
[1,2,4]triazolo[4,3-a]quinoxaline as Novel Scaffold in the Imiqualines Family: Candidates with Cytotoxic Activities on Melanoma Cell Lines
 , and
, and
Abstract
: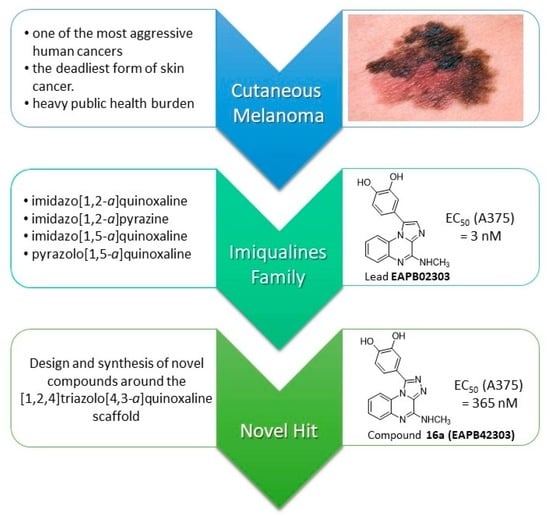
1. Introduction
2. Results and Discussion
2.1. Background
2.2. Previous Lab Strategy
2.3. Alternative Strategies
2.4. Selected General Chemical Strategy and Diversification
2.5. Predicted Physicochemical Parameters
2.6. In Vitro Cell Growth Inhibition Assay
3. Materials and Methods
3.1. Chemistry
3.1.1. General
3.1.2. Procedures for Compounds’ Syntheses
- Procedure for the Suzuki–Miyaura cross-coupling reactions in position 8.
- Procedure for condensation with aldehydes and compound 2.
- Procedure for the cyclization with chloranil.
- Procedure for the introduction of the methylamino group.
- Procedure for the introduction of the amino group.
- Procedure for the cleavage of catechol protections.
3.2. Cell lines and Culture Techniques
3.2.1. General
3.2.2. In Vitro Cytotoxicity Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Abbreviations
| MAPK | Mitogen-activated protein kinases |
| BRAF | B-rapidly accelerated fibrosarcoma |
| NRAS | Neuroblastoma RAS viral oncogene |
| NF1 | Neurofibromatosis-1 |
| MEK | Mitogen-activated protein kinase kinase |
| PD-1 | Programmed cell death protein-1 |
| CTLA-4 | Cytotoxique T lymphocyte antigen-4 protein |
| VEGFR-2 | Vascular endothelial growth factor receptor-2 |
| BAX | Bcl-2-associated X protein |
| hPBMCs | Human peripheral blood mononuclear cells |
| hERG | Human ether-à-go-go-related gene |
| SILAC | Stable isotope labeling by amino acids in cell culture |
| logS | Aqueous solubility |
| logP | Partition coefficient |
| logD | Distribution coefficient |
| pKa | Ionization constant |
| MW | Micro-wave |
| NBS | N-bromosuccinimide |
| NMR | Nuclear magnetic resonance |
| NOESY | Nuclear Overhauser effect epectroscopy |
| EtOH | Ethanol |
| ACN | Acetonitrile |
| RT | Room temperature |
| DME | Dimethoxyethane |
| DMF | Dimethylformamide |
| 2D NOESY | Two-dimensional nuclear Overhauser effect spectroscopy |
| ADME | Absorption distribution metabolism elimination |
| PSA | Polar surface area |
| SAR | Structure–activity relationship |
| EC50 | Half-maximal effective concentration |
| TLC | Thin-layer chromatography |
| UV | Ultra-violet |
| LC-MS | Liquid chromatography mass spectroscopy |
| ESI | Electrospray ionization |
| UPLC/MS | Ultra-performance liquid chromatography/mass spectroscopy |
| Da | Dalton |
| HPLC | High-performance liquid chromatography |
| HRMS | High-resolution mass spectroscopy |
| V | Volt |
| ppm | Parts per million |
| DMSO | Dimethylsulfoxide |
| s | Singlet |
| d | Doublet |
| t | Triplet |
| q | Quartet |
| m | Multiplet |
| Mw | Molecular weight |
| MeOH | Methanol |
| EtOAc | Ethyl acetate |
| DMEM | Dulbecco’s Modified Eagle Medium |
| FBS | Fetal bovine serum |
| MTT | Thiazolyl blue tetrazolium bromide |
| SDS | Sodium dodecyl sulfate |
| MDA-MB-231 | Human triple-negative breast adenocarcinoma cell line |
| HepG2 | Hepatocellular carcinoma (HCC) type |
| HCT116 | Human colorectal carcinoma-116 |
| MCF-7 | Michigan Cancer Foundation-7 breast cancer |
| DNA | Desoxyribonucleic acid |
| HL60 | Human acute myelocytic leukemia |
References
- Shain, A.H.; Bastian, B.C. From Melanocytes to Melanomas. Nat. Rev. Cancer 2016, 16, 345–358. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Network. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arozarena, I.; Wellbrock, C. Phenotype Plasticity as Enabler of Melanoma Progression and Therapy Resistance. Nat. Rev. Cancer 2019, 19, 377–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strashilov, S.; Yordanov, A. Aetiology and Pathogenesis of Cutaneous Melanoma: Current Concepts and Advances. Int. J. Mol. Sci. 2021, 22, 6395. [Google Scholar] [CrossRef]
