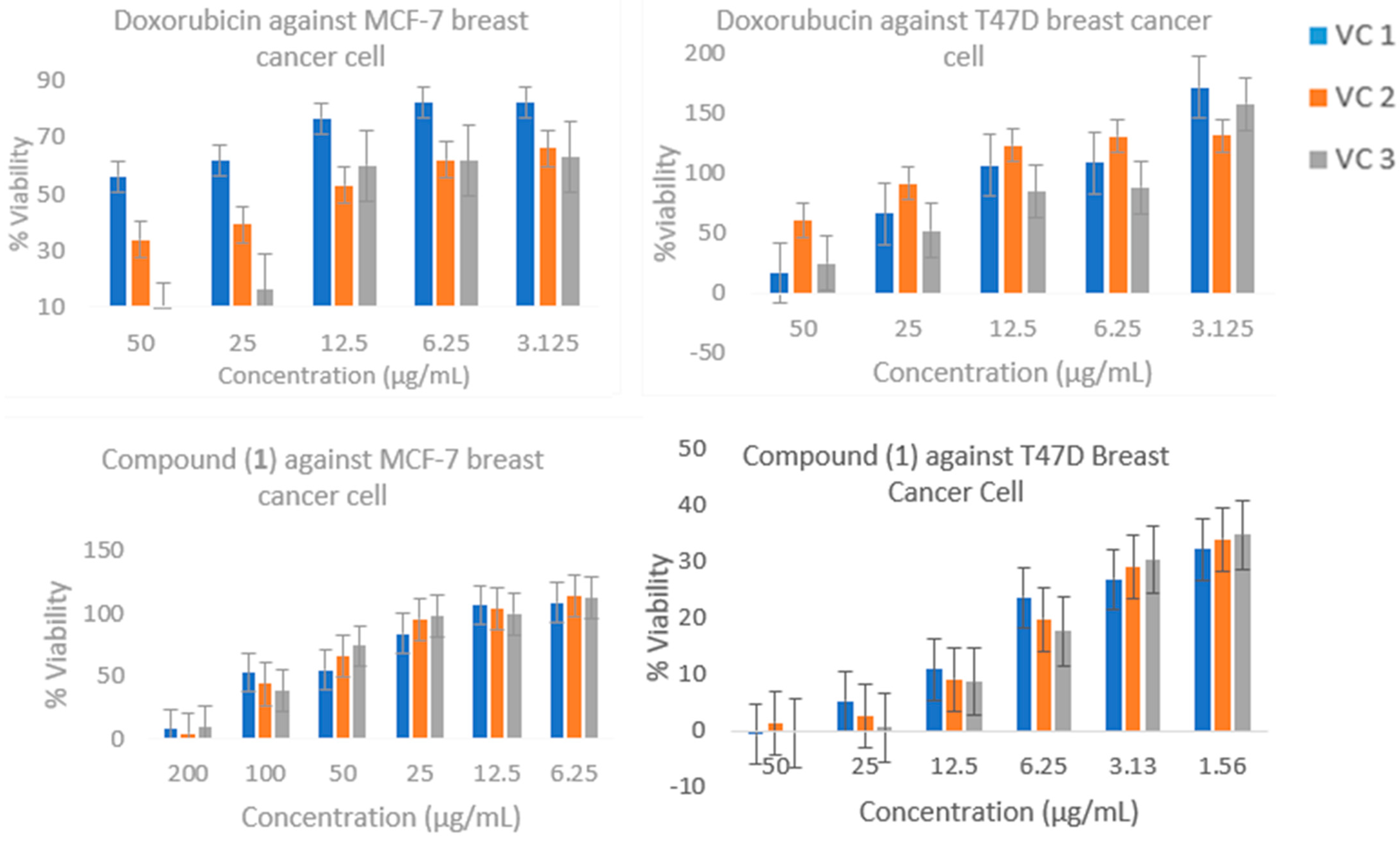
2.4. Cytotoxic Activity
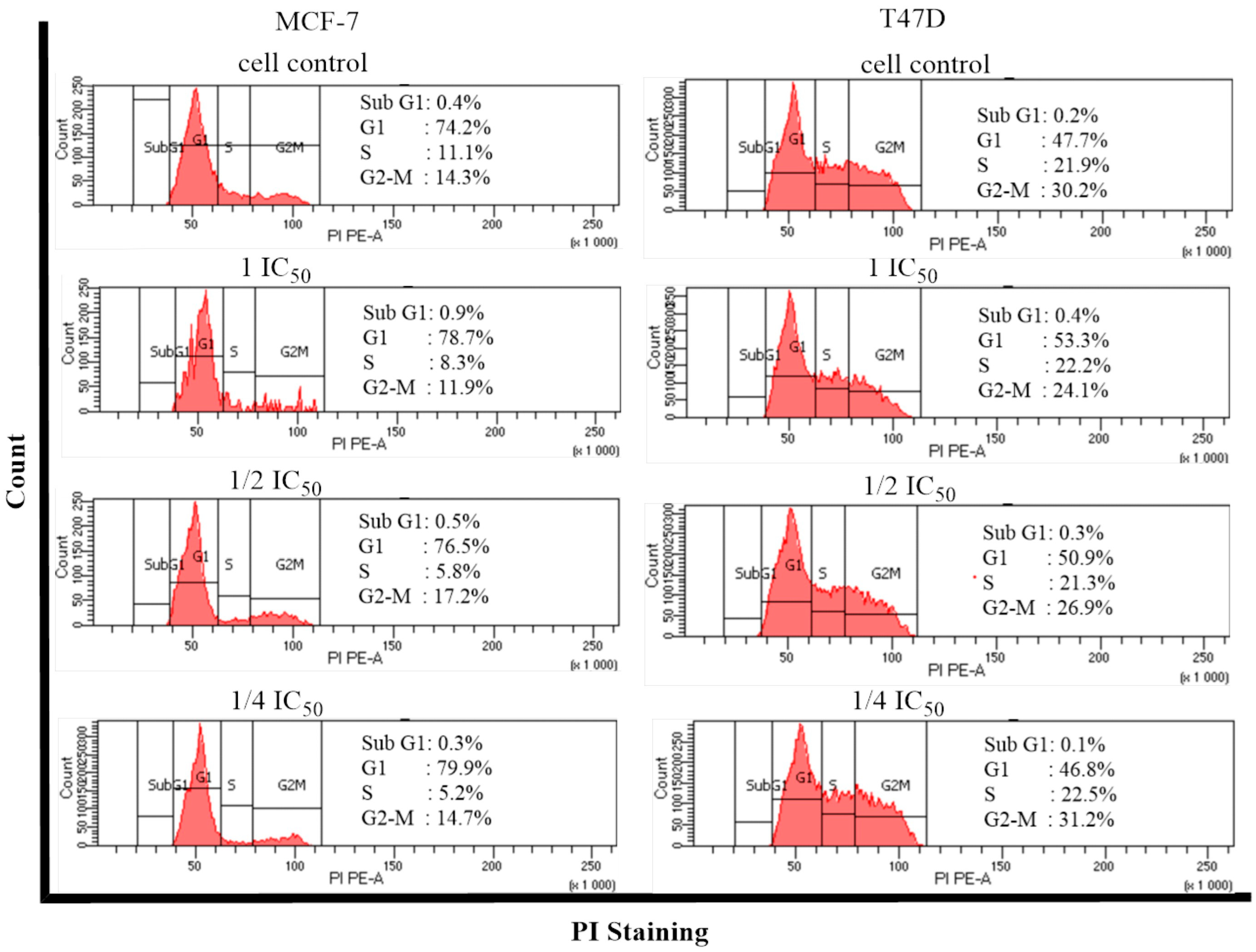
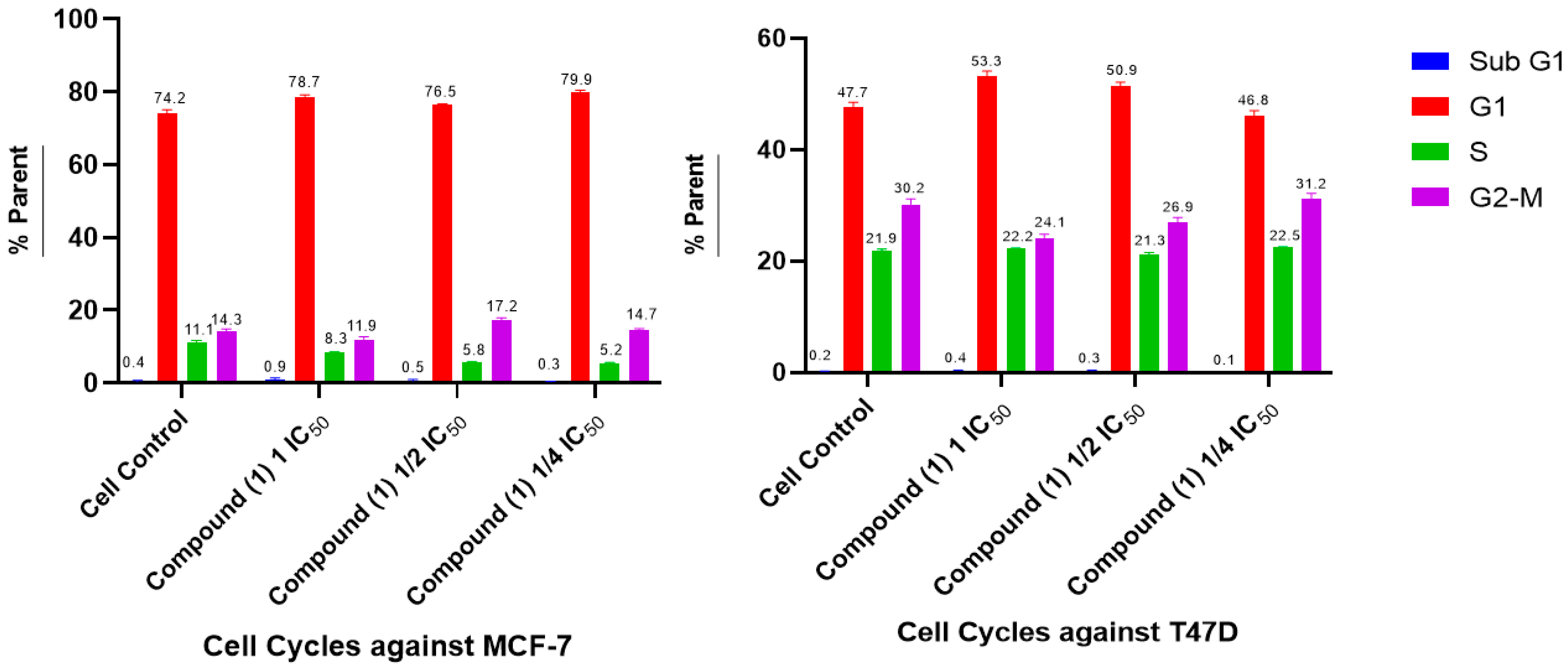
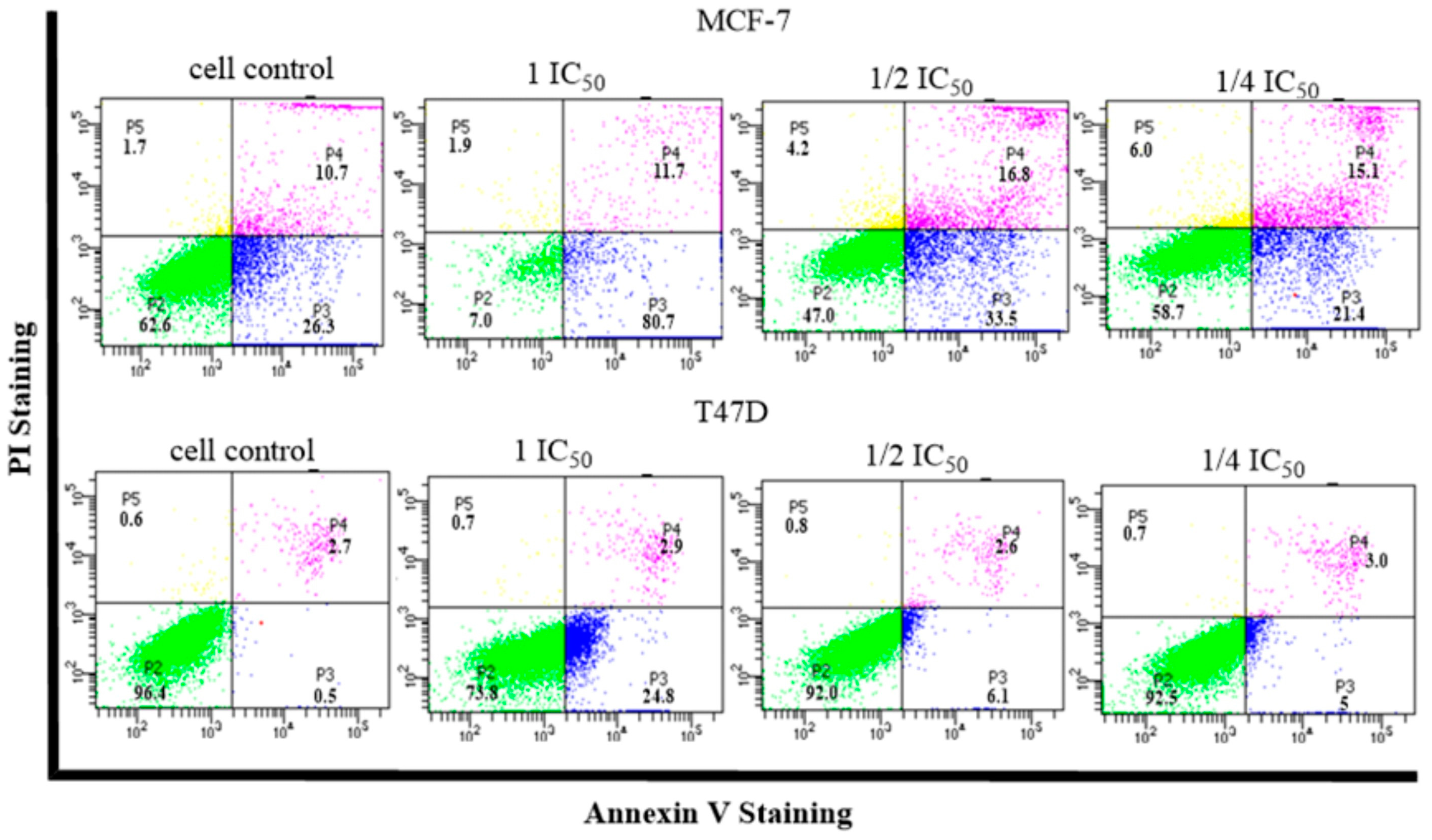
