
Monitoring Pharmaceuticals and Personal Care Products in Drinking Water Samples by the LC-MS/MS Method to Estimate Their Potential Health Risk

Abstract
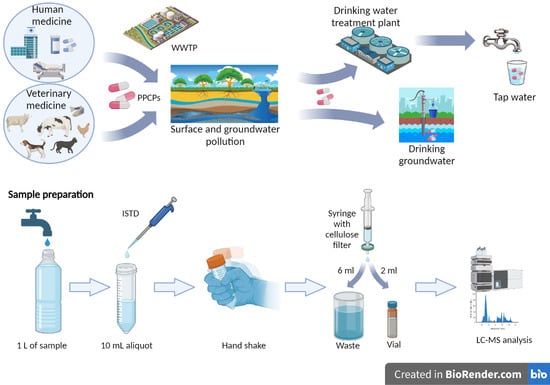
:
1. Introduction
2. Results and Discussion
2.1. Optimization of HPLC-MS/MS
2.2. Validation of Method
2.2.1. Selectivity
2.2.2. Limit of Detection (LOD) and Limit of Quantitation (LOQ)
2.2.3. Linear and Working Range
2.2.4. Precision
2.2.5. Accuracy
2.3. Results of Drug Monitoring in Drinking Water Samples
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Sample Collection
3.3. Stock Solutions, Calibration Standards, and Quality Control (QC) Samples
3.4. Chromatographic Conditions
3.5. Preparation of Blank Samples
3.6. Preparation of Laboratory Control Samples (LCS)
3.7. Preparation of Fortified Matrices for Validation
3.8. Method Validation
3.8.1. Selectivity
3.8.2. Limit of Detection and Limit of Quantification
3.8.3. Linear and Working Range
3.8.4. Precision
3.8.5. Accuracy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Gosset, A.; Wiest, L.; Fildier, A.; Libert, C.; Giroud, B.; Hammada, M.; Hervé, M.; Sibeud, E.; Vulliet, E.; Polomé, P.; et al. Ecotoxicological Risk Assessment of Contaminants of Emerging Concern Identified by “Suspect Screening” from Urban Wastewater Treatment Plant Effluents at a Territorial Scale. Sci. Total Environ. 2021, 778, 146275. [Google Scholar] [CrossRef] [PubMed]
- Ohoro, C.R.; Adeniji, A.O.; Okoh, A.I.; Okoh, O.O. Distribution and Chemical Analysis of Pharmaceuticals and Personal Care Products (PPCPs) in the Environmental Systems: A Review. Int. J. Environ. Res. Public Health 2019, 16, 3026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gwenzi, W.; Kanda, A.; Danha, C.; Muisa-Zikali, N.; Chaukura, N. Occurrence, Human Health Risks, and Removal of Pharmaceuticals in Aqueous Systems: Current Knowledge and Future Perspectives. Appl. Water Sci. Vol. 1 Fundam. Appl. 2021, 1, 63–101. [Google Scholar] [CrossRef]
- Santos, A.V.; Couto, C.F.; Lebron, Y.A.R.; Moreira, V.R.; Foureaux, A.F.S.; Reis, E.O.; Santos, L.V.d.S.; de Andrade, L.H.; Amaral, M.C.S.; Lange, L.C. Occurrence and Risk Assessment of Pharmaceutically Active Compounds in Water Supply Systems in Brazil. Sci. Total Environ. 2020, 746, 141011. [Google Scholar] [CrossRef]
- Marsik, P.; Rezek, J.; Židková, M.; Kramulová, B.; Tauchen, J.; Vaněk, T. Non-Steroidal Anti-Inflammatory Drugs in the Watercourses of Elbe Basin in Czech Republic. Chemosphere 2017, 171, 97–105. [Google Scholar] [CrossRef]
- Pereira, A.; Silva, L.; Laranjeiro, C.; Pena, A. Assessment of Human Pharmaceuticals in Drinking Water Catchments, Tap and Drinking Fountain Waters. Appl. Sci. 2021, 11, 7062. [Google Scholar] [CrossRef]
- Paíga, P.; Santos, L.H.M.L.M.; Delerue-Matos, C. Development of a Multi-Residue Method for the Determination of Human and Veterinary Pharmaceuticals and Some of Their Metabolites in Aqueous Environmental Matrices by SPE-UHPLC–MS/MS. J. Pharm. Biomed. Anal. 2017, 135, 75–86. [Google Scholar] [CrossRef] [Green Version]