- Algazi, A.P.; Soon, C.W.; Daud, A.I. Treatment of Cutaneous Melanoma: Current Approaches and Future Prospects. Cancer Manag. Res. 2010, 2, 197–211. [Google Scholar] [CrossRef] [Green Version]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved Survival with Vemurafenib in Melanoma with BRAF V600E Mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [Green Version]
- Dummer, R.; Ascierto, P.A.; Gogas, H.J.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; Gutzmer, R.; et al. Overall Survival in Patients with BRAF-Mutant Melanoma Receiving Encorafenib plus Binimetinib versus Vemurafenib or Encorafenib (COLUMBUS): A Multicentre, Open-Label, Randomised, Phase 3 Trial. Lancet Oncol. 2018, 19, 1315–1327. [Google Scholar] [CrossRef]
- Davis, J.; Wayman, M. Encorafenib and Binimetinib Combination Therapy in Metastatic Melanoma. J. Adv. Pract. Oncol. 2022, 13, 450–455. [Google Scholar] [CrossRef]
- Hauschild, A.; Grob, J.-J.; Demidov, L.V.; Jouary, T.; Gutzmer, R.; Millward, M.; Rutkowski, P.; Blank, C.U.; Miller, W.H.; Kaempgen, E.; et al. Dabrafenib in BRAF-Mutated Metastatic Melanoma: A Multicentre, Open-Label, Phase 3 Randomised Controlled Trial. Lancet Lond. Engl. 2012, 380, 358–365. [Google Scholar] [CrossRef]
- Robert, C.; Karaszewska, B.; Schachter, J.; Rutkowski, P.; Mackiewicz, A.; Stroiakovski, D.; Lichinitser, M.; Dummer, R.; Grange, F.; Mortier, L.; et al. Improved Overall Survival in Melanoma with Combined Dabrafenib and Trametinib. N. Engl. J. Med. 2015, 372, 30–39. [Google Scholar] [CrossRef] [Green Version]
- Ribas, A.; Gonzalez, R.; Pavlick, A.; Hamid, O.; Gajewski, T.F.; Daud, A.; Flaherty, L.; Logan, T.; Chmielowski, B.; Lewis, K.; et al. Combination of Vemurafenib and Cobimetinib in Patients with Advanced BRAF(V600)-Mutated Melanoma: A Phase 1b Study. Lancet Oncol. 2014, 15, 954–965. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Combined BRAF and MEK Inhibition versus BRAF Inhibition Alone in Melanoma. N. Engl. J. Med. 2014, 371, 1877–1888. [Google Scholar] [CrossRef] [Green Version]
- Ott, P.A.; Hodi, F.S.; Robert, C. CTLA-4 and PD-1/PD-L1 Blockade: New Immunotherapeutic Modalities with Durable Clinical Benefit in Melanoma Patients. Clin. Cancer Res. 2013, 19, 5300–5309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirkwood, J.M.; Tarhini, A.A.; Panelli, M.C.; Moschos, S.J.; Zarour, H.M.; Butterfield, L.H.; Gogas, H.J. Next Generation of Immunotherapy for Melanoma. J. Clin. Oncol. 2008, 26, 3445–3455. [Google Scholar] [CrossRef] [PubMed]
- Belardelli, F.; Ferrantini, M.; Proietti, E.; Kirkwood, J.M. Interferon-Alpha in Tumor Immunity and Immunotherapy. Cytokine Growth Factor Rev. 2002, 13, 119–134. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA. Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Arnold, M.; Singh, D.; Laversanne, M.; Vignat, J.; Vaccarella, S.; Meheus, F.; Cust, A.E.; de Vries, E.; Whiteman, D.C.; Bray, F. Global Burden of Cutaneous Melanoma in 2020 and Projections to 2040. JAMA Dermatol. 2022, 158, 495–503. [Google Scholar] [CrossRef]
- Barbaric, J.; Sekerija, M.; Agius, D.; Coza, D.; Dimitrova, N.; Demetriou, A.; Safaei Diba, C.; Eser, S.; Gavric, Z.; Primic-Zakelj, M.; et al. Disparities in Melanoma Incidence and Mortality in South-Eastern Europe: Increasing Incidence and Divergent Mortality Patterns. Is Progress around the Corner? Eur. J. Cancer 2016, 55, 47–55. [Google Scholar] [CrossRef]
- Allemani, C.; Matsuda, T.; Di Carlo, V.; Harewood, R.; Matz, M.; Nikšić, M.; Bonaventure, A.; Valkov, M.; Johnson, C.J.; Estève, J.; et al. Global Surveillance of Trends in Cancer Survival 2000–14 (CONCORD-3): Analysis of Individual Records for 37 513 025 Patients Diagnosed with One of 18 Cancers from 322 Population-Based Registries in 71 Countries. Lancet 2018, 391, 1023–1075. [Google Scholar] [CrossRef] [Green Version]
- Garbe, C.; Amaral, T.; Peris, K.; Hauschild, A.; Arenberger, P.; Basset-Seguin, N.; Bastholt, L.; Bataille, V.; del Marmol, V.; Dréno, B.; et al. European Consensus-Based Interdisciplinary Guideline for Melanoma. Part 1: Diagnostics: Update 2022. Eur. J. Cancer 2022, 170, 236–255. [Google Scholar] [CrossRef]