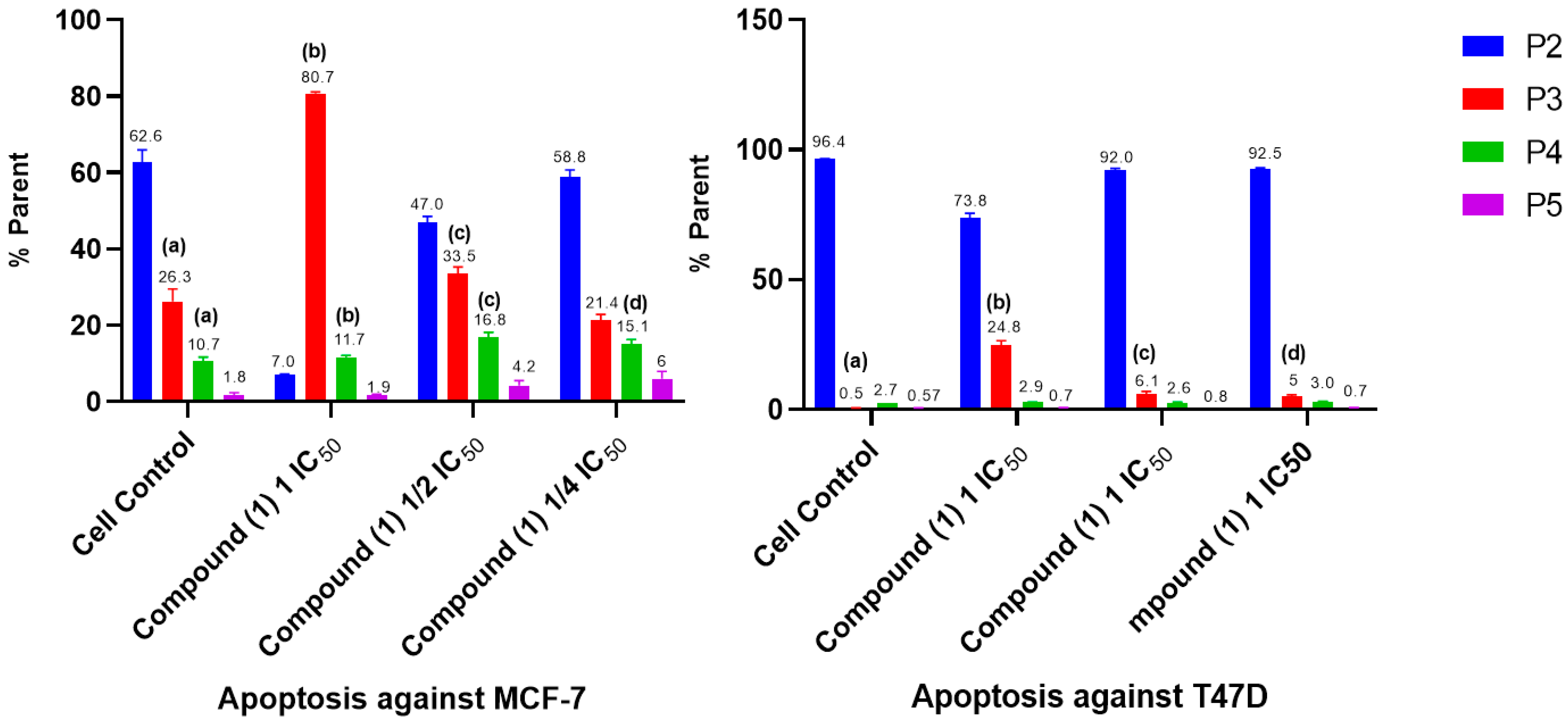
2.5. Cell Cycle and Apoptosis
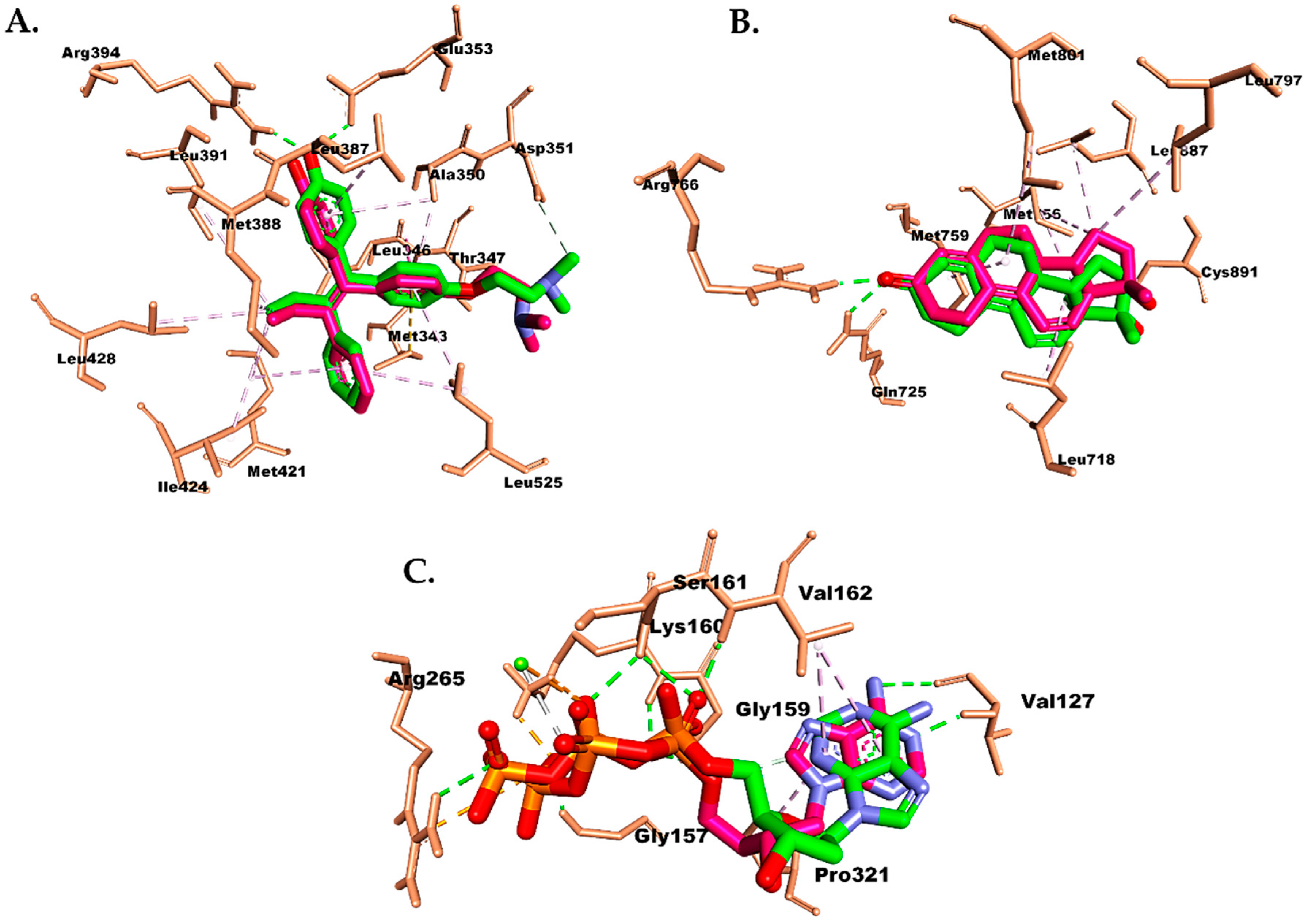
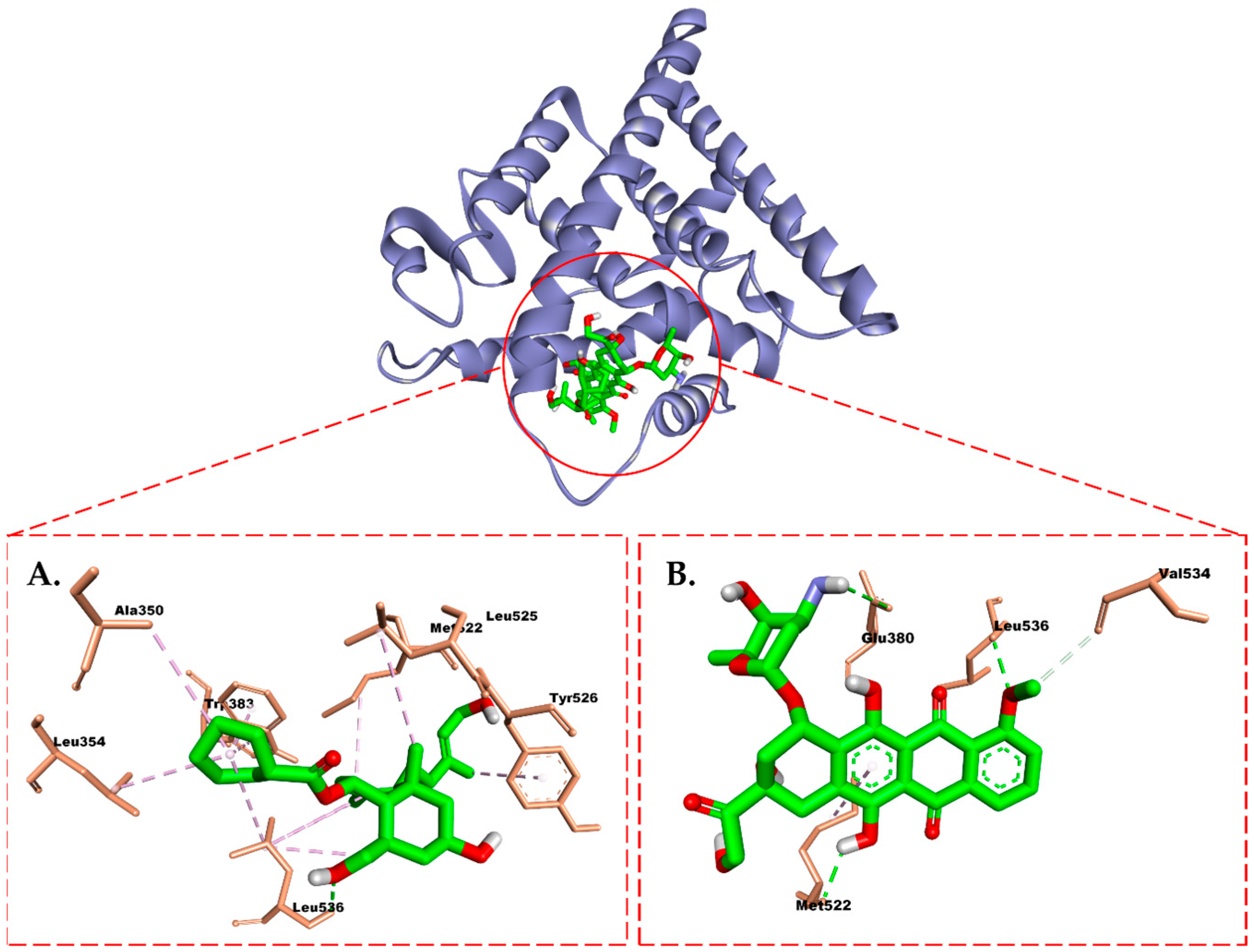
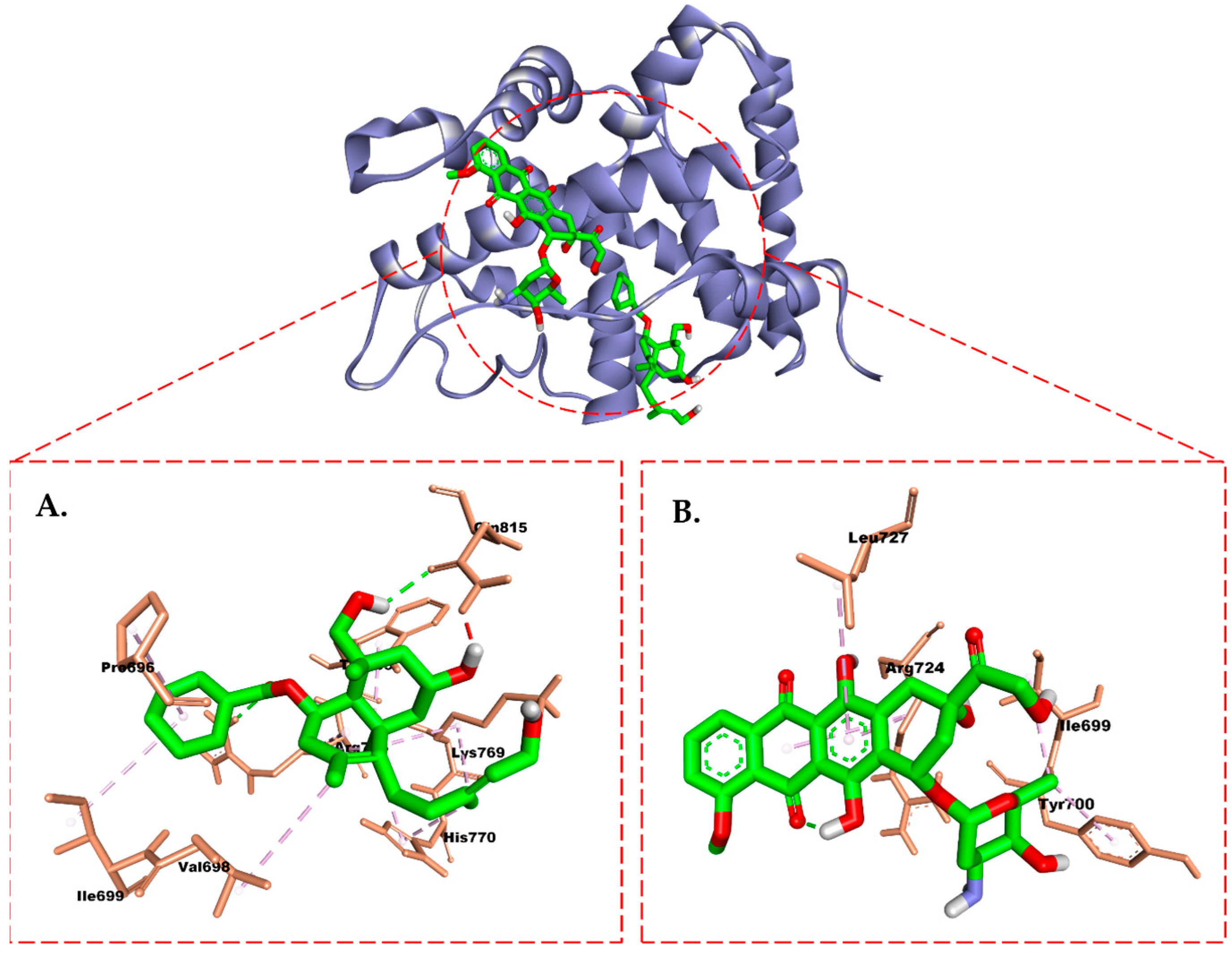
