

Finding a Novel Chalcone–Cinnamic Acid Chimeric Compound with Antiproliferative Activity against MCF-7 Cell Line Using a Free-Wilson Type Approach
 ,
,  , , , , and
, , , , and
Abstract
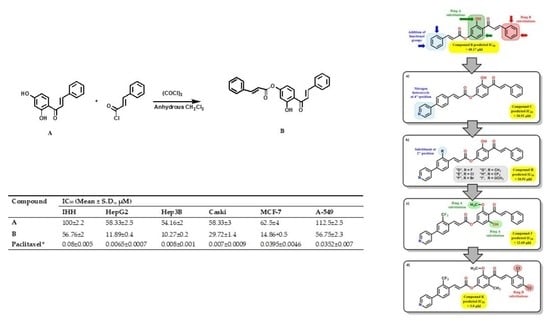
:
1. Introduction
2. Results
2.1. QSAR Study
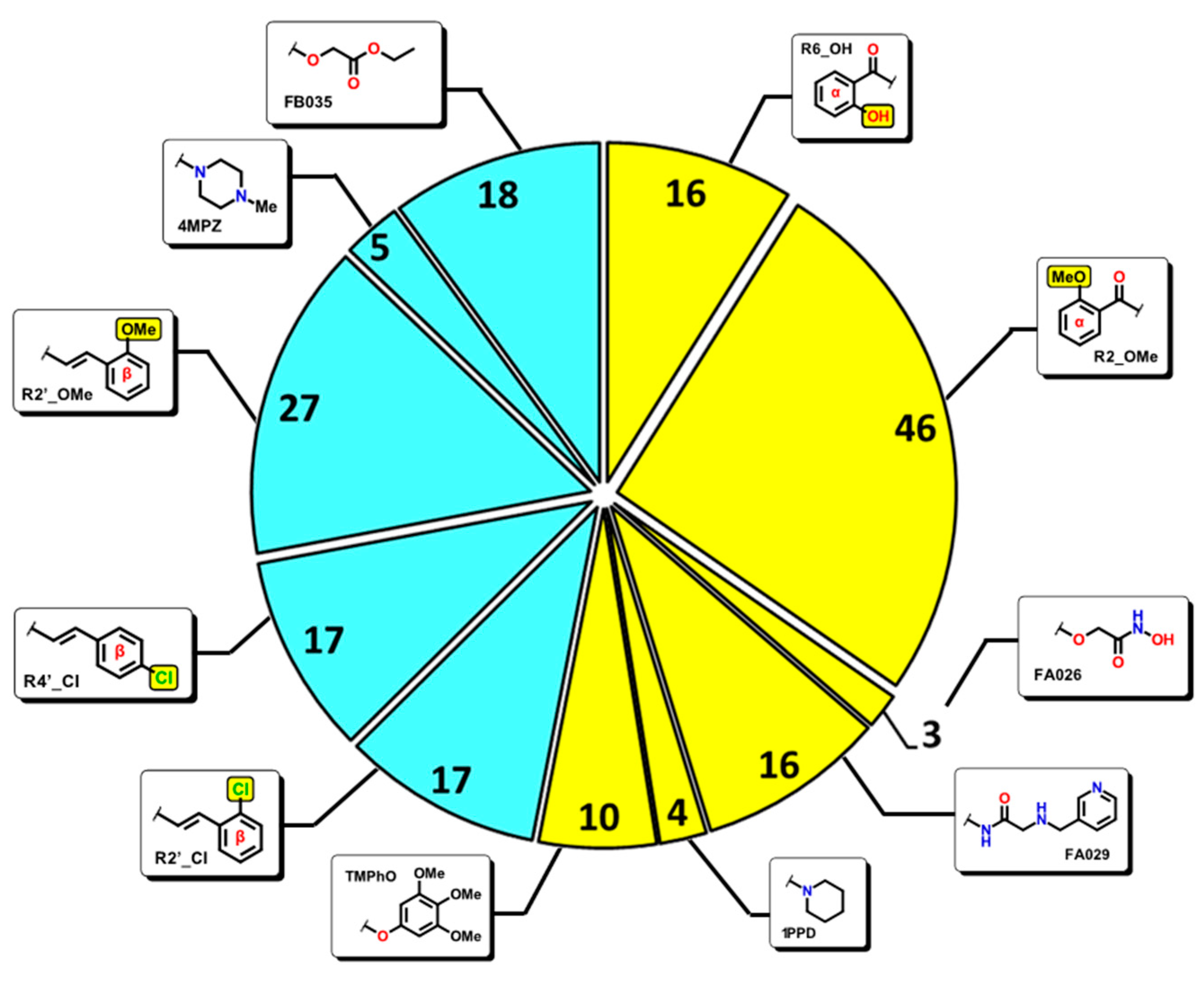
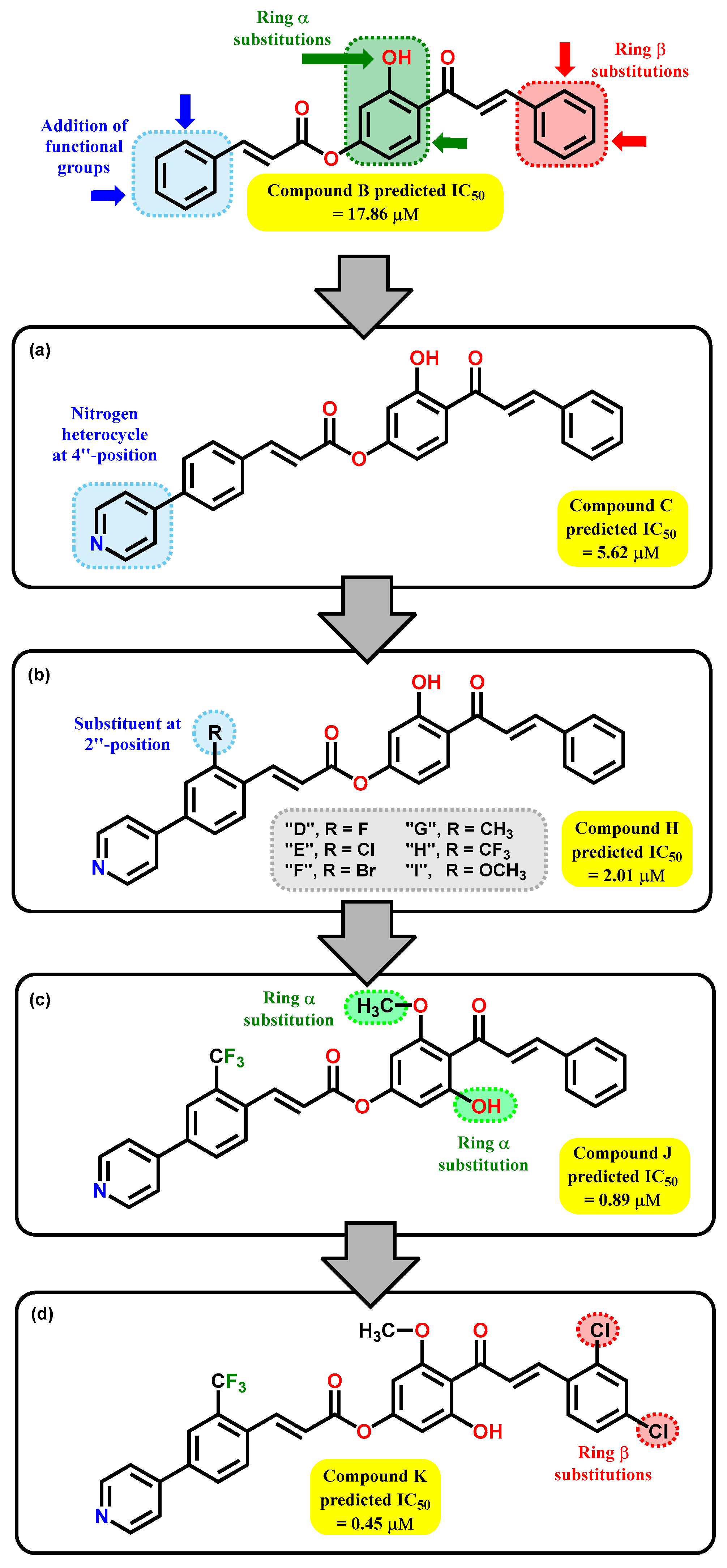
2.2. QSAR Interpretation
2.3. Design and Synthesis of 2′-Hydroxy-4′-cinnamate Chalcone
2.4. Biological Activity
2.5. In Silico ADME/tox Studies of A–K
3. Material and Methods