- Sacchetto, L.; Zanetti, R.; Comber, H.; Bouchardy, C.; Brewster, D.H.; Broganelli, P.; Chirlaque, M.D.; Coza, D.; Galceran, J.; Gavin, A.; et al. Trends in Incidence of Thick, Thin and in Situ Melanoma in Europe. Eur. J. Cancer 2018, 92, 108–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patinote, C.; Cirnat, N.; Bonnet, P.-A.; Deleuze-Masquéfa, C. Fused Azolo-Quinoxalines: Candidates for Medicinal Chemistry. A Review of Their Biological Applications. Curr. Med. Chem. 2021, 28, 712–749. [Google Scholar] [CrossRef] [PubMed]
- Ali, I.; Lee, J.; Go, A.; Choi, G.; Lee, K. Discovery of Novel [1,2,4]Triazolo[4,3-a]Quinoxaline Aminophenyl Derivatives as BET Inhibitors for Cancer Treatment. Bioorg. Med. Chem. Lett. 2017, 27, 4606–4613. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.K.; Taghour, M.S.; Metwaly, A.M.; Belal, A.; Mehany, A.B.M.; Elhendawy, M.A.; Radwan, M.M.; Yassin, A.M.; El-Deeb, N.M.; Hafez, E.E.; et al. Design, Synthesis, Molecular Modeling and Anti-Proliferative Evaluation of Novel Quinoxaline Derivatives as Potential DNA Intercalators and Topoisomerase II Inhibitors. Eur. J. Med. Chem. 2018, 155, 117–134. [Google Scholar] [CrossRef] [PubMed]
- Omar, A.M.; Alswah, M.; Ahmed, H.E.A.; Bayoumi, A.H.; El-Gamal, K.M.; El-Morsy, A.; Ghiaty, A.; Afifi, T.H.; Sherbiny, F.F.; Mohammed, A.S.; et al. Antimicrobial Screening and Pharmacokinetic Profiling of Novel Phenyl-[1,2,4]Triazolo[4,3-a]Quinoxaline Analogues Targeting DHFR and E. Coli DNA Gyrase B. Bioorganic Chem. 2020, 96, 103656. [Google Scholar] [CrossRef]
- Issa, D.A.E.; Habib, N.S.; Wahab, A.E.A. Design, Synthesis and Biological Evaluation of Novel 1,2,4-Triazolo and 1,2,4-Triazino[4,3-a]Quinoxalines as Potential Anticancer and Antimicrobial Agents. MedChemComm 2015, 6, 202–211. [Google Scholar] [CrossRef]
- El-Sawy, E.R.; Bassyouni, F.A.; Abu-Bakr, S.H.; Rady, H.M.; Abdlla, M.M. Synthesis and Biological Activity of Some New 1-Benzyl and 1-Benzoyl-3-Heterocyclic Indole Derivatives. Acta Pharm. Zagreb Croat. 2010, 60, 55–71. [Google Scholar] [CrossRef] [Green Version]
- Henen, M.A.; El Bialy, S.A.A.; Goda, F.E.; Nasr, M.N.A.; Eisa, H.M. [1,2,4]Triazolo[4,3-a]Quinoxaline: Synthesis, Antiviral, and Antimicrobial Activities. Med. Chem. Res. 2012, 21, 2368–2378. [Google Scholar] [CrossRef]
- Sekhar, K.V.G.C.; Rao, V.S.; Kumar, D. Synthesis of Triazoloquinoxalines as Antitubercular Agents. Bull. Korean Chem. Soc. 2011, 32, 2657–2660. [Google Scholar] [CrossRef] [Green Version]
- Corona, P.; Vitale, G.; Loriga, M.; Paglietti, G.; La Colla, P.; Collu, G.; Sanna, G.; Loddo, R. 4-Substituted Anilino Imidazo[1,2-a] and Triazolo[4,3-a]Quinoxalines. Synthesis and Evaluation of in Vitro Biological Activity. Eur. J. Med. Chem. 2006, 41, 1102–1107. [Google Scholar] [CrossRef] [PubMed]
- El-Tombary, A.A.; El-Hawash, S.A.M. Synthesis, Antioxidant, Anticancer and Antiviral Activities of Novel Quinoxaline Hydrazone Derivatives and Their Acyclic C-Nucleosides. Med. Chem. Shariqah United Arab Emir. 2014, 10, 521–532. [Google Scholar] [CrossRef]
- El-Attar, M.A.Z.; Elbayaa, R.Y.; Shaaban, O.G.; Habib, N.S.; Abdel Wahab, A.E.; Abdelwahab, I.A.; El-Hawash, S.A.M. Synthesis of Pyrazolo-1,2,4-Triazolo[4,3-a]Quinoxalines as Antimicrobial Agents with Potential Inhibition of DHPS Enzyme. Future Med. Chem. 2018, 10, 2155–2175. [Google Scholar] [CrossRef]
- El-Attar, A.Z.; Shaaban, O.G.; Elbayaa, R.Y.; Habib, N.S.; El-Hawash, S.A.; Wahab, A.E.A. Design and Synthesis of Some New 1,2,4-Triazolo[4,3-a]Quinoxaline Derivatives as Potential Antimicrobialagents. Med. Chem. 2015, 5, 11. [Google Scholar] [CrossRef]
- Suresh, M.; Lavanya, P.; Sudhakar, D.; Vasu, K.; Rao, C.V. Synthesis and Biological Activity of 8-Chloro-[1,2,4]Triazolo [4,3-a]Quinoxalines. J. Chem. Pharm. Res. 2010, 2, 497–504. [Google Scholar]