- Papagiannaki, D.; Morgillo, S.; Bocina, G.; Calza, P.; Binetti, R. Occurrence and Human Health Risk Assessment of Pharmaceuticals and Hormones in Drinking Water Sources in the Metropolitan Area of Turin in Italy. Toxics 2021, 9, 88. [Google Scholar] [CrossRef]
- Klančar, A.; Trontelj, J.; Roškar, R. Development of a Multi-Residue Method for Monitoring 44 Pharmaceuticals in Slovene Surface Water by SPE-LC-MS/MS. Water Air Soil Pollut. 2018, 229, 192. [Google Scholar] [CrossRef]
- Brieudes, V.; Lardy-Fontan, S.; Lalere, B.; Vaslin-Reimann, S.; Budzinski, H. Validation and Uncertainties Evaluation of an Isotope Dilution-SPE-LC–MS/MS for the Quantification of Drug Residues in Surface Waters. Talanta 2016, 146, 138–147. [Google Scholar] [CrossRef]
- Ferrer-Aguirre, A.; Romero-González, R.; Vidal, J.L.M.; Frenich, A.G. Simple and Quick Determination of Analgesics and Other Contaminants of Emerging Concern in Environmental Waters by On-Line Solid Phase Extraction Coupled to Liquid Chromatography–Tandem Mass Spectrometry. J. Chromatogr. A 2016, 1446, 27–33. [Google Scholar] [CrossRef]
- United States Environmental Protection Agency. Method 1694: Pharmaceuticals and Personal Care Products in Water, Soil, Sediment, and Biosolids by HPLC/MS/MS; United States Environmental Protection Agency: Washington, DC, USA, 2007.
- Meng, Y.; Liu, W.; Liu, X.; Zhang, J.; Peng, M.; Zhang, T. A Review on Analytical Methods for Pharmaceutical and Personal Care Products and Their Transformation Products. J. Environ. Sci. 2021, 101, 260–281. [Google Scholar] [CrossRef]
- Barceló, D.; Petrovic, M. Challenges and Achievements of LC-MS in Environmental Analysis: 25 Years On. TrAC Trends Anal. Chem. 2007, 26, 2–11. [Google Scholar] [CrossRef]
- Buchberger, W.W. Novel Analytical Procedures for Screening of Drug Residues in Water, Wastewater, Sediment and Sludge. Anal. Chim. Acta 2007, 593, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Jakimska, A.; Śliwka-Kaszyńska, M.; Reszczyńska, J.; Namieśnik, J.; Kot-Wasik, A. Elucidation of Transformation Pathway of Ketoprofen, Ibuprofen, and Furosemide in Surface Water and Their Occurrence in the Aqueous Environment Using UHPLC-QTOF-MS. Anal. Bioanal. Chem. 2014, 406, 3667–3680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wabaidur, S.M.; AlOthman, Z.A.; Siddiqui, M.R.; Mohsin, K.; Bousiakou, L.G.; Karikas, G.A. UPLC-MS Method for the Simultaneous Determination of Naproxen, Fluvastatin and Ibuprofen in Wastewater Samples. J. Ind. Eng. Chem. 2015, 24, 302–307. [Google Scholar] [CrossRef]
- Campos-Mañas, M.C.; Plaza-Bolaños, P.; Sánchez-Pérez, J.A.; Malato, S.; Agüera, A. Fast Determination of Pesticides and Other Contaminants of Emerging Concern in Treated Wastewater Using Direct Injection Coupled to Highly Sensitive Ultra-High Performance Liquid Chromatography-Tandem Mass Spectrometry. J. Chromatogr. A 2017, 1507, 84–94. [Google Scholar] [CrossRef]