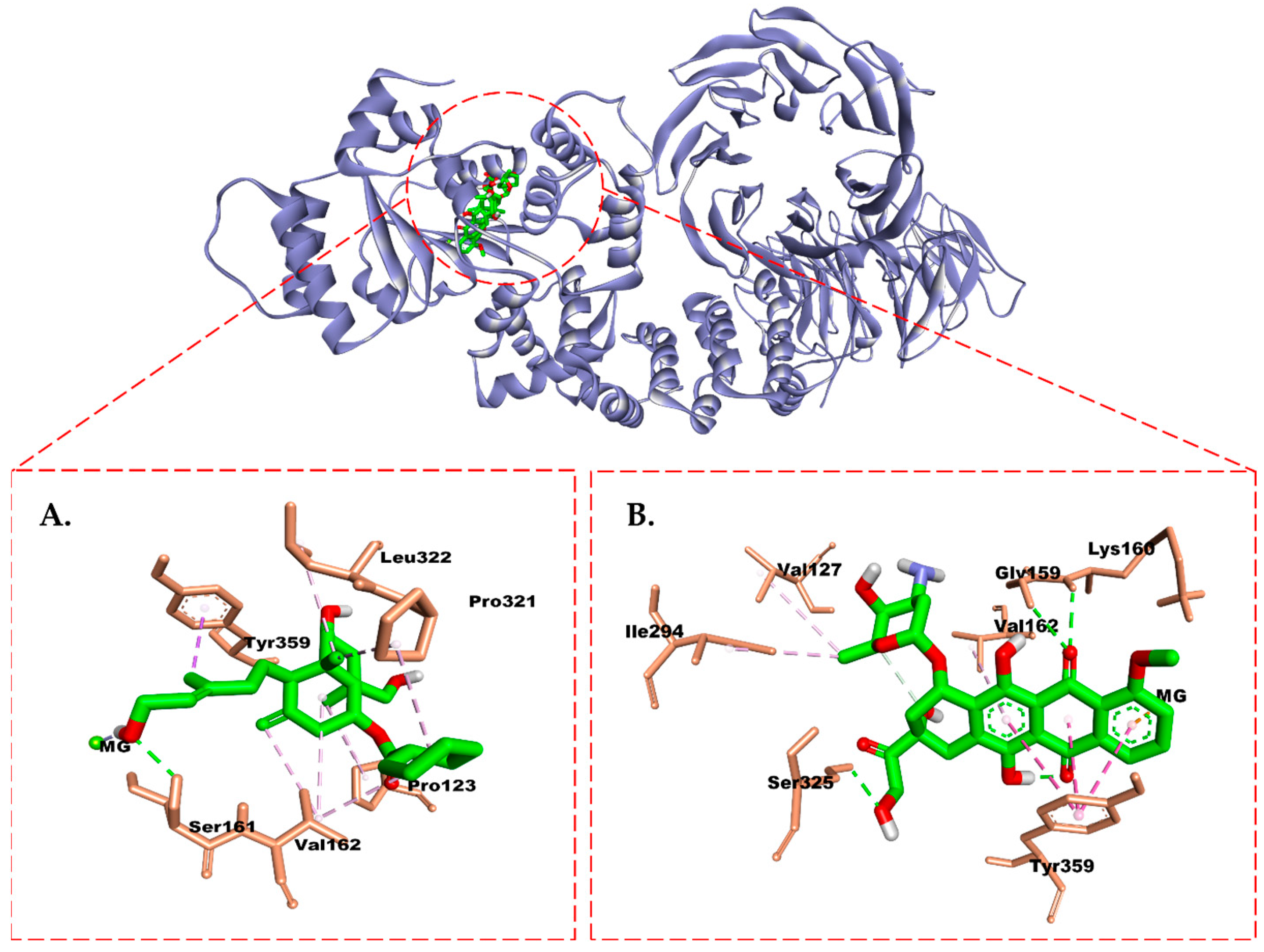
2.6. Molecular Docking Study
3. Discussion
- a.
- Isolation and Identification of Compound (1)
- b.
- Cytotoxic Test
- c.
- Cell Cycle and Apoptosis
4. Materials and Methods
4.1. General Procedure
4.2. Plant Material
4.3. Extraction and Separation
4.4. Thin-Layer Chromatography
4.5. Cytotoxic Activity Assay
4.6. Cell Cycle and Apoptosis Assay
4.7. Molecular Docking Study
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- World Health Organization 2019. Available online: https://www.who.int/ (accessed on 28 June 2023).