- Debbert, S.L.; Hintz, M.J.; Bell, C.J.; Earl, K.R.; Forsythe, G.E.; Häberli, C.; Keiser, J. Activities of Quinoxaline, Nitroquinoxaline, and [1,2,4]Triazolo[4,3-a]Quinoxaline Analogs of MMV007204 against Schistosoma Mansoni. Antimicrob. Agents Chemother. 2021, 65, e01370-20. [Google Scholar] [CrossRef]
- Liu, X.-K.; Ma, L.-X.; Wei, Z.-Y.; Cui, X.; Zhan, S.; Yin, X.-M.; Piao, H.-R. Synthesis and Positive Inotropic Activity of [1,2,4]Triazolo[4,3-a] Quinoxaline Derivatives Bearing Substituted Benzylpiperazine and Benzoylpiperazine Moieties. Molecules 2017, 22, 273. [Google Scholar] [CrossRef] [Green Version]
- Alswah, M.; Ghiaty, A.; El-Morsy, A.; El-Gamal, K. Synthesis and Biological Evaluation of Some [1,2,4]Triazolo[4,3-a]Quinoxaline Derivatives as Novel Anticonvulsant Agents. ISRN Org. Chem. 2013, 2013, 587054. [Google Scholar] [CrossRef] [Green Version]
- Wagle, S.; Adhikari, A.V.; Kumari, N.S. Synthesis of Some New 4-Styryltetrazolo[1,5-a]Quinoxaline and 1-Substituted-4-Styryl[1,2,4]Triazolo[4,3-a]Quinoxaline Derivatives as Potent Anticonvulsants. Eur. J. Med. Chem. 2009, 44, 1135–1143. [Google Scholar] [CrossRef] [PubMed]
- Colotta, V.; Catarzi, D.; Varano, F.; Lenzi, O.; Filacchioni, G.; Martini, C.; Trincavelli, L.; Ciampi, O.; Traini, C.; Pugliese, A.M.; et al. Synthesis, Ligand-Receptor Modeling Studies and Pharmacological Evaluation of Novel 4-Modified-2-Aryl-1,2,4-Triazolo[4,3-a]Quinoxalin-1-One Derivatives as Potent and Selective Human A3 Adenosine Receptor Antagonists. Bioorg. Med. Chem. 2008, 16, 6086–6102. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.-K.; Gong, G.-H.; Li, G.-; Jin, M.-; Cao, L.-H.; Quan, Z.-S. Discovery and Evaluation of Novel Synthetic 5-Alkyl-4-Oxo-4,5-Dihydro-[1,2,4]Triazolo[4,3-a]Quinoxaline-1-Carbox-Amide Derivatives as Anti-Inflammatory Agents. J. Enzym. Inhib. Med. Chem. 2020, 35, 85–95. [Google Scholar] [CrossRef] [Green Version]
- Ezzat, H.G.; Bayoumi, A.H.; Sherbiny, F.F.; El-Morsy, A.M.; Ghiaty, A.; Alswah, M.; Abulkhair, H.S. Design, Synthesis, and Molecular Docking Studies of New [1,2,4]Triazolo[4,3-a]Quinoxaline Derivatives as Potential A2B Receptor Antagonists. Mol. Divers. 2021, 25, 291–306. [Google Scholar] [CrossRef]
- El-Adl, K.; El-Helby, A.-G.A.; Sakr, H.; Elwan, A. [1,2,4]Triazolo[4,3- a ]Quinoxaline and [1,2,4]Triazolo[4,3- a ]Quinoxaline-1-Thiol-Derived DNA Intercalators: Design, Synthesis, Molecular Docking, in Silico ADMET Profiles and Anti-Proliferative Evaluations. New J. Chem. 2021, 45, 881–897. [Google Scholar] [CrossRef]
- El-Adl, K.; El-Helby, A.G.A.; Sakr, H.; Elwan, A. Design, Synthesis, Molecular Docking and Anti-Proliferative Evaluations of [1,2,4]Triazolo[4,3-a]Quinoxaline Derivatives as DNA Intercalators and Topoisomerase II Inhibitors. Bioorganic Chem. 2020, 105, 104399. [Google Scholar] [CrossRef] [PubMed]
- Elwan, A.; Sakr, H.; El-Helby, A.-G.A.; El-morsy, A.; Abdelgawad, M.A.; Ghoneim, M.M.; El-Sherbiny, M.; El-Adl, K. Triazoloquinoxalines-Based DNA Intercalators-Topo II Inhibitors: Design, Synthesis, Docking, ADMET and Anti-Proliferative Evaluations. J. Enzym. Inhib. Med. Chem. 2022, 37, 1556–1567. [Google Scholar] [CrossRef] [PubMed]
- Deleuze-Masquéfa, C.; Gerebtzoff, G.; Subra, G.; Fabreguettes, J.-R.; Ovens, A.; Carraz, M.; Strub, M.-P.; Bompart, J.; George, P.; Bonnet, P.-A. Design and Synthesis of Novel Imidazo[1,2-a]Quinoxalines as PDE4 Inhibitors. Bioorg. Med. Chem. 2004, 12, 1129–1139. [Google Scholar] [CrossRef] [PubMed]
- Moarbess, G.; Deleuze-Masquefa, C.; Bonnard, V.; Gayraud-Paniagua, S.; Vidal, J.-R.; Bressolle, F.; Pinguet, F.; Bonnet, P.-A. In Vitro and in Vivo Anti-Tumoral Activities of Imidazo[1,2-a]Quinoxaline, Imidazo[1,5-a]Quinoxaline, and Pyrazolo[1,5-a]Quinoxaline Derivatives. Bioorg. Med. Chem. 2008, 16, 6601–6610. [Google Scholar] [CrossRef] [PubMed]
- Moarbess, G.; El-Hajj, H.; Kfoury, Y.; El-Sabban, M.E.; Lepelletier, Y.; Hermine, O.; Deleuze-Masquéfa, C.; Bonnet, P.-A.; Bazarbachi, A. EAPB0203, a Member of the Imidazoquinoxaline Family, Inhibits Growth and Induces Caspase-Dependent Apoptosis in T-Cell Lymphomas and HTLV-I–Associated Adult T-Cell Leukemia/Lymphoma. Blood 2008, 111, 3770–3777. [Google Scholar] [CrossRef]