3.1. Calculation of the Molecular Descriptors
3.2. DataSet
3.3. Construction of QSAR Model
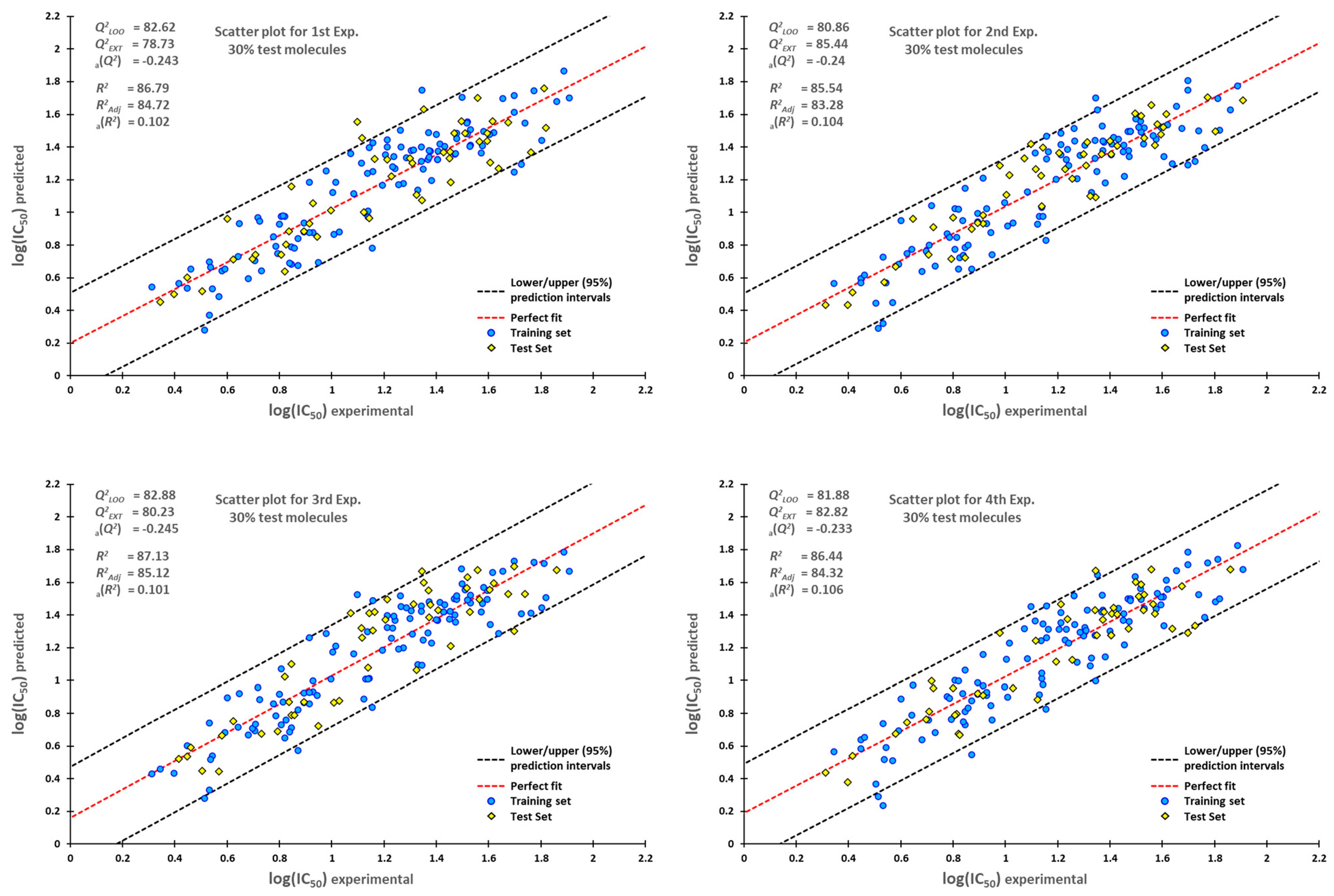
3.4. Statistical Validation
3.5. External Validation
3.6. Chemistry
3.6.1. Synthesis of 2′,4′-Dihydroxychalcone (A)
3.6.2. Synthesis of 2′-Hydroxy-4′-cinnamate Chalcone (B)
3.7. Antiproliferative Activity
3.8. In Silico ADME/tox
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Organización Mundial de la Salud. 2018. Available online: https://www.who.int/es/news-room/fact-sheets/detail/cancer (accessed on 20 April 2018).