- González-Curbelo, M.Á.; Socas-Rodríguez, B.; Herrera-Herrera, A.V.; González-Sálamo, J.; Hernández-Borges, J.; Rodriguez-Delgado, M.A. Evolution and Applications of the QuEChERS Method. TrAC Trends Anal. Chem. 2015, 71, 169–185. [Google Scholar] [CrossRef]
- Cerqueira, M.B.R.; Guilherme, J.R.; Caldas, S.S.; Martins, M.L.; Zanella, R.; Primel, E.G. Evaluation of the QuEChERS Method for the Extraction of Pharmaceuticals and Personal Care Products from Drinking-Water Treatment Sludge with Determination by UPLC-ESI-MS/MS. Chemosphere 2014, 107, 74–82. [Google Scholar] [CrossRef]
- Aristizabal-Ciro, C.; Botero-Coy, A.M.; López, F.J.; Peñuela, G.A. Monitoring Pharmaceuticals and Personal Care Products in Reservoir Water Used for Drinking Water Supply. Environ. Sci. Pollut. Res. 2017, 24, 7335–7347. [Google Scholar] [CrossRef] [Green Version]
- Afonso-Olivares, C.; Čadková, T.; Sosa-Ferrera, Z.; Santana-Rodríguez, J.J.; Nováková, L. Simplified Solid-Phase Extraction Procedure Combined with Liquid Chromatography Tandem–Mass Spectrometry for Multiresidue Assessment of Pharmaceutical Compounds in Environmental Liquid Samples. J. Chromatogr. A 2017, 1487, 54–63. [Google Scholar] [CrossRef]
- Ngubane, N.P.; Naicker, D.; Ncube, S.; Chimuka, L.; Madikizela, L.M. Determination of Naproxen, Diclofenac and Ibuprofen in Umgeni Estuary and Seawater: A Case of Northern Durban in KwaZulu–Natal Province of South Africa. Reg. Stud. Mar. Sci. 2019, 29, 100675. [Google Scholar] [CrossRef]
- Petrie, B.; Youdan, J.; Barden, R.; Kasprzyk-Hordern, B. Multi-Residue Analysis of 90 Emerging Contaminants in Liquid and Solid Environmental Matrices by Ultra-High-Performance Liquid Chromatography Tandem Mass Spectrometry. J. Chromatogr. A 2016, 1431, 64–78. [Google Scholar] [CrossRef] [PubMed]
- Yao, W.; Ge, J.; Hu, Q.; Ma, J.; Yuan, D.; Fu, X.; Qi, Y.; Volmer, D.A. An Advanced LC–MS/MS Protocol for Simultaneous Detection of Pharmaceuticals and Personal Care Products in the Environment. Rapid Commun. Mass Spectrom. 2022, 37, e9397. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-Q.; Hu, L.-X.; Zhao, J.-H.; Han, Y.; Liu, Y.-S.; Zhao, J.-L.; Yang, B.; Ying, G.-G. Suspect, Non-target and Target Screening of Pharmaceuticals and Personal Care Products (PPCPs) in a Drinking Water System. Sci. Total Environ. 2021, 808, 151866. [Google Scholar] [CrossRef]
- Wee, S.Y.; Ismail, N.A.H.; Haron, D.E.M.; Yusoff, F.M.; Praveena, S.M.; Aris, A.Z. Pharmaceuticals, Hormones, Plasticizers, and Pesticides in Drinking Water. J. Hazard. Mater. 2022, 424, 127327. [Google Scholar] [CrossRef]
- Dodávky léčivých přípravků v ČR v jednotlivých letech, Státní ústav pro kontrolu léčiv. Available online: https://www.sukl.cz/dodavky-leciv-v-ceske-republice-v-jednotlivych-letech (accessed on 26 June 2023).