- Link, W. Anti-Cancer Drugs: Discovery, Development and Therapy. In International Manual of Oncology Practice; de Mello, R.A., Tavares, Á., Mountzios, G., Eds.; Springer International Publishing: Cham, Germany, 2015; pp. 81–94. ISBN 978-3-319-21682-9. [Google Scholar]
- Sofowora, A.; Ogunbodede, E.; Onayade, A. The role and place of medicinal plants in the strategies for disease prevention. Afr. J. Tradit. Complement. Altern. Med. 2013, 10, 210–229. [Google Scholar] [CrossRef] [PubMed]
- Sasidharan, S.; Chen, Y.; Saravanan, D.; Sundram, K.M.; Yoga Latha, L. Extraction, Isolation and Characterization of Bioactive Compounds from Plants’ Extracts. Afr. J. Tradit. Complement. Altern. Med. 2011, 8, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Kawasaki, M.; Hayashi, T.; Arisawa, M.; Morita, N.; Berganza, L.H. 8-Hydroxytricetin 7-glucuronide, a β-glucuronidase inhibitor from Scoparia dulcis. Phytochemistry 1988, 27, 3709–3711. [Google Scholar] [CrossRef]
- Hayashi, K.; Hayashi, T.; Morita, N. Cytotoxic and antitumour activity of scopadulcic acid from Scoparia dulcis L. Phytotherapy Res. 1992, 6, 6–9. [Google Scholar] [CrossRef]
- Ahsan, M.; Islam, S.N.; Gray, A.I.; Stimson, W.H. Cytotoxic Diterpenes from Scoparia dulcis. J. Nat. Prod. 2003, 66, 958–961. [Google Scholar] [CrossRef]
- Phan, M.G.; Phan, T.S.; Matsunami, K.; Otsuka, H. Chemical and Biological Evaluation on Scopadulane-Type Diterpenoids from Scoparia dulcis of Vietnamese Origin. Chem. Pharm. Bull. 2006, 54, 546–549. [Google Scholar] [CrossRef] [Green Version]
- Wankhar, W.; Srinivasan, S.; Sundareswaran, L.; Wankhar, D.; Rajan, R.; Sheeladevi, R. Role of Scoparia dulcis linn on noise-induced nitric oxide synthase (NOS) expression and neurotransmitter assessment on motor function in Wistar albino rats. Biomed. Pharmacother. 2017, 86, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Coulibaly, A.Y.; Hashim, R.; Sulaiman, S.F.; Sulaiman, O.; Ang, L.Z.P.; Ooi, K.L. Bioprospecting Medicinal Plants for Antioxidant Components. Asian Pac. J. Trop. Med. 2014, 7, S553–S559. [Google Scholar] [CrossRef] [Green Version]
- Krishnamurthy, P.T.; Bajaj, J.; Sharma, A.; Manimaran, S.; Ravanappa, P.K.B.; Pottekad, V. Hepatoprotective Activity of Terpenoids and Terpenoid Fractions of Scoparia dulcis L. Orient. Pharm. Exp. Med. 2010, 10, 263–270. [Google Scholar]
- Babincová, M.; Schronerová, K.; Sourivong, P. Antiulcer activity of water extract of Scoparia dulcis. Fitoterapia 2008, 79, 587–588. [Google Scholar] [CrossRef]
- Paul, M.; Vasudevan, K. Scoparia dulcis: A Review on Its Phytochemical and Pharmacological Profile. Innoriginal Int. J. Sci. 2017, 4, 17–21. [Google Scholar]
- Valsalakumari, P.; Narayanan, N. Antiproliferative Activity of Scoparia dulcis Linn. Ethanolic Extract on Cell Line. Res. J. Pharm. Biol. Chem. Sci. 2017, 8, 427–431. [Google Scholar]
- Fuentes, R.G.; Toume, K.; Arai, M.A.; Sadhu, S.K.; Ahmed, F.; Ishibashi, M. Scopadulciol, Isolated from Scoparia dulcis, induces β-Catenin Degradation and Overcomes Tumor Necrosis Factor-Related Apoptosis Ligand Resistance in AGS Human Gastric Adenocarcinoma Cells. J. Nat. Prod. 2015, 78, 864–872. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.-H.; Chen, T.-Y.; Lu, R.-W.; Chen, S.-T.; Chang, C.-C. Benzoxazinoids from Scoparia dulcis (sweet broomweed) with antiproliferative activity against the DU-145 human prostate cancer cell line. Phytochemistry 2012, 83, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Mishra, M.R.; Behera, R.K.; Jha, S.; Panda, A.K.; Mishra, A.; Pradhan, D.K.; Choudary, P.R. A Brief Review on Phytoconstituents and Ethnopharmacology of Scoparia dulcis Linn. (Scrophulariaceae). Int. J. Phytomed. 2011, 3, 422. [Google Scholar]
- Hayashi, K.; Lee, J.-B.; Maitani, Y.; Toyooka, N.; Nemoto, H.; Hayashi, T. The Role of a HSV Thymidine Kinase Stimulating Substance, Scopadulciol, in Improving the Efficacy of Cancer Gene Therapy. J. Gene Med. 2006, 8, 1056–1067. [Google Scholar] [CrossRef]
- Nishino, H.; Hayashi, T.; Arisawa, M.; Satomi, Y.; Iwashima, A. Antitumor-Promoting Activity of Scopadulcic Acid B, Isolated from the Medicinal Plant Scoparia dulcis L. Oncology 1993, 50, 100–103. [Google Scholar] [CrossRef]
- Hasnawati; Wahyuono, S.; Susidarti, R.A.; Santosa, D. Phytochemical Constituents and Cytotoxic Activity from Scoparia dulcis Linn of Indonesia Origin. Rasayan J. Chem. 2022, 15, 933–936. [Google Scholar] [CrossRef]
- Ahsan, M.; Haque, M.R.; Islam, S.; Gray, A.I.; Hasan, C.M. New labdane diterpenes from the aerial parts of Scoparia dulcis L. Phytochem. Lett. 2012, 5, 609–612. [Google Scholar] [CrossRef]
- Liu, Q.; Yang, Q.-M.; Hu, H.-J.; Yang, L.; Yang, Y.-B.; Chou, G.-X.; Wang, Z.-T. Bioactive Diterpenoids and Flavonoids from the Aerial Parts of Scoparia dulcis. J. Nat. Prod. 2014, 77, 1594–1600. [Google Scholar] [CrossRef]
- Hayashi, T.; Okamura, K.; Kawasaki, M.; Morita, N. Production of diterpenoids by cultured cells from two chemotypes of Scoparia dulcis. Phytochemistry 1993, 33, 353–356. [Google Scholar] [CrossRef]
- Zhang, T.; Fang, M.; Fu, Z.-M.; Du, H.-C.; Yuan, H.; Xia, G.-Y.; Feng, J.; Yin, G.-Y. Expression of PI3-K, PKB and GSK-3 β in the Skeletal Muscle Tissue of Gestational Diabetes Mellitus. Asian Pac. J. Trop. Med. 2014, 7, 309–312. [Google Scholar] [CrossRef]
- Ebada, S.S.; Edrada, R.A.; Lin, W.; Proksch, P. Methods for isolation, purification and structural elucidation of bioactive secondary metabolites from marine invertebrates. Nat. Protoc. 2008, 3, 1820–1831. [Google Scholar] [CrossRef]
- Weerapreeyakul, N.; Nonpunya, A.; Barusrux, S.; Thitimetharoch, T.; Sripanidkulchai, B. Evaluation of the anticancer potential of six herbs against a hepatoma cell line. Chin. Med. 2012, 7, 15. [Google Scholar] [CrossRef] [Green Version]