- U.S. Department of Health and Human Services; National Institutes of Health; National Cancer Institute. Understanding Cancer. 2023. Available online: https://www.cancer.gov (accessed on 20 April 2018).
- Ferlay, J.; Ervik, M.; Lam, F.; Colombet, M.; Mery, L.; Piñeros, M. Global Cancer Observatory: Cancer Today. Int. Agency Res. Cancer 2020, 3, 2019. [Google Scholar]
- International Agency for Research on Cancer Mexico. The Global Cancer Observatory, 283, 2018–2019. Available online: http://gco.iarc.fr/today/data/factsheets/populations/484-mexico-fact-sheets.pdf (accessed on 18 December 2022).
- Mathew, A.B.; Mathew, G.E.; Joy, M.; Nafna, E.K.; Suresh, J. Monoamine oxidase inhibitory activity of methoxy-substituted chalcones. Int. J. Biol. Macromol. 2017, 104, 1321–1329. [Google Scholar] [CrossRef]
- Lou, C.; Guangming Yang, G.; Hao Cai, H.; Zou, M.; Xu, Z.; Li, Y.; Zhao, F.; Li, W.; Li Tong, L.; Wang, M.; et al. 2′,4′-Dihydroxychalcone-induced apoptosis of human gastric cancer MGC-803 cells via down-regulation of survivin mRNA. Toxicol. In Vitro 2010, 24, 1333–1337. [Google Scholar] [CrossRef]
- Sahu, K.; Balbhadra, N.S.; Choudhary, S.J.; Kohli, V.D. Exploring Pharmacological Significance of Chalcone Scaffold: A Review. Curr. Med. Chem. 2012, 19, 209–225. [Google Scholar] [CrossRef]
- Cabral, B.L.S.; da Silva, A.C.G.; de Avila, R.I.; Cortez, A.P.; Luzin, R.M.; Liao, L.M.; de Souza Gil, E.; Sanz, G.; Vaz, B.G.; Sabino, J.R.; et al. A novel chalcone derivative, LQFM064, induces breast cancer cells death via p53, p21, KIT and PDGFRA. Eur. J. Pharm. Sci. 2017, 107, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Damodar, K.; Kim, J.K.; Jun, J.G. Synthesis and pharmacological properties of naturally occurring prenylated and pyranochalcones as potent anti-inflammatory agents. Chin. Chem. Lett. 2016, 27, 698–702. [Google Scholar] [CrossRef]
- Sánchez-Carranza, J.N.; Alvarez, L.; Marquina-Bahena, S.; Salas-Vidal, E.; Cuevas, V.; Jiménez, E.W.; Veloz, G.R.A.; Carraz, M.; González-Maya, L. Phenolic Compounds Isolated from Caesalpinia coriaria Induce S and G2/M Phase Cell Cycle Arrest Differentially and Trigger Cell Death by Interfering with Microtubule Dynamics in Cancer Cell Lines. Molecules 2017, 22, 666. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, E.; Webster, J.; Do, T.; Kline, R.; Snider, L.; Hauser, Q.; Higginbottom, G.; Campbell, A.; Ma, L.; Stefan, P. Hydroxylated chalcones with dual properties: Xanthine oxidase inhibitors and radical scavengers. Bioorg. Med. Chem. 2015, 24, 578–587. [Google Scholar] [CrossRef] [Green Version]
- Iftikhar, S.; Khan, S.; Bilal, A.; Manzoor, S.; Abdullah, M.; Emwas, A.; Salim, S.; Gao, X.; Chotana, G.A.; Faisal, A.; et al. Synthesis and evaluation of modified chalcone based p53 stabilizing agents. Bioorg. Med. Chem. Lett. 2017, 27, 4101–4106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, L.; Yang, Z.; Li, C.; Zhu, Z.; Shen, X.; Hu, L. Design, synthesis and SAR study of hydroxychalcone inhibitors of human β-secretase (BACE1). J. Enzyme Inhib. Med. Chem. 2011, 26, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Niu, C.; Tuerxuntayi, A.; Li, G.; Kabas, M.; Dong, C.Z.; Aisa, H.A. Design, synthesis and bioactivity of chalcones and its analogues. Chin. Chem. Lett. 2017, 28, 1533–1538. [Google Scholar] [CrossRef]
- Zhuang, C.; Zhang, W.; Sheng, C.; Zhang, W.; Xing, C.; Miao, Z. Chalcone: A Privileged Structure in Medicinal Chemistry. Chem. Rev. 2017, 117, 7762–7810. [Google Scholar] [CrossRef] [PubMed]
- Das, M.; Manna, K. Chalcone Scaffold in Anticancer Armamentarium: A Molecular Insight. J. Toxicol. 2016, 2016, 7651047. [Google Scholar] [CrossRef] [Green Version]
- Zhang, E.H.; Wang, R.F.; Guo, S.-Z.; Liu, B. An Update on Antitumor Activity of Naturally Occurring Chalcones. Evid. Based Complement. Altern. Med. 2013, 2013, 815621. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.H.; Wang, R.; Shi, Y.P. Flavonoids in the poisonous plant oxytropis falcata. J. Nat. Prod. 2010, 73, 1398–1403. [Google Scholar] [CrossRef]
- Ghani, N.A.; Ahmat, N.H.; Ismail, I.Z.; Zawawi, N.K.N.A. Chemical Constituents and Cytotoxic Activity of Polyanthia cauliflora var. cauliflora. J. Med. Plant Res. 2012, 6, 74–82. [Google Scholar] [CrossRef] [Green Version]
- Marquina, S.; Maldonado-Santiago, M.; Sanchez-Carranza, J.N.; Antunez-Mojica, M.Y.; Gonzalez-Maya, L.; Razo-Hernandez, R.S.; Alavarez, L. Design, synthesis and QSAR study of 2′-hydroxy-4′-alkoxy chalcone derivatives that exert cytotoxic activity by the mitochondrial apoptotic pathway. Bioorg. Med. Chem. 2019, 27, 43–54. [Google Scholar] [CrossRef]