- Mompelat, S.; Le Bot, B.; Thomas, O. Occurrence and Fate of Pharmaceutical Products and By-Products, from Resource to Drinking Water. Environ. Int. 2009, 35, 803–814. [Google Scholar] [CrossRef]
- Caban, M.; Lis, E.; Kumirska, J.; Stepnowski, P. Determination of Pharmaceutical Residues in Drinking Water in Poland Using a New SPE-GC-MS(SIM) Method Based on Speedisk Extraction Disks and DIMETRIS Derivatization. Sci. Total Environ. 2015, 538, 402–411. [Google Scholar] [CrossRef] [PubMed]
- Becerra-Herrera, M.; Honda, L.; Richter, P. Ultra-High-Performance Liquid Chromatography—Time-of-Flight High Resolution Mass Spectrometry to Quantify Acidic Drugs in Wastewater. J. Chromatogr. A 2015, 1423, 96–103. [Google Scholar] [CrossRef]
- Liu, S.; Ying, G.-G.; Zhao, J.-L.; Chen, F.; Yang, B.; Zhou, L.-J.; Lai, H.-J. Trace Analysis of 28 Steroids in Surface Water, Wastewater and Sludge sSamples by Rapid Resolution Liquid Chromatography–Electrospray Ionization Tandem Mass Spectrometry. J. Chromatogr. A 2011, 1218, 1367–1378. [Google Scholar] [CrossRef]
- Zhou, L.-J.; Ying, G.-G.; Liu, S.; Zhao, J.-L.; Chen, F.; Zhang, R.-Q.; Peng, F.-Q.; Zhang, Q.-Q. Simultaneous Determination of Human and Veterinary Antibiotics in Various Environmental Matrices by Rapid Resolution Liquid Chromatography–Electrospray Ionization Tandem Mass Spectrometry. J. Chromatogr. A 2012, 1244, 123–138. [Google Scholar] [CrossRef]
- Hong, B.; Yu, S.; Zhou, M.; Li, J.; Ding, J.; Niu, Y. Development of a PH-Paralleling Approach of Quantifying Six-Category Pharmaceuticals in Surface Water Using SPE-HPLC-MS/MS. Watershed Ecol. Environ. 2021, 3, 1–16. [Google Scholar] [CrossRef]
- Cai, M.-Q.; Wang, R.; Feng, L.; Zhang, L.-Q. Determination of Selected Pharmaceuticals in Tap Water and Drinking Water Treatment Plant by High-Performance Liquid Chromatography-Triple Quadrupole Mass Spectrometer in Beijing, China. Environ. Sci. Pollut. Res. 2015, 22, 1854–1867. [Google Scholar] [CrossRef] [PubMed]
- Pérez, S.; Barceló, D. Fate and Occurrence of X-ray Contrast Media in the Environment. Anal. Bioanal. Chem. 2007, 387, 1235–1246. [Google Scholar] [CrossRef]
- Wang, C.; Shi, H.; Adams, C.D.; Gamagedara, S.; Stayton, I.; Timmons, T.; Ma, Y. Investigation of Pharmaceuticals in Missouri Natural and Drinking Water Using High Performance Liquid Chromatography-Tandem Mass Spectrometry. Water Res. 2011, 45, 1818–1828. [Google Scholar] [CrossRef]
- Azzouz, A.; Ballesteros, E. Influence of Seasonal Climate Differences on the Pharmaceutical, Hormone and Personal Care Product Removal Efficiency of a Drinking Water Treatment Plant. Chemosphere 2013, 93, 2046–2054. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Fontela, M.; Galceran, M.T.; Ventura, F. Occurrence and Removal of Pharmaceuticals and Hormones through Drinking Water Treatment. Water Res. 2011, 45, 1432–1442. [Google Scholar] [CrossRef]
- Sodré, F.F.; Sampaio, T.R. Development and Application of a SPE-LC-QTOF Method for the Quantification of Micropollutants of Emerging Concern in Drinking Waters from the Brazilian Capital. Emerg. Contam. 2020, 6, 72–81. [Google Scholar] [CrossRef]
- Fontanals, N.; Boleda, M.R.; Borrull, F.; Marcé, R.M.; Lacorte, S. Ceramic Passive Samplers for Determining Pharmaceuticals and Drugs of Abuse in River and Drinking Water. Sci. Total Environ. 2023, 889, 164267. [Google Scholar] [CrossRef]
- Peng, Y.; Hall, S.; Gautam, L. Drugs of Abuse in Drinking Water—A Review of Current Detection Methods, Occurrence, Elimination and Health Risks. TrAC Trends Anal. Chem. 2016, 85, 232–240. [Google Scholar] [CrossRef]
- Lee, S.-H.; Kim, K.-H.; Lee, M.; Lee, B.-D. Detection Status and Removal Characteristics of Pharmaceuticals in Wastewater Treatment Effluent. J. Water Process. Eng. 2019, 31, 100828. [Google Scholar] [CrossRef]
- Water Treatment and Pathogen Control—M. W. LeChevallier, Kwok-Keung Au—Knihy Google. Available online: https://books.google.cz/books?hl=cs&lr=&id=ImGUVFKp9vwC&oi=fnd&pg=PR9&ots=vkfcjHsvfH&sig=MF-WR4siXz-QU8n6I_5u7pNAgYQ&redir_esc=y#v=onepage&q&f=false (accessed on 26 June 2023).