- Kaneko, D.; Ninomiya, M.; Yoshikawa, R.; Ono, Y.; Sonawane, A.D.; Tanaka, K.; Nishina, A.; Koketsu, M. Synthesis of [1,2,4]Triazolo[4,3-a]Quinoxaline-1,3,4-Oxadiazole Derivatives as Potent Antiproliferative Agents via a Hybrid Pharmacophore Approach. Bioorganic Chem. 2020, 104, 104293. [Google Scholar] [CrossRef]
- Alanazi, M.M.; Mahdy, H.A.; Alsaif, N.A.; Obaidullah, A.J.; Alkahtani, H.M.; Al-Mehizia, A.A.; Alsubaie, S.M.; Dahab, M.A.; Eissa, I.H. New Bis([1,2,4]Triazolo)[4,3-a:3′,4′-c]Quinoxaline Derivatives as VEGFR-2 Inhibitors and Apoptosis Inducers: Design, Synthesis, in Silico Studies, and Anticancer Evaluation. Bioorganic Chem. 2021, 112, 104949. [Google Scholar] [CrossRef]
- Pandiri, M.; Nukala, S.K.; Dasari, G.; Badithapuram, V.; Bandari, S. Design and Synthesis of Some New N-Phenyl-[1,2,4]Triazolo[4,3-a]Quinoxaline-1-Sulfonamide Derivatives and Their Anti-Cancer Activity. Russ. J. Gen. Chem. 2021, 91, 2280–2285. [Google Scholar] [CrossRef]
- Deleuze-Masquefa, C.; Bonnet, P.-A.; Cuq, P.; Patinote, C. New Imidazo[1,2-a]quinoxalines and Derivates Thereof for the Treatment of Cancer. U.S. Patent WO/2016/107895, 7 July 2016. [Google Scholar]
- Deleuze-Masquefa, C.; Moarbess, G.; Bonnet, P.-A.; Pinguet, F.; Bazarbachi, A.; Bressolle, F. Imidazo[1,2-a]Quinoxalines and Derivatives Thereof for Treating Cancers. U.S. Patent WO 2009/043934 A1, 9 April 2009. [Google Scholar]
- Patinote, C.; Deleuze-Masquéfa, C.; Kaddour, K.H.; Vincent, L.-A.; Larive, R.; Zghaib, Z.; Guichou, J.-F.; Assaf, M.D.; Cuq, P.; Bonnet, P.-A. Imidazo[1,2-a]Quinoxalines for Melanoma Treatment with Original Mechanism of Action. Eur. J. Med. Chem. 2021, 212, 113031. [Google Scholar] [CrossRef]
- Bou Karroum, N.; Moarbess, G.; Guichou, J.-F.; Bonnet, P.-A.; Patinote, C.; Bouharoun-Tayoun, H.; Chamat, S.; Cuq, P.; Diab-Assaf, M.; Kassab, I.; et al. Novel and Selective TLR7 Antagonists among the Imidazo[1,2-a]Pyrazines, Imidazo[1,5-a]Quinoxalines, and Pyrazolo[1,5-a]Quinoxalines Series. J. Med. Chem. 2019, 62, 7015–7031. [Google Scholar] [CrossRef]
- Chouchou, A.; Patinote, C.; Cuq, P.; Bonnet, P.-A.; Deleuze-Masquéfa, C. Imidazo[1,2-a]Quinoxalines Derivatives Grafted with Amino Acids: Synthesis and Evaluation on A375 Melanoma Cells. Molecules 2018, 23, 2987. [Google Scholar] [CrossRef] [Green Version]
- Patinote, C.; Bou Karroum, N.; Moarbess, G.; Deleuze-Masquefa, C.; Hadj-Kaddour, K.; Cuq, P.; Diab-Assaf, M.; Kassab, I.; Bonnet, P.-A. Imidazo[1,2-a]Pyrazine, Imidazo[1,5-a]Quinoxaline and Pyrazolo[1,5-a]Quinoxaline Derivatives as IKK1 and IKK2 Inhibitors. Eur. J. Med. Chem. 2017, 138, 909–919. [Google Scholar] [CrossRef] [PubMed]
- Hajj, R.E.; Youness, H.B.; Lachaud, L.; Bastien, P.; Masquefa, C.; Bonnet, P.-A.; Hajj, H.E.; Khalifeh, I. EAPB0503: An Imiquimod Analog with Potent in Vitro Activity against Cutaneous Leishmaniasis Caused by Leishmania Major and Leishmania Tropica. PLoS Negl. Trop. Dis. 2018, 12, e0006854. [Google Scholar] [CrossRef]
- Nabbouh, A.I.; Hleihel, R.S.; Saliba, J.L.; Karam, M.M.; Hamie, M.H.; Wu, H.-C.J.M.; Berthier, C.P.; Tawil, N.M.; Bonnet, P.-A.A.; Deleuze-Masquefa, C.; et al. Imidazoquinoxaline Derivative EAPB0503: A Promising Drug Targeting Mutant Nucleophosmin 1 in Acute Myeloid Leukemia. Cancer 2017, 123, 1662–1673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saliba, J.; Deleuze-Masquéfa, C.; Iskandarani, A.; El Eit, R.; Hmadi, R.; Mahon, F.-X.; Bazarbachi, A.; Bonnet, P.-A.; Nasr, R. EAPB0503, a Novel Imidazoquinoxaline Derivative, Inhibits Growth and Induces Apoptosis in Chronic Myeloid Leukemia Cells. Anticancer Drugs 2014, 25, 624–632. [Google Scholar] [CrossRef]