- Talia, J.M.; Tonn, C.E.; Debattista, N.B.; Pappano, N.B. Antibacterial Efficacy of Dihydroxylated Chalcones in Binary and Ternary Combinations with Nalidixic Acid and Nalidix Acid-Rutin against Escherichia coli ATCC 25 922. Indian J. Microbiol. 2012, 52, 638–641. [Google Scholar] [CrossRef] [Green Version]
- Lou, C.; Wang, M.; Yang, G.; Cai, H.; Li, Y.; Zhao, F.; Yang, H.; Cai, B. Preliminary studies on anti-tumor activity of 2′,4′-dihydroxychalcone isolated from Herba Oxytropis in human gastric cancer MGC-803 cells. Toxicol. In Vitro 2009, 23, 906–910. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Picó, A.; León-González, A.J.; Martín-Cordero, C.; Daga, R.R. Screening for natural anticancer agents using a fission yeast bioassay. Phytochem. Lett. 2014, 8, 184–189. [Google Scholar] [CrossRef]
- Siti Azima, A.M.; Noriham, A.; Manshoor, N. Phenolics, antioxidants and color properties of aqueous pigmented plant extracts: Ardisia colorata var. elliptica, Clitoria ternatea, Garcinia mangostana and Syzygium cumini. J. Funct. Foods 2017, 38, 232–241. [Google Scholar] [CrossRef]
- Dai, J.; Mumper, R.J. Plant Phenolics: Extraction, analysis and their antioxidant and anticancer properties. Molecules 2010, 15, 7313–7352. [Google Scholar] [CrossRef]
- Bezerra, G.S.N.; Pereira, M.A.V.; Ostrosky, E.A.; Barbosa, E.G.; de Moura, M.D.F.V.; Ferrari, M.; Aragão, C.F.S.; Gomes, A.P.B. Compatibility study between ferulic acid and excipients used in cosmetic formulations by TG/DTG, DSC and FTIR. J. Therm. Anal. Calorim. 2017, 127, 1683–1691. [Google Scholar] [CrossRef]
- Sairam, T.V. Home Remedies: A Handbook of Herbal Cures for Common Ailments; Penguin Books: Delhi, India, 1998. [Google Scholar]
- Sharma, P. Cinnamic acid derivatives: A new chapter in various pharmacological activities. Research. J. Chem. Pharm. 2011, 3, 403–423. [Google Scholar]
- Kim, J.H.; Campbell, B.C.; Mahomey, N.E.; Chan, K.L.; Molyneux, R.J. Identification of antifungal phenolics for control of using Aspergills flavus using Sacchromyces cerevisiae in a model target-gene bioassay. J. Agric. Food Chem. 2004, 52, 7814–7821. [Google Scholar] [CrossRef] [PubMed]
- Meunier, B. Hybrid Molecules with a Dual Mode of Action: Dream or Reality? Acc. Chem. Res. 2008, 41, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Batovska, D.I.; Todorova, I.T. Trends in utilization of the pharmacological potential of chalcones. Curr. Clin. Pharmacol. 2010, 5, 1–29. [Google Scholar] [CrossRef]
- Benigni, R.; Giuliani, A.; Franke, R.; Gruska, A. Quantitative structure-activity relationships of mutagenic and carcinogenic aromatic amines. Chem. Rev. 2000, 100, 3697–3714. [Google Scholar] [CrossRef]
- Razo-Hernández, R.S.; Pineda-Urbina, K.; Velazco-Medel, M.A.; Villanueva-García, M.; Sumaya-Martínez, M.T.; Martínez-Martínez, F.J.; Gómez-Sandoval, Z. QSAR study of the DPPH• radical scavenging activity of coumarin derivatives and xanthine oxidase inhibition by molecular docking. Cent. Eur. J. Chem. 2014, 12, 1067–1080. [Google Scholar] [CrossRef]
- Mehmood, R.; Sadiq, A.; Alsantali, R.I.; Mughal, E.U.; Alsharif, M.A.; Naeem, N.; Javid, A.; Al-Rooqi, M.M.; Chaudhry, G.-S.; Ahmed, S.A. Synthesis and Evaluation of 1,3,5-Triaryl-2-pyrazoline Derivatives as Potent Dual Inhibitors of Urease and α-Glucosidase Together with Their Cytotoxic, Molecular Modeling and Drug-Likeness Studies. ACS Omega 2022, 7, 3775–3795. [Google Scholar] [CrossRef]
- Mehmood, R.; Mughal, E.U.; Elkaeed, E.B.; Obaid, R.J.; Nazir, Y.; Al-Ghulikah, H.A.; Naeem, N.; Al-Rooqi, M.M.; Ahmed, S.A.; Shah, S.W.; et al. Synthesis of Novel 2,3-Dihydro-1,5-benzothiazepines as α-Glucosidase Inhibitors: In Vitro, in Vivo, Kinetic, SAR, Molecular Docking, and QSAR Studies. ACS Omega 2022, 7, 30215–30232. [Google Scholar] [CrossRef] [PubMed]
- Qiu, H.Y.; Wang, F.; Wang, X.; Sun, W.X.; Qi, J.-L.; Pang, Y.J.; Yang, R.-W.; Lu, G.H.; Wang, X.M.; Yang, Y.H. Design, Synthesis, and Biological Evaluation of Chalcone-Containing Shikonin Derivatives as Inhibitors of Tubulin Polymerization. Chem. Med. Chem. 2017, 12, 399–406. [Google Scholar] [CrossRef]
- Peng, F.; Meng, C.W.; Zhou, Q.M.; Chen, J.P.; Xiong, L. Cytotoxic Evaluation against Breast Cancer Cells of Isoliquiritigenin Analogues from Spatholobus suberectus and Their Synthetic Derivatives. J. Nat. Prod. 2016, 79, 248–251. [Google Scholar] [CrossRef]
- Sathya, S.; Jonathan, D.R.; Pandiammal, S.; Revathi, B.K.; Usha, G. Synthesis and Screening for Anticancer Activity of a series of Novel Chalcone derivatives on MCF-7 Cell Line. Int. J. ChemTech Res. 2017, 10, 995–1004. Available online: https://sphinxsai.com/2017/ch_vol10_no2/3/(995-1004)V10N2CT.pdf (accessed on 2 February 2023).