- Okoye, C.O.; Okeke, E.S.; Okoye, K.C.; Echude, D.; Andong, F.A.; Chukwudozie, K.I.; Okoye, H.U.; Ezeonyejiaku, C.D. Occurrence and Fate of Pharmaceuticals, Personal Care Products (PPCPs) and Pesticides in African Water Systems: A Need for Timely Intervention. Heliyon 2022, 8, e09143. [Google Scholar] [CrossRef] [PubMed]
- Houtman, C.J.; Broek, R.T.; de Jong, K.; Pieterse, B.; Kroesbergen, J. A Multicomponent Snapshot of Pharmaceuticals and Pesticides in the River Meuse Basin. Environ. Toxicol. Chem. 2013, 32, 2449–2459. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MS conditions | Heat block temperature | 150 °C | |||||||
| Column voltage | 0.75 kV | ||||||||
| Desolvation temperature | 600 °C | ||||||||
| Desolvation gas flow | 800 L hod−1 | ||||||||
| Cone gas flow | 150 L hod−1 | ||||||||
| Nebulizer gas | 7.0 bar | ||||||||
| Collision gas flow | 0.19 L min−1 | ||||||||
| MRM window | 0.4 min | ||||||||
| LC conditions | Column | Acquity UPLC BEH C18 (2.1 mm × 100 mm × 1.7 µm) | |||||||
| Column temperature | 40 °C | ||||||||
| Injection volume | 50 µL | ||||||||
| Flow rate | 0.4 mL min−1 | ||||||||
| Total run time | 9 min | ||||||||
| Mobile phase | A | 0.01% formic acid in Milli-Q water | |||||||
| B | methanol | ||||||||
| Gradient profile | Time (min) | 0 | 0.5 | 5.0 | 5.1 | 7.0 | 7.1 | 9.0 | |
| A (%) | 98 | 98 | 5 | 0 | 0 | 98 | 98 | ||
| B (%) | 2 | 2 | 95 | 100 | 100 | 2 | 2 | ||
| Analyte | Average [ng L−1] | SD [ng L−1] | LOD [ng L−1] | LOQ [ng L−1] |
|---|---|---|---|---|
| Anastrozole | 9.57 | 0.26 | 0.96 | 2.89 |
| Atenolol | 11.47 | 0.88 | 3.30 | 9.89 |
| Azathioprine | 9.87 | 0.48 | 1.80 | 5.39 |
| Bezafibrate | 8.88 | 0.86 | 3.22 | 9.66 |
| Buprenorphine | 12.04 | 0.40 | 1.51 | 4.52 |
| Butorphanol | 11.92 | 1.12 | 4.21 | 12.62 |
| Caffeine | 90.94 | 15.60 | 58.45 | 175.36 |
| Capecitabine | 88.04 | 17.02 | 63.78 | 191.35 |
| Carbamazepine | 9.54 | 0.36 | 1.36 | 4.07 |
| Citalopram | 13.39 | 1.37 | 5.14 | 15.41 |
| Clofibric acid | 66.05 | 12.59 | 47.19 | 141.55 |
| Cyclobenzaprine | 67.31 | 7.70 | 28.85 | 86.56 |
| Cyclophosphamide | 9.26 | 0.56 | 2.10 | 6.30 |
| Diazepam | 9.79 | 0.19 | 0.72 | 2.16 |
| Diclofenac | 8.12 | 2.27 | 8.50 | 25.49 |
| Enalapril | 9.89 | 0.46 | 1.71 | 5.14 |
| Fluoxetine | 93.57 | 17.08 | 64.00 | 192.01 |
| Flutamide | 8.69 | 0.55 | 2.05 | 6.16 |
| Furosemide | 8.14 | 1.47 | 5.49 | 16.48 |
| Gabapentin | 88.59 | 2.97 | 11.14 | 33.41 |
| Gemfibrozil | 10.59 | 1.05 | 3.92 | 11.75 |
| Hydrochlorothiazide | 8.44 | 1.38 | 5.18 | 15.54 |
| Chloramfenicol | 11.42 | 1.39 | 5.19 | 15.58 |
| Ifosfamide | 10.02 | 0.62 | 2.31 | 6.93 |
| Indomethacin | 8.44 | 0.70 | 2.62 | 7.85 |
| Iomeprol | 8.13 | 1.09 | 4.10 | 12.30 |
| Iopamidol | 9.96 | 0.92 | 3.46 | 10.38 |
| Iopromide | 157.95 | 25.87 | 96.93 | 290.78 |
| Ketoprofen | 7.79 | 0.58 | 2.18 | 6.55 |
| Lincomycin | 10.28 | 0.64 | 2.41 | 7.22 |
| Loperamide | 145.68 | 19.76 | 74.04 | 222.11 |
| Metoprolol | 11.55 | 0.90 | 3.36 | 10.09 |
| Metronidazole | 9.90 | 0.19 | 0.70 | 2.09 |
| Mycophenolate Mofetil | 8.10 | 1.50 | 5.62 | 16.86 |