- Skayneh, H.; Jishi, B.; Hleihel, R.; Hamie, M.; El Hajj, R.; Deleuze-Masquefa, C.; Bonnet, P.-A.; El Sabban, M.; El Hajj, H. EAPB0503, an Imidazoquinoxaline Derivative Modulates SENP3/ARF Mediated SUMOylation, and Induces NPM1c Degradation in NPM1 Mutant AML. Int. J. Mol. Sci. 2022, 23, 3421. [Google Scholar] [CrossRef]
- Khier, S.; Gattacceca, F.; Messaoudi, S.E.; Lafaille, F.; Deleuze-Masquéfa, C.; Bompart, J.; Cooper, J.-F.; Solassol, I.; Pinguet, F.; Bonnet, P.-A.; et al. Metabolism and Pharmacokinetics of EAPB0203 and EAPB0503, Two Imidazoquinoxaline Compounds Previously Shown to Have Antitumoral Activity on Melanoma and T-Lymphomas. Drug Metab. Dispos. 2010, 38, 1836–1847. [Google Scholar] [CrossRef] [Green Version]
- Sarges, R.; Howard, H.R.; Browne, R.G.; Lebel, L.A.; Seymour, P.A.; Koe, B.K. 4-Amino[1,2,4]Triazolo[4,3-a]Quinoxalines. A Novel Class of Potent Adenosine Receptor Antagonists and Potential Rapid-Onset Antidepressants. J. Med. Chem. 1990, 33, 2240–2254. [Google Scholar] [CrossRef]
- Makino, K.; Sakata, G.; Morimoto, K.; Ochiai, Y. A Facile Synthesis of Novel Tricyclic Compounds, Tetrazoloquinoxalines and 1,2,4-Triazoloquinoxalines. Heterocycles 1985, 23, 2025. [Google Scholar] [CrossRef]
- Lee, S.; Cil, O.; Diez-Cecilia, E.; Anderson, M.O.; Verkman, A.S. Nanomolar-Potency 1,2,4-Triazoloquinoxaline Inhibitors of the Kidney Urea Transporter UT-A1. J. Med. Chem. 2018, 61, 3209–3217. [Google Scholar] [CrossRef] [PubMed]
- Al-Marhabi, A.R.; Abbas, H.-A.S.; Ammar, Y.A. Synthesis, Characterization and Biological Evaluation of Some Quinoxaline Derivatives: A Promising and Potent New Class of Antitumor and Antimicrobial Agents. Molecules 2015, 20, 19805–19822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guirado, A.; López Sánchez, J.I.; Ruiz-Alcaraz, A.J.; Bautista, D.; Gálvez, J. Synthesis and Biological Evaluation of 4-Alkoxy-6,9-Dichloro[1,2,4]Triazolo[4,3-a]Quinoxalines as Inhibitors of TNF-α and IL-6. Eur. J. Med. Chem. 2012, 54, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Abbass, E.M.; Khalil, A.K.; Mohamed, M.M.; Eissa, I.H.; El-Naggar, A.M. Design, Efficient Synthesis, Docking Studies, and Anticancer Evaluation of New Quinoxalines as Potential Intercalative Topo II Inhibitors and Apoptosis Inducers. Bioorganic Chem. 2020, 104, 104255. [Google Scholar] [CrossRef]
- el-Hawash, S.A.; Habib, N.S.; Fanaki, N.H. Quinoxaline Derivatives. Part II: Synthesis and Antimicrobial Testing of 1,2,4-Triazolo[4,3-a]Quinoxalines, 1,2,4-Triazino[4,3-a]Quinoxalines and 2-Pyrazolylquinoxalines. Die Pharm. 1999, 54, 808–813. [Google Scholar]
- EI-Bendary, E.R.; Goda, F.E.; Maarouf, A.R.; Badria, F.A. Synthesis and Antimicrobial Evaluation of 3-Hydrazino-Quinoxaline Derivatives and Their Cyclic Analoaues. Sci. Pharm. 2004, 72, 175–185. [Google Scholar] [CrossRef] [Green Version]
- Ajani, O.; Nwinyi, O. Synthesis and Evaluation of Antimicrobial Activity of Phenyl and Furan-2-Yl[1,2,4] Triazolo[4,3-a]Quinoxalin-4(5H)-One and Their Hydrazone Precursors. Can. J. Pure Appl. Sci. 2009, 3, 983–992. [Google Scholar]
- McQuaid, L.A.; Smith, E.C.R.; South, K.K.; Mitch, C.H.; Schoepp, D.D.; True, R.A.; Calligaro, D.O.; O’Malley, P.J.; Lodge, D.; Ornstein, P.L. Synthesis and Excitatory Amino Acid Pharmacology of a Series of Heterocyclic-Fused Quinoxalinones and Quinazolinones. J. Med. Chem. 1992, 35, 3319–3324. [Google Scholar] [CrossRef]
- Shiho, D.; Tagami, S. Studies on Compounds Related to Pyrazine. II. The Reaction of 3-Substituted-2-Hydrazinoquinoxalines with Carbonyl Compounds. J. Am. Chem. Soc. 1960, 82, 4044–4054. [Google Scholar] [CrossRef]
- Potts, K.T.; Schneller, S.W. 1,2,4-Triazoles. XX. Pyrolytic Decomposition of Ketone Hydrazones Derived from Pyrid-2-Ylhydrazine and Related Bases. Some Further Examples of the s-Triazolo[4,3-α]Pyrazine and s-Triazolo[4,3-a]Quinoxaline Series. J. Heterocycl. Chem. 1968, 5, 485–495. [Google Scholar] [CrossRef]