- Samirana, P.O.; Murti, Y.B.; Jenie, R.I.; Setyowati, E.P. Antibacterial and cytotoxic activities of supernatant and mycelium extracts from fermentation of fungal symbiont Trichoderma reesei TV221. J. Appl. Pharm. Sci. 2021, 11, 090–099. [Google Scholar] [CrossRef]
- Badisa, R.B.; Darling-Reed, S.F.; Joseph, P.; Cooperwood, J.S.; Latinwo, L.M.; Goodman, C.B. Selective cytotoxic activities of two novel synthetic drugs on human breast carcinoma MCF-7 cells. Anticancer Res. 2009, 29, 2993–2996. [Google Scholar] [PubMed]
- Schwartz, G.K.; Shah, M.A. Targeting the Cell Cycle: A New Approach to Cancer Therapy. J. Clin. Oncol. 2005, 23, 9408–9421. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, G.I.; Harper, J.W. Anticancer drug targets: Cell cycle and checkpoint control. J. Clin. Investig. 1999, 104, 1645–1653. [Google Scholar] [CrossRef] [Green Version]
- Nunez, R. DNA Measurement and Cell Cycle Analysis by Flow Cytometry. Curr. Issues Mol. Biol. 2001, 3, 67–70. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.S.; Linette, G.P.; Stec, J.; Ross, M.S.; Anwar, S.; Boguniewicz, A. DNA ploidy and cell cycle analysis in breast cancer. Pathol. Patterns Rev. 2003, 120, S72–S84. [Google Scholar] [CrossRef]
- Drummond, C. The Mechanismof Anti-Tumour Activity of DNA Binding Agent SN 280409; The University of Auckland: Auckland, New Zealand, 2007. [Google Scholar]
- Taylor, W.R.; Stark, G.R. Regulation of the G2/M transition by p53. Oncogene 2001, 20, 1803–1815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, Y.; Miyoshi, N. Cell death induction by isothiocyanates and their underlying molecular mechanisms. Biofactors 2006, 26, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Krysko, D.V.; Berghe, T.V.; D’herde, K.; Vandenabeele, P. Apoptosis and necrosis: Detection, discrimination and phagocytosis. Methods 2008, 44, 205–221. [Google Scholar] [CrossRef]
- Fink, S.L.; Cookson, B.T. Apoptosis, Pyroptosis, and Necrosis: Mechanistic Description of Dead and Dying Eukaryotic Cells. Infect. Immun. 2005, 73, 1907–1916. [Google Scholar] [CrossRef] [Green Version]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Saraste, A.; Pulkki, K. Morphologic and biochemical hallmarks of apoptosis. Cardiovasc. Res. 2000, 45, 528–537. [Google Scholar] [CrossRef]
- Schafer, J.M.; Lee, E.S.; O’Regan, R.M.; Yao, K.; Jordan, V.C. Rapid development of tamoxifen-stimulated mutant p53 breast tumors (T47D) in athymic mice. Clin. Cancer Res. 2000, 6, 4373–4380. [Google Scholar]
- Prunet, C.; Lemaire-Ewing, S.; Ménétrier, F.; Néel, D.; Lizard, G. Activation of caspase-3-dependent and -independent pathways during 7-ketocholesterol- and 7β-hydroxycholesterol-induced cell death: A morphological and biochemical study. J. Biochem. Mol. Toxicol. 2005, 19, 311–326. [Google Scholar] [CrossRef]
- Fulda, S.; Gorman, A.M.; Hori, O.; Samali, A. Cellular Stress Responses: Cell Survival and Cell Death. Int. J. Cell Biol. 2010, 2010, 1–23. [Google Scholar] [CrossRef] [Green Version]
- Wilken, R.; Veena, M.S.; Wang, M.B.; Srivatsan, E.S. Curcumin: A review of anti-cancer properties and therapeutic activity in head and neck squamous cell carcinoma. Mol. Cancer 2011, 10, 12. [Google Scholar] [CrossRef] [Green Version]
- Nacht, A.S.; Ferrari, R.; Zaurin, R.; Scabia, V.; Carbonell-Caballero, J.; Le Dily, F.; Quilez, J.; Leopoldi, A.; Brisken, C.; Beato, M.; et al. C/EBPα mediates the growth inhibitory effect of progestins on breast cancer cells. EMBO J. 2019, 38, e101426. [Google Scholar] [CrossRef] [PubMed]
- Truong, T.H.; Dwyer, A.R.; Diep, C.H.; Hu, H.; Hagen, K.M.; A Lange, C. Phosphorylated Progesterone Receptor Isoforms Mediate Opposing Stem Cell and Proliferative Breast Cancer Cell Fates. Endocrinology 2018, 160, 430–446. [Google Scholar] [CrossRef] [Green Version]
- Mohammed, H.; Russell, I.A.; Stark, R.; Rueda, O.M.; Hickey, T.E.; Tarulli, G.A.; Serandour, A.A.A.; Birrell, S.N.; Bruna, A.; Saadi, A.; et al. Progesterone receptor modulates ERα action in breast cancer. Nature 2015, 523, 313–317. [Google Scholar] [CrossRef] [Green Version]
- Shiau, A.K.; Barstad, D.; Loria, P.M.; Cheng, L.; Kushner, P.J.; Agard, D.A.; Greene, G.L. The Structural Basis of Estrogen Receptor/Coactivator Recognition and the Antagonism of This Interaction by Tamoxifen. Cell 1998, 95, 927–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matias, P.M.; Donner, P.; Coelho, R.; Thomaz, M.; Peixoto, C.; Macedo, S.; Otto, N.; Joschko, S.; Scholz, P.; Wegg, A.; et al. Structural Evidence for Ligand Specificity in the Binding Domain of the Human Androgen Receptor: Implications for Pathogenic Gene Mutations. J. Biol. Chem. 2000, 275, 26164–26171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, F.; Liao, M.; Qin, R.; Zhu, S.; Peng, C.; Fu, L.; Chen, Y.; Han, B. Regulated cell death (RCD) in cancer: Key pathways and targeted therapies. Signal Transduct. Target. Ther. 2022, 7, 286. [Google Scholar] [CrossRef]
- Li, P.; Zhou, L.; Zhao, T.; Liu, X.; Zhang, P.; Liu, Y.; Zheng, X.; Li, Q. Caspase-9: Structure, mechanisms and clinical application. Oncotarget 2017, 8, 23996–24008. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zhou, M.; Hu, Q.; Bai, X.-C.; Huang, W.; Scheres, S.H.W.; Shi, Y. Mechanistic insights into caspase-9 activation by the structure of the apoptosome holoenzyme. Proc. Natl. Acad. Sci. USA 2017, 114, 1542–1547. [Google Scholar] [CrossRef]
- Sharifi, S.; Barar, J.; Hejazi, M.S.; Samadi, N. Doxorubicin Changes Bax /Bcl-xL Ratio, Caspase-8 and 9 in Breast Cancer Cells. Adv. Pharm. Bull. 2015, 5, 351–359. [Google Scholar] [CrossRef] [Green Version]