- Ivković, B.M.; Nikolic, K.; Ilić, B.B.; Žižak, Ž.S.; Novaković, R.B.; Čudina, O.A.; Vladimirov, S.M. Phenylpropiophenone derivatives as potential anticancer agents: Synthesis, biological evaluation and quantitative structure–activity relationship study. Eur. J. Med. Chem. 2013, 63, 239–255. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, M.F.A.; Shaykoon, M.S.A.; Abdelrahman, M.H.; Elsadek, B.E.M.; Elsadek, B.E.M.; Aboraia, A.S.; Abuo-Rahma, G.E.D.A.A. Design, synthesis, docking studies and biological evaluation of novel chalcone derivatives as potential histone deacetylase inhibitors. Bioorg. Chem. 2017, 72, 32–41. [Google Scholar] [CrossRef]
- Shenvi, S.; Kumar, K.; Hatti, K.S.; Rijesh, K.; Diwakar, L.; Reddy, G.C. Synthesis, anticancer and antioxidant activities of 2,4,5-trimethoxy chalcones and analogues from asaronaldehyde: Structure–activity relationship. Eur. J. Med. Chem. 2013, 62, 435–442. [Google Scholar] [CrossRef]
- Fikroh, R.A.; Matsjeh, S.; Anwar, C. Synthesis and Anticancer Activity of 2′-hydroxy-2-bromo-4, 5-dimetoxychalcone against Breast Cancer (MCF-7) Cell Line. Molekul 2020, 15, 34–39. [Google Scholar] [CrossRef]
- Durgapal, S.D.; Soni, R.; Umar, S.; Suresh, B.; Soman, S.S. 3-Aminomethyl pyridine chalcone derivatives: Design, synthesis, DNA binding and cytotoxic studies. Chem. Biol. Drug Des. 2018, 92, 1279–1287. [Google Scholar] [CrossRef] [PubMed]
- Hashim, F.; Wan Mohamed Zin, W.M.K.; Mohamed, M.; Mohd Norhadi Shah, N.S.; Tuan Johari, S.A.T.; Daud, A.I.; Rahamathullah, R. Morphological analysis of MCF-7 cells treated with chalcone derivatives. Front. Pharmacol. 2018, 9. [Google Scholar] [CrossRef]
- Mellado, M.; Reyna-Jeldes, M.; Weinstein-Oppenheimer, C.; Coddou, C.; Jara-Gutierrez, C.; Villena, J.; Aguilar, L.F. Inhibition of Caco-2 and MCF-7 cancer cells using chalcones: Synthesis, biological evaluation and computational study. Nat. Prod. Res. 2022, 36, 4404–4410. [Google Scholar] [CrossRef]
- Hussain, A.; Grootveld, M.; Arroo, R.; Beresford, K.; Ruparelia, K.; Fretwell, L. Effects of novel phenolic chalcone derivatives upon MCF-7 Cell viability. Planta Med. 2016, 82, P929. [Google Scholar] [CrossRef]
- Ma, X.; Wang, D.; Wei, G.; Zhou, Q.; Gan, X. Synthesis and anticancer activity of chalcone–quinoxalin conjugates. Synth. Commun. 2021, 51, 1363–1372. [Google Scholar] [CrossRef]
- Liu, X.; Go, M.L. Antiproliferative activity of chalcones with basic functionalities. Bioorg. Med. Chem. 2007, 15, 7021–7034. [Google Scholar] [CrossRef] [PubMed]
- Alswah, M.; Bayoumi, A.H.; Elgamal, K.; Elmorsy, A.; Ihmaid, S.; Ahmed, H.E.A. Design, Synthesis and Cytotoxic Evaluation of Novel Chalcone Derivatives Bearing Triazolo[4,3-a]-quinoxaline Moieties as Potent Anticancer Agents with Dual EGFR Kinase and Tubulin Polymerization Inhibitory Effects. Molecules 2018, 23, 48. [Google Scholar] [CrossRef] [Green Version]
- Abu Bakar, A.; Akhtar, M.N.; Mohd Ali, N.; Yeap, S.K.; Quah, C.K.; Loh, W.-S.; Alitheen, N.B.; Zareen, S.; Ul-Haq, Z.; Shah, S.A.A. Design, Synthesis and Docking Studies of Flavokawain B Type Chalcones and Their Cytotoxic Effects on MCF-7 and MDA-MB-231 Cell Lines. Molecules 2018, 23, 616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, Y.-F.; Liu, X.-J.; Yuan, X.-Y.; Liu, W.-B.; Li, Y.-R.; Yu, G.-X.; Tian, X.-Y.; Zhang, Y.-B.; Song, J.; Li, W.; et al. Design, Synthesis, and Anticancer Activity Studies of Novel Quinoline-Chalcone Derivatives. Molecules 2021, 26, 4899. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Xu, S.; Wu, C.; Liu, X.; Tao, H.; Huang, Y.; Liu, Y.; Zheng, P.; Zhu, W. Design, synthesis and activity of novel sorafenib analogues bearing chalcone unit. Bioorg. Med. Chem. Lett. 2016, 26, 5450–5454. [Google Scholar] [CrossRef]
- Wang, G.; Liu, W.; Gong, Z.; Huang, Y.; Li, Y.; Peng, Z. Design, synthesis, biological evaluation and molecular docking studies of new chalcone derivatives containing diaryl ether moiety as potential anticancer agents and tubulin polymerization inhibitors. Bioorg. Chem. 2020, 95, 103565. [Google Scholar] [CrossRef]