- Alswah, M.; Bayoumi, A.H.; Elgamal, K.; Elmorsy, A.; Ihmaid, S.; Ahmed, H.E.A. Design, Synthesis and Cytotoxic Evaluation of Novel Chalcone Derivatives Bearing Triazolo[4,3-a]-Quinoxaline Moieties as Potent Anticancer Agents with Dual EGFR Kinase and Tubulin Polymerization Inhibitory Effects. Molecules 2017, 23, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uçar, S.; Eşsiz, S.; Daştan, A. Bromination of Quinoxaline and Derivatives: Effective Synthesis of Some New Brominated Quinoxalines. Tetrahedron 2017, 73, 1618–1632. [Google Scholar] [CrossRef]
- Garrison, A.T.; Abouelhassan, Y.; Norwood, V.M.; Kallifidas, D.; Bai, F.; Nguyen, M.T.; Rolfe, M.; Burch, G.M.; Jin, S.; Luesch, H.; et al. Structure-Activity Relationships of a Diverse Class of Halogenated Phenazines That Targets Persistent, Antibiotic-Tolerant Bacterial Biofilms and Mycobacterium Tuberculosis. J. Med. Chem. 2016, 59, 3808–3825. [Google Scholar] [CrossRef]
- Conda-Sheridan, M.; Marler, L.; Park, E.-J.; Kondratyuk, T.P.; Jermihov, K.; Mesecar, A.D.; Pezzuto, J.M.; Asolkar, R.N.; Fenical, W.; Cushman, M. Potential Chemopreventive Agents Based on the Structure of the Lead Compound 2-Bromo-1-Hydroxyphenazine, Isolated from Streptomyces Species, Strain CNS284. J. Med. Chem. 2010, 53, 8688–8699. [Google Scholar] [CrossRef] [Green Version]
- Jørgensen, M.; Bruun, A.; Rasmussen, L.; Larsen, M. Dérivés De Triazolopyrazine Et Leur Utilisation Pour Le Traitement De Troubles Neurologiques Et Psychiatriques. U.S. Patent WO2013034755, 14 March 2013. [Google Scholar]
- Gupta, R.C.; Kumar, P.; Mane, U.R.; Mohanan, A.; Munshi, S.; Nadkarni, S.S.; Tandon, R. 2-Propene-1-Ones as HSP 70 Inducers. U.S. Patent WO2005097746A2, 20 October 2005. [Google Scholar]
- Andrés, J.-I.; Buijnsters, P.; De Angelis, M.; Langlois, X.; Rombouts, F.; Trabanco, A.A.; Vanhoof, G. Discovery of a New Series of [1,2,4]Triazolo[4,3-a]Quinoxalines as Dual Phosphodiesterase 2/Phosphodiesterase 10 (PDE2/PDE10) Inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 785–790. [Google Scholar] [CrossRef]
- Bouillon, C.; Paolantoni, D.; Rote, J.C.; Bessin, Y.; Peterson, L.W.; Dumy, P.; Ulrich, S. Degradable Hybrid Materials Based on Cationic Acylhydrazone Dynamic Covalent Polymers Promote DNA Complexation through Multivalent Interactions. Chem.–Eur. J. 2014, 20, 14705–14714. [Google Scholar] [CrossRef]
- Chen, S.; Sun, L.; Koya, K.; Tatsuta, N.; Xia, Z.; Korbut, T.; Du, Z.; Wu, J.; Liang, G.; Jiang, J.; et al. Syntheses and Antitumor Activities of N′1,N′3-Dialkyl-N′1,N′3-Di-(Alkylcarbonothioyl) Malonohydrazide: The Discovery of Elesclomol. Bioorg. Med. Chem. Lett. 2013, 23, 5070–5076. [Google Scholar] [CrossRef]
- Krishnan, V.; Chowdary, K.; Dubey, P.K. Studies in the Syntheses of S-Triazolo[4,3-a]Quinoxalines. Indian J. Chem. 1999, 38B, 45–51. [Google Scholar] [CrossRef]
- Cheng, D.; Yan, X.; Shen, J.; Pu, Y.; Xu, X.; Yan, J. Synthesis of 2,4-Diarylquinoline Derivatives via Chloranil-Promoted Oxidative Annulation and One-Pot Reaction. Synthesis 2020, 52, 1833–1840. [Google Scholar] [CrossRef]
- Delaney, J.S. ESOL: Estimating Aqueous Solubility Directly from Molecular Structure. J. Chem. Inf. Comput. Sci. 2004, 44, 1000–1005. [Google Scholar] [CrossRef]
- Avdeef, A.; Fuguet, E.; Llinàs, A.; Ràfols, C.; Bosch, E.; Völgyi, G.; Verbić, T.; Boldyreva, E.; Takács-Novák, K. Equilibrium Solubility Measurement of Ionizable Drugs–Consensus Recommendations for Improving Data Quality. ADMET DMPK 2016, 4, 117–178. [Google Scholar] [CrossRef] [Green Version]
- Shoghi, E.; Fuguet, E.; Bosch, E.; Ràfols, C. Solubility-PH Profiles of Some Acidic, Basic and Amphoteric Drugs. Eur. J. Pharm. Sci. 2013, 48, 291–300. [Google Scholar] [CrossRef]
- Mtewa, A.; Ngwira, K.; Lampiao, F.; Weisheit; Tolo, C.; Ogwang, P.; Sesaazi, D. Fundamental Methods in Drug Permeability, PKa, LogP and LogDx Determination. J. Drug Res. Dev. 2019, 5, 146. [Google Scholar]
- Bhal, S.K.; Kassam, K.; Peirson, I.G.; Pearl, G.M. The Rule of Five Revisited: Applying Log D in Place of Log P in Drug-Likeness Filters. Mol. Pharm. 2007, 4, 556–560. [Google Scholar] [CrossRef] [PubMed]