| Naproxen | 79.33 | 6.37 | 23.87 | 71.60 |
| Oxazepam | 9.56 | 0.32 | 1.18 | 3.55 |
| Paclitaxel | 64.46 | 12.44 | 46.60 | 139.81 |
| Paracetamol | 9.25 | 1.20 | 4.49 | 13.48 |
| Piroxicam | 9.93 | 0.31 | 1.15 | 3.45 |
| Propranolol | 9.42 | 0.63 | 2.35 | 7.04 |
| Salbutamol | 11.96 | 0.94 | 3.50 | 10.51 |
| Sertraline | 73.65 | 14.35 | 53.78 | 161.34 |
| Sotalol | 10.97 | 0.81 | 3.03 | 9.09 |
| Sulfamethazine | 9.68 | 0.24 | 0.90 | 2.71 |
| Sulfamethoxazole | 9.46 | 0.55 | 2.05 | 6.14 |
| Terbutaline | 11.93 | 1.37 | 5.13 | 15.38 |
| Thebaine | 11.92 | 1.38 | 5.18 | 15.55 |
| Tramadol | 11.54 | 1.09 | 4.08 | 12.24 |
| Trimethoprim | 8.04 | 1.19 | 4.47 | 13.40 |
| Valsartan | 9.09 | 1.27 | 4.77 | 14.31 |
| Warfarin | 8.04 | 0.51 | 1.92 | 5.75 |
| Zolpidem | 9.29 | 0.68 | 2.55 | 7.64 |
| Analyte | Concentration in Samples [μg L−1] | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TW1 | TW2 | TW3 | TW4 | TW5 | TW6 | TW7 | TW8 | TW9 | TW10 | TW11 | TW12 | TW13 | GW1 | GW2 | |
| Caffeine | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | <LOQ |
| Carbamazepine | 0.180 | 0.049 | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | 0.412 | n. d. | 0.047 | 0.058 | 0.058 | 0.047 |
| Gabapentin | n. d. | 0.204 | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | 0.185 | 0.071 | 0.252 |
| Gemfibrozil | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | 0.096 | n. d. | n. d. | n. d. | n. d. | 0.095 | 0.083 |
| Chloramphenicol | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | 0.024 | n. d. |
| Iomeprol | n. d. | 0.029 | 0.082 | 0.067 | 0.055 | 0.087 | 0.070 | 0.075 | 0.102 | 0.393 | 0.082 | 0.032 | 0.044 | 0.088 | 0.099 |
| Iopamidol | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | 0.014 | 0.188 |
| Sotalol | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | 0.011 |
| Sulfamethazine | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | 0.011 |
| Sulfamethoxazole | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | 0.035 |
| Tramadol | <LOQ | <LOQ | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | 0.036 |
| Valsartan | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | n. d. | 0.021 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Molnarova, L.; Halesova, T.; Vaclavikova, M.; Bosakova, Z. Monitoring Pharmaceuticals and Personal Care Products in Drinking Water Samples by the LC-MS/MS Method to Estimate Their Potential Health Risk. Molecules 2023, 28, 5899. https://doi.org/10.3390/molecules28155899
Molnarova L, Halesova T, Vaclavikova M, Bosakova Z. Monitoring Pharmaceuticals and Personal Care Products in Drinking Water Samples by the LC-MS/MS Method to Estimate Their Potential Health Risk. Molecules. 2023; 28(15):5899. https://doi.org/10.3390/molecules28155899
Chicago/Turabian StyleMolnarova, Lucia, Tatana Halesova, Marta Vaclavikova, and Zuzana Bosakova. 2023. "Monitoring Pharmaceuticals and Personal Care Products in Drinking Water Samples by the LC-MS/MS Method to Estimate Their Potential Health Risk" Molecules 28, no. 15: 5899. https://doi.org/10.3390/molecules28155899