- Todeschini, R.; Ballabio, D.; Consonni, V.; Mauri, A.M.P. MobyDigs User Manual Version 1-2004; Milano Chemometrics and QSAR Research Group: Milano, Italy, 2004. [Google Scholar]
- Biau, D.J. In Brief: Standard Deviation and Standard Error. Clin. Orthop. Relat. Res. 2011, 469, 2661–2664. [Google Scholar] [CrossRef] [Green Version]
- Golbraikh, A.; Tropsha, A. Beware of q2! J. Mol. Graph. Model. 2002, 20, 269–276. [Google Scholar] [CrossRef]
- Alexander, D.L.J.; Tropsha, A.; Winkler, D.A. Beware of R2: Simple, Unambiguous Assessment of the Prediction Accuracy of QSAR and QSPR Models. J. Chem. Inf. Model. 2015, 55, 1316–1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todeschini, R.; Consonni, V.; Maiocchi, A. The K correlation index: Theory development and its application in chemometrics. Chemom. Intell. Lab. Syst. 1999, 46, 13–29. [Google Scholar] [CrossRef]
- Todeschini, R. Data Correlation Number of significant principal components and shape of molecules. The K correlation index. Anal. Chim. Acta 1997, 48, 419–430. [Google Scholar] [CrossRef]
- Todeschini, R.; Consonni, V.; Pavan, M. MobyDigs: Software for regression and classification models by genetic algorithms. In Nature-Inspired Methods in Chemometrics: Genetic Algorithms and Artificial Neural Networks; Leardi, R., Ed.; Elsevier: Amsterdam, The Netherlands, 2003. [Google Scholar]
- Todeschini, R.; Consonni, V.; Mauri, A.; Pava, M. Detecting “bad” regression models: Multicriteria fitness functions in regression analysis. Anal. Chim. Acta 2004, 515, 199–208. [Google Scholar] [CrossRef]
- Balaban, A.T. Chemical graphs. Theoret. Chim. Acta 1979, 53, 355–375. [Google Scholar] [CrossRef]
- Konstantinova, E.V.; Skorobogatov, V.A. Molecular hypergraphs: The new representation of nonclassical molecular structures with polycentric delocalized bonds. J. Chem. Inf. Comput. Sci. 1995, 35, 472–478. [Google Scholar] [CrossRef]
- Mihalic, M.; Nikolic, S.; Trinajstic, N. Comparative study of molecular descriptors derived from the distance matrix. J. Chem. Inf. Comput. Sci. 1992, 32, 28–37. [Google Scholar] [CrossRef]
- Robinson, D.D.; Barlow, T.W.; Richards, W.G. Reduced Dimensional Representations of Molecular Structure. J. Chem. Inf. Comput. Sci. 1997, 37, 939–942. [Google Scholar] [CrossRef]
- Arteca, G.A. Molecular Shape Descriptors. In Reviews in Computational Chemistry; Lipkowitz, K.B., Boyd, D., Eds.; VCH Publishers: New York, NY, USA, 1991; Volume 9, pp. 191–253. [Google Scholar]
- Bird, C.W. The application of a new aromaticity index to six-membered ring heterocycles. Tetrahedron 1986, 42, 89–92. [Google Scholar] [CrossRef]
- Sangpheak, S.; Mueller, M.; Darai, N.; Wolschann, P.; Suwattanasophon, C.; Ruga, R.; Chavasiri, W.; Seetaha, S.; Kiattawee Choowongkomon, K.; Nawee Kungwan, N.; et al. Computational screening of chalcones acting against topoisomerase IIα and their cytotoxicity towards cancer cell lines. J. Enzyme Inhib. Med. Chem. 2019, 34, 134–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Y.; Lee, I.-S. Effects of Microbial Transformation on the Biological Activities of Prenylated Chalcones from Angelica keiskei. Foods 2022, 11, 543. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Kishan, N.; Biswas, S.; Gourishetti, K.; Kamal, M.; Chamallamudi, M.R. Anti-metastatic and Anticancer Potentials of Synthesized Chalcones in B16-F10 Melanoma Cells Induced Metastatic Lung Cancer in C57BL/6 Mice. Indian J. Pharm. Educ. Res. 2021, 55, S742–S750. [Google Scholar] [CrossRef]
- Palacios-Can, F.J.; Silva-Sánchez, J.; León-Rivera, I.; Tlahuext, H.; Pastor, N.; Razo-Hernández, R.S. Identification of a Family of Glycoside Derivatives Biologically Active against Acinetobacter baumannii and Other MDR Bacteria Using a QSPR Model. Pharmaceuticals 2023, 16, 250. [Google Scholar] [CrossRef]
- Lee, S.; Lee, I.H.; Kim, H.J.; Chang, G.S.; Chung, J.E.; No, K.T. The PreADME Approach: Web-based program for rapid prediction of physico-chemical, drug absorption and drug-like properties. Euro QSAR 2003, 2003, 418–420. [Google Scholar]