- Boedtkjer, E.; Pedersen, S.F. The Acidic Tumor Microenvironment as a Driver of Cancer. Annu. Rev. Physiol. 2020, 82, 103–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vistoli, G.; Pedretti, A. 5.24-Molecular Fields to Assess Recognition Forces and Property Spaces. In Comprehensive Medicinal Chemistry II; Taylor, J.B., Triggle, D.J., Eds.; Elsevier: Oxford, UK, 2007; pp. 577–602. ISBN 978-0-08-045044-5. [Google Scholar]
- Manallack, D.T. The PKa Distribution of Drugs: Application to Drug Discovery. Perspect. Med. Chem. 2007, 1, 25–38. [Google Scholar] [CrossRef]
- Courbet, A.; Bec, N.; Constant, C.; Larroque, C.; Pugniere, M.; Messaoudi, S.E.; Zghaib, Z.; Khier, S.; Deleuze-Masquefa, C.; Gattacceca, F. Imidazoquinoxaline Anticancer Derivatives and Imiquimod Interact with Tubulin: Characterization of Molecular Microtubule Inhibiting Mechanisms in Correlation with Cytotoxicity. PLoS ONE 2017, 12, e0182022. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Overall Yields | pKa1 | pKa2 | LogP | PSA | LogD (at pH 5.6) | LogS (mol/L) (at pH 5.6) | Aqueous Solubility (mg/mL at pH 7) |
|---|---|---|---|---|---|---|---|---|
| EAPB02303 | - | 4.32 | - | 3.55 | 82.68 | 3.50 | −4.26 | 1.70 × 10−2 |
| 4 | 85% | 2.09 | −4.03 | 2 | 55.11 | 2 | −3.21 | 12.0 × 10−2 |
| 8a | 2% | 1.94 | −4.23 | 2.86 | 73.57 | 2.85 | −4.34 | 1.52 × 10−2 |
| 8b | 6% | 1.91 | −4.24 | 3.53 | 64.34 | 3.50 | −4.62 | 7.23 × 10−3 |
| 8c | 7% | 1.98 | −4.17 | 3.91 | 55.11 | 3.65 | −4.76 | 4.84 × 10−3 |
| 8d | 50% | 1.81 | −4.34 | 2.07 | 82.80 | 2.05 | −4.00 | 3.64 × 10−2 |
| 16a | 59% | 1.58 | −4.04 | 4.74 | 73.57 | 4.75 | −5.36 | 1.45 × 10−3 |
| 16b | 60% | 1.56 | −4.11 | 5.23 | 64.34 | 5.25 | −5.55 | 8.67 × 10−4 |
| 16c | 29% | 1.74 | −4.06 | 5.24 | 55.11 | 5.25 | −5.47 | 9.23 × 10−4 |
| 16d | 14% | 1.31 | −4.21 | 4.33 | 82.80 | 4.30 | −5.23 | 2.17 × 10−3 |
| 16e | 24% | 2.51 | −0.34 | 3.83 | 68.00 | 3.80 | −4.60 | 6.98 × 10−3 |
| 16f | 37% | 2.49 | −3.77 | 3.37 | 55.11 | 3.30 | −4.50 | 9.50 × 10−3 |
| 16g | 17% | 2.55 | −3.69 | 3.22 | 55.11 | 3.25 | −3.96 | 2.79 × 10−2 |
| 16h | 57% | 2.49 | −3.77 | 3.06 | 55.11 | 3.10 | −3.88 | 3.35 × 10−2 |
| 16i | 39% | 2.50 | −3.67 | 1.69 | 55.11 | 1.70 | −3.06 | 19.0 × 10−2 |
| 16j | 46% | 1.29 | −4.26 | 3.58 | 87.56 | 3.55 | −4.77 | 5.47 × 10−3 |
| 17a | 4% | 1.31 | −4.26 | 3.91 | 95.57 | 3.90 | −4.65 | 7.07 × 10−3 |
| Compounds | Imiqualines References | Cell Viability at 10 µM (%) |
|---|---|---|
| 4 | EAPB4003 | 6.0 ± 1.2 |
| 8a | EAPB4003-8j | 96.9 ± 8.1 |
| 8b | EAPB4003-8k | 95.0 ± 3.4 |
| 8c | EAPB4003-8l | 50.7 ± 2.2 |
| 8d | EAPB4003-8m | 90.6 ± 4.8 |
| 16a | EAPB42203 | 2.9 ± 0.1 |
| 16b | EAPB4503 | 11.3 ± 6.9 |
| 16c | EAPB4403 | 46.7 ± 2.1 |
| 16d | EAPB42703 | 41.5 ± 0.8 |
| 16e | EAPB43803 | 56.8 ± 7.0 |
| 16f | EAPB4203 | 33.5 ± 13.3 |
| 16g | EAPB43603 | 76.6 ± 16.5 |
| 16h | EAPB4103 | 66.2 ± 5.9 |
| 16i | EAPB4303 | 85.4 ± 13.3 |
| 16j | EAPB42203 | 98.6 ± 11.58 |
| 17a | EAPB42303 | 5.5 ± 1.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patinote, C.; Raevens, S.; Baumann, A.; Pellegrin, E.; Bonnet, P.-A.; Deleuze-Masquéfa, C. [1,2,4]triazolo[4,3-a]quinoxaline as Novel Scaffold in the Imiqualines Family: Candidates with Cytotoxic Activities on Melanoma Cell Lines. Molecules 2023, 28, 5478. https://doi.org/10.3390/molecules28145478
Patinote C, Raevens S, Baumann A, Pellegrin E, Bonnet P-A, Deleuze-Masquéfa C. [1,2,4]triazolo[4,3-a]quinoxaline as Novel Scaffold in the Imiqualines Family: Candidates with Cytotoxic Activities on Melanoma Cell Lines. Molecules. 2023; 28(14):5478. https://doi.org/10.3390/molecules28145478
Chicago/Turabian StylePatinote, Cindy, Sandy Raevens, Amélie Baumann, Eloise Pellegrin, Pierre-Antoine Bonnet, and Carine Deleuze-Masquéfa. 2023. "[1,2,4]triazolo[4,3-a]quinoxaline as Novel Scaffold in the Imiqualines Family: Candidates with Cytotoxic Activities on Melanoma Cell Lines" Molecules 28, no. 14: 5478. https://doi.org/10.3390/molecules28145478