- Backer, C.A.; van den Brink, B. Flora of Java (Spermatophytes Only); Wolters-Noordhoff N.V.: Groningen, The Netherlands, 1965; Volume II, p. 512. [Google Scholar]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Van Engeland, M.; Ramaekers, F.C.S.; Schutte, B.; Reutelingsperger, C.P.M. A Novel Assay to Measure Loss of Plasma Membrane Asymmetry during Apoptosis of Adherent Cells in Culture. Cytometry 1996, 24, 131–139. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.I.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Cousins, K.R. ChemDraw Ultra 9.0. CambridgeSoft, 100 CambridgePark Drive, Cambridge, MA 02140. www.cambridgesoft.com. See Web site for pricing options. J. Am. Chem. Soc. 2005, 127, 4115–4116. [Google Scholar] [CrossRef]
- Froimowitz, M. HyperChem(TM): A Software Package for Computational Chemistry and Molecular Modeling. BioTechniques 1993, 14, 1010–1013. [Google Scholar] [PubMed]
- Arfan, A.; Muliadi, R.; Malina, R.; Trinovitasari, N.; Asnawi, A. Docking and Dynamics Studies: Identifying the Binding Ability of Quercetin Analogs to the ADP-Ribose Phosphatase of SARS CoV-2. J. Kartika Kim. 2023, 5, 145–151. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Citra, S.N.A.L.; Arfan, A.; Alroem, A.; Bande, L.S.; Irnawati, I.; Arba, M. Docking-based workflow and ADME prediction of some compounds in Curcuma longa and Andrographis paniculata as polymerase PA-PB1 inhibitors of influenza A/H5N1 virus. J. Res. Pharm. 2023, 27, 221–231. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1H (δppm) | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| H-1 | 1.13 m; 1.58 m | 1.76 m; 1.19 br d (13.9) | 1.82 m; 1.29 br d (13.3) | - | - |
| H-2 | 4.20 m | 1.75 m; 1.58 m | 1.77m; 1.67 m | - | - |
| H-3 | 1.63 m | 1.73 m; 1.21 br d (13.6) | 1.86 m; 1.77 m | - | - |
| H-5 | 2.06 d (2.5) | 2.47 br d (2.2) | 2.88 d (2.0) | 2.06 br d (2.4) | 2.06 br d (2.3) |
| H-6 | 5.60 s | 5.51 t (2.4) | 5.30 t (2.4) | 5.63 br d (2.4) | 5.63 br d (2.0) |
| H-7 | 2.33 m | 4.16 d (2.4) | 5.29 d (2.5) | 2.40 m | 2.36 m |
| H-9 | 1.80 m | 1.88 br s | 1.97 m | - | - |
| H-11 | 1.74 m | 1.91 dd (10.6; 4.1); 1.77 m | 1.81 m | - | - |
| H-12 | 1.83 m | 2.11 br t (11.1); 1.85 m | 1.96 m; 1.83 m | - | - |
| H-14 | 5.12 d (10) | 5.43 t (6.9) | 5.43 t (6.9) | 5.42 br t (6.8) | 5.35 br t (7.0) |
| H-15 | 3.66 m; 3.50 dt (5;10) | 4.13 d (6.9) | 4.19 d (6.9) | 4.16 d (6.8) | 4.60 d (7.0) |
| H-16 | 1.24 s | 1.68 s | 1.71 s | 1.68 s | 1.71 s |
| H-17 | 4.71 s | 5.03 d (2.1); 4.90 d (2.1) | 5.26 d (1.8); 5.07 d (1.8) | 4.73 s | 4.73 s |
| H-18 | 3.59 d (11); 3.10 d (11) | 3.66 d (11.4); 3.10 d (11.4) | 1.35 s | 3.60 d (11.0); 3.15 d (11.0) | 3.61 d (10.9); 3.16 d (10.6) |
| H-19 | 0.88 s | 0.89 s | 0.91 s | 0.92 s | |
| H-20 | 1.44 s | 1.43 s | 1.49 s | 1.46 s | 1.47 s |
| H-2′6′ | 8.01 d (7.5) | 8.01 d (7.4) | 8.03 d (7.4) | 8.04 d (7.3) | 8.04 d (7.6) |
| H-3′5′ | 7.43 t (7.5) | 7.45 t (7.4) | 7.46 t (7.4) | 7.44 t (7.3) | 7.45 t (7.6) |
| H-4′ | 7.54 t (7.5) | 7.57 t (7.4) | 7.58 t (7.4) | 7.56 t (7.4) | 7.56 t (7.6) |
| H-2″ | - | - | 2.09 s | - | 2.31 t (7.2) |
| H-3″-9″ | - | - | - | - | 1.2 –1.70 18 H |
| H-10″ | - | - | - | - | 0.90 t (7.2) |
| 13C | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| C-1 | 38.1 | 38.7 | 38.3 | 38.7 | 38.7 |
| C-2 | 75.2 | 18.5 | 18.2 | 18.7 | 18.8 |
| C-3 | 38.1 | 38.0 | 40.2 | 38.6 | 38.6 |
| C-4 | 38.8 | 38.6 | 46.8 | 38.3 | 38.3 |
| C-5 | 41.1 | 35.9 | 38.9 | 41.5 | 41.6 |
| C-6 | 71.7 | 74.7 | 73.9 | 71.8 | 71.8 |
| C-7 | 36.9 | 75.7 | 75.2 | 37.1 | 37.1 |
| C-8 | 144.4 | 145.2 | 140.4 | 144.5 | 144.5 |
| C-9 | 57.5 | 56.6 | 56.7 | 57.6 | 57.6 |
| C-10 | 38.5 | 38.4 | 38.2 | 38.9 | 38.9 |
| C-11 | 24.7 | 26.6 | 26.0 | 24.4 | 24.4 |
| C-12 | 31.5 | 38.7 | 38.2 | 38.3 | 38.3 |
| C-13 | 148.9 | 140.6 | 140.0 | 140.3 | 142.6 |
| C-14 | 111.0 | 123.1 | 123.1 | 123.4 | 118.6 |
| C-15 | 65.8 | 59.4 | 59.4 | 59.6 | 61.4 |
| C-16 | 29.8 | 16.6 | 16.7 | 16.8 | 16.9 |
| C-17 | 113.2 | 118.3 | 121.9 | 113.2 | 113.2 |
| C-18 | 71.2 | 70.8 | 182.1 | 71.5 | 71.5 |
| C-19 | 20.5 | 20.9 | 19.7 | 20.5 | 20.5 |
| C-20 | 26.0 | 26.0 | 25.7 | 26.1 | 26.1 |
| C=O | 166.4 | 166.4 | 165.6 | 166.5 | 166.5 |
| C-1′ | 130.7 | 130.3 | 130.0 | 131.0 | 131.0 |
| C-2′6′ | 129.7 | 129.7 | 129.8 | 129.9 | 129.9 |
| C-3′5′ | 128.5 | 128.5 | 128.5 | 128.7 | 128.7 |
| C-4′ | 132.9 | 133.1 | 133.2 | 133.1 | 133.1 |
| C-1″ | - | - | 21.3 | - | 174.1 |
| C-2″ | - | - | - | - | 38.4 |
| C-3″-9″ | - | - | - | - | 25.1–38.9 |
| C-10″ | - | - | - | - | 15.0 |
| NO | Sample | IC50 (μg/mL) | Selectivity Index (SI) | |||
|---|---|---|---|---|---|---|
| MCF-7 | T47D | Vero | MCF-7 | T47D | ||
| 1 | Compound (1) | 70.56 ± 1.54 a | <3.125 ± 0.43 a | 36.50 ± 1.41 a | 0.51 | 12.67 |
| 2 | Doxorubicin | 16.62 ± 5.85 b | 35.08 ± 3.14 b | 69.52 ± 6.80 b | 4.2 | 1.45 |
| Compound | Binding Energy (kcal/mol) | ||
|---|---|---|---|
| Estrogen Receptor (PDB ID 3ERT) | Progesterone Receptor (PDB ID 1E3K) | Caspase-9 (PDB ID 5WVE) | |
| TMX (Tamoxifen) | −9.8 | - | - |
| R18 | - | −11.5 | - |
| DTP | - | - | −9.9 |
| 2α-hydroxyscopadiol | −8.2 | −7.3 | −9.0 |
| Doxorubicin | −7.7 | −5.6 | −9.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hasnawati, H.; Wahyuono, S.; Susidarti, R.A.; Santosa, D.; Arfan, A. A New Diterpenoid of Indonesian Scoparia dulcis Linn: Isolation and Cytotoxic Activity against MCF-7 and T47D Cell Lines. Molecules 2023, 28, 5960. https://doi.org/10.3390/molecules28165960
Hasnawati H, Wahyuono S, Susidarti RA, Santosa D, Arfan A. A New Diterpenoid of Indonesian Scoparia dulcis Linn: Isolation and Cytotoxic Activity against MCF-7 and T47D Cell Lines. Molecules. 2023; 28(16):5960. https://doi.org/10.3390/molecules28165960
Chicago/Turabian StyleHasnawati, Hasnawati, Subagus Wahyuono, Ratna Asmah Susidarti, Djoko Santosa, and Arfan Arfan. 2023. "A New Diterpenoid of Indonesian Scoparia dulcis Linn: Isolation and Cytotoxic Activity against MCF-7 and T47D Cell Lines" Molecules 28, no. 16: 5960. https://doi.org/10.3390/molecules28165960