- Zerroug, A.; Belaidi, S.; BenBrahim, I.; Sinha, L.; Chtita, S. Virtual screening in drug-likeness and structure/activity relationship of pyridazine derivatives as Anti-Alzheimer drugs. J. King Saud Univ. Sci. 2017, 31, 595–601. [Google Scholar] [CrossRef]
- Pardridge, W.M. Drug transport across the blood-brain barrier. J. Cereb. Blood Flow Metab. 2012, 32, 1959–1972. [Google Scholar] [CrossRef]
- Saneja, A.; Vaibhav Khare, V.; Alam, N.; Dubey, R.-D.; Gupta, N.P. Advances in P-glycoprotein-based approaches for delivering anticancer drugs: Pharmacokinetic perspective and clinical relevance. Expert Opin. Drug Deliv. 2014, 11, 121–138. [Google Scholar] [CrossRef]
- Talete. DRAGON for Windows (Software for Molecular Descriptor Calculations); Talete: Milan, Italy, 2006. [Google Scholar]
- Veerasamy, R.; Rajak, H.; Jain, A.; Sivadasan, S.; Varghese, C.P.; Agrawal, R.K. Validation of QSAR Models—Strategies and Importance. Int. J. Drug Des. Discov. 2011, 2, 511–519. [Google Scholar]
- Todeschini, I.E.F.R. The Data Analysis Handbook; Elsevier Science: Amsterdam, The Netherlands, 1994. [Google Scholar]
- Jovanovic, B.; Miller, C. Subset Selection in Regression; CRC Press: London, UK, 1990. [Google Scholar]
- Basu, A.; Saito, K.; Meyer, K.; Ranta, B.R.; Friedman, S.; Chang, Y.L.; Ray, R. Stellate cell apoptosis by a soluble mediator from immortalized human hepatocytes. Apoptosis 2006, 11, 1391–1400. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal hemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef] [PubMed] [Green Version]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 (Mean ± S.D., µM) | IC50 Pred | |||||
|---|---|---|---|---|---|---|---|
| IHH | HepG2 | Hep3B | Caski | A-549 | MCF-7 | MCF-7 | |
| A | 100 ± 2.2 | 58.33 ± 2.5 | 54.16 ± 2 | 58.33 ± 3 | 112.5 ± 2.5 | 62.5 ± 4 | 55.95 |
| A1 | 140.47 ± 4.5 | 109.52 ± 3.0 | 90.47 ± 4.0 | 114.28 ± 3.0 | 185.71 ± 7.0 | 88 ± 5.0 | 1.23 |
| B | 56.76 ± 2 | 11.89 ± 0.4 | 10.27 ± 0.2 | 29.72 ± 1.4 | 56.75 ± 2.3 | 14.86 ± 0.5 | 17.86 |
| Paclitaxel * | 0.08 ± 0.005 | 0.0065 ± 0.0007 | 0.008 ± 0.001 | 0.007 ± 0.0009 | 0.0352 ± 0.007 | 0.0395 ± 0.0046 | - |
| Molecule | MLOGP a | TPSA b | BBB c | Pgp Substrate d | GI Absorption e | Lipinski Violations | Ghose Violations | Veber Violations | Mutagenicity | Carcinogenicity | Hepatotoxicity |
|---|---|---|---|---|---|---|---|---|---|---|---|
| A | 2.17 | 57.53 | Yes | No | High | 0 | 0 | 0 | Inactive | Inactive | Inactive |
| B | 3.22 | 63.6 | Yes | No | High | 0 | 0 | 0 | Inactive | Inactive | Inactive |
| C | 2.79 | 76.49 | No | No | High | 0 | 2 | 0 | Inactive | Inactive | Inactive |
| D | 3.15 | 76.49 | No | No | High | 0 | 2 | 0 | Inactive | Inactive | Active |
| E | 3.24 | 76.49 | No | No | High | 0 | 3 | 0 | Inactive | Inactive | Inactive |
| F | 3.34 | 76.49 | No | No | Low | 1 | 3 | 0 | Inactive | Inactive | Active |
| G | 2.98 | 76.49 | No | No | High | 0 | 2 | 0 | Inactive | Inactive | Inactive |
| H | 3.52 | 76.49 | No | Yes | Low | 1 | 3 | 0 | Inactive | Inactive | Inactive |
| I | 2.43 | 85.72 | No | No | High | 0 | 3 | 0 | Inactive | Inactive | Inactive |
| J | 3.16 | 85.72 | No | Yes | Low | 1 | 3 | 1 | Inactive | Inactive | Inactive |
| K | 3.79 | 85.72 | No | Yes | Low | 1 | 3 | 1 | Inactive | Inactive | Active |
| Molecular Descriptors | Descriptor Family | Program |
|---|---|---|
| ZM1, ZM1V, ZM2, ZM2V, Qindex, SNar, HNar, GNar, Xt, Dz, Ram, Pol, LPRS, VDA, MSD, SMTI, SMTIV, GMTI, GMTIV, Xu, SPI, W, WA, Har, Har2, QW, TI1, TI2, STN, HyDp, RHyDp, w, ww, Rww, D/D, Wap, WhetZ, Whetm, Whetv, Whete, Whetp, J, JhetZ, Jhetm, Jhetv, Jhete, Jhetp, MAXDN, MAXDP, DELS, TIE, S0K, S1K, S2K, S3K, PHI, BLI, PW2, PW3, PW4, PW5, PJI2, CSI, ECC, AECC, DECC, MDDD, UNIP, CENT, VAR, BAC, Lop, ICR, D/Dr03, D/Dr04, D/Dr05, D/Dr06, D/Dr07, D/Dr08, D/Dr09, D/Dr10, D/Dr11, D/Dr12, T(N..N), T(N..O), T(N..S), T(N..F), T(N..Cl), T(N..Br), T(N..I), T(O..O), T(O..S), T(O..F), T(O..Cl), T(O..Br), T(S..S), T(S..F), T(S..Cl), T(S..Br),T(F..F), T(F..Cl), T(F..Br), T(Cl..Cl), T(Cl..Br), T(Br..Br) | Topological | DRAGON |
| W3D, J3D, H3D, AGDD, DDI, ADDD, G1, G2, RGyr, SPAN, SPAM, MEcc, SPH, ASP, FDI, PJI3, L/Bw, SEig, HOMA, RCI, AROM, HOMT, DISPm, QXXm, QYYm, QZZm, DISPv, QXXv, QYYv, QZZv, DISPe, QXXe, QYYe, QZZe, DISPp, QXXp, QYYp, QZZp, G(N..N), G(N..O), G(N..S), G(N..P), G(N..F), G(N..Cl), G(N..Br), G(N..I), G(O..O), G(O..S), G(O..P), G(O..F), G(O..Cl), G(O..Br), G(O..I), G(S..S), G(S..P), G(S..F), G(S..Cl), G(S..Br), G(S..I), G(P..P), G(P..F), G(P..Cl), G(P..Br), G(P..I), G(F..F), G(F..Cl), G(F..Br), G(F..I), G(Cl..Cl), G(Cl..Br), G(Cl..I), G(Br..Br), G(Br..I), G(I..I) | Geometrical | DRAGON |
| nCp, nCs, nCt, nCq, nCrs, nCrt, nCrq, nCar, nCbH, nCb-, nCconj, nR=Cp, nR=Cs, nR=Ct, nR#CH/X, nR#C-, nRCOOH, nArCOOH, nRCOOR, nArCOOR, nRCONH2, nRCONHR, nArCONHR, nRCONR2, nArCONR2, nROCON, nArOCON, nArCOSR, nRCO, nArCO, nCONN, nN=C-N<, nC(=N)N2, nRC=N, nArC=N, nArCNO, nRNH2, nArNH2, nRNHR, nArNHR, nRNR2, nArNR2, nN-N, nRCN, nArCN nN+, nNq, nRNHO, nArNHO, nArNO2, nN(CO)2, nC=N-N<, nROH, nArOH, nOHp, nOHs, nOHt, nROR, nArOR, nRSR, nSO, nS(=O)2, nSO2OH, nSO3OH, nSO2N, nCH2RX, nR=CRX, nCHRX2, nCRX3, nArX, nCXr, nCXr=, nCconjX, nOxiranes, nAzetidines, nBeta-Lactams, nPyrrolidines, nOxolanes, nPyrroles, nPyrazoles, nImidazoles, nFuranes, nThiophenes, nIsoxazoles, nThiazoles, nTriazoles, nPyridines, nPyridazines, nPyrimidines, nPyrazines, nHDon, nHAcc, nHBonds | Functional Groups | DRAGON |
| C-001, C-002, C-003, C-004, C-005, C-006, C-007, C-008, C-009, C-010, C-011, C-012, C-013, C-014, C-015, C-016, C-017, C-018, C-019, C-020, C-021, C-022, C-024, C-025, C-026, C-027, C-028, C-029, C-030, C-031, C-032, C-033, C-034, C-035, C-036, C-037, C-038, C-039, C-040, C-041, C-042, C-043, C-044, H-046, H-047, H-048, H-049, H-050, H-051, H-052, H-053, H-054, O-056, O-057, O-058, O-059, O-060, O-061, N-066, N-067, N-068, N-069, N-070, N-071, N-072, N-073, N-074, N-075, N-076, N-077, N-078, N-079, F-081, F-082, F-083, F-084, F-085, Cl-086, Cl-089, Cl-090, Br-091, Br-092, Br-094, S-106, S-107, S-108, S-109, S-110 | Atom-centred fragments | DRAGON |
| Ui, Hy, Hy2, AMR, TPSA(NO), TPSA(Tot), MLOGP, MLOGP2, ALOGP, ALOGP2 | Molecular properties | DRAGON |
| R2_F, R4_F, R2_Cl, R3_Cl, R4_Cl, R2_Br, R4_Br, R2_Me, R3_Me, R4_Me, R5_Me, R2_OH, R4_OH, R6_OH, R2_OMe, R3_OMe, R4_OMe, R5_OMe, R6_OMe, R2_CF3, R3_CF3, R4_CF3, R5_CF3, R4_Et, R3_OCF3, R3_Bn, R5_Bn, R3_NO2, R2_OAc, R5_OAc, R3_CO2Ph, R4_FA026, R4_FA027, R4_FA028, R4_OC6H13, R4_FA029, R5_(4′MPPD), R4_(4′PPZ), R4_(1PPD), R4_(4′EPPD), R4_(4′MPPZ), R4_(4BiPPD), R4_FA031, R4_FA032, R4_FA033, R4_FA030, R2_TMPhO, R′2_F, R′3_F, R′4_F, R′5_F, R′6_F, R′2_Cl, R′3_Cl, R′4_Cl, R′5_Cl, R′2_Br, R′3_Br, R′4_Br, R′5_Br, R′2_Me, R′3_Me, R′4_Me, R′5_Me, R′2_CF3, R′4_CF3, R′2_OH, R′4_OH, R′2_OMe, R′3_OMe, R′4_OMe, R′5_OMe, R′3_NO2, R′4_NO2, R′5_NO2, R′3_CN, R′4_CN, R′3_OC6H13, R′4_SMe, R′4_(4MPPZ), R′4_FB034, R′4_FB035, R′4_nPr, R′3_NH2, R′4_NMe2 | Indicative variables | N/A |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ventura-Salazar, I.A.Y.; Palacios-Can, F.J.; González-Maya, L.; Sánchez-Carranza, J.N.; Antunez-Mojica, M.; Razo-Hernández, R.S.; Alvarez, L. Finding a Novel Chalcone–Cinnamic Acid Chimeric Compound with Antiproliferative Activity against MCF-7 Cell Line Using a Free-Wilson Type Approach. Molecules 2023, 28, 5486. https://doi.org/10.3390/molecules28145486
Ventura-Salazar IAY, Palacios-Can FJ, González-Maya L, Sánchez-Carranza JN, Antunez-Mojica M, Razo-Hernández RS, Alvarez L. Finding a Novel Chalcone–Cinnamic Acid Chimeric Compound with Antiproliferative Activity against MCF-7 Cell Line Using a Free-Wilson Type Approach. Molecules. 2023; 28(14):5486. https://doi.org/10.3390/molecules28145486
Chicago/Turabian StyleVentura-Salazar, Isis A. Y., Francisco J. Palacios-Can, Leticia González-Maya, Jessica Nayelli Sánchez-Carranza, Mayra Antunez-Mojica, Rodrigo Said Razo-Hernández, and Laura Alvarez. 2023. "Finding a Novel Chalcone–Cinnamic Acid Chimeric Compound with Antiproliferative Activity against MCF-7 Cell Line Using a Free-Wilson Type Approach" Molecules 28, no. 14: 5486. https://doi.org/10.3390/molecules28145486