2.2. The Total Intermolecular Interaction Energy Values
The computed values for the supermolecular interaction energy and the total SAPT interaction energy are given in
Table 1. When focusing on the MP2 supermolecular energies, it can be seen that structures of Types 3 and 3a had the lowest values, ranging from ca.
to ca.
kJ mol
: these could generally be considered the most stable structures. Next, in terms of stability, were the Type 1 structures that involved heavier halogen atoms in the HX unit: their interaction energies ranged from ca.
to ca.
kJ mol
. Type 1 structures with light halogen atoms in HX, and Type 2 structures, had the highest interaction energy values: ca.
to ca.
kJ mol
. The Type 2 structures were bound the weakest.
The data in
Table 1 indicate that the total interaction energies for all Type 2 structures were very close to one another. The same can be observed for Group 3. Independent of the ingredients of the complex, and excluding complexes containing HF, the energies ranged from ca.
to ca.
kJ mol
for Type 3 structures, and from ca.
to ca.
kJ mol
for Type 2 structures. Meanwhile, the energies for Type 1 structures were very different, and clearly increased when going from HF to HI (with the exception of HXeF). The cause for this different trend seems to have been the difference in the origin of the interaction.
As depicted in
Figure 1, all the structure types, except Type 1, were formed representing hydrogen bonding. For Type 3 and 3a structures, the hydrogen bond was strong, because it was formed between the strongly negative Y
moiety of the noble-gas molecule and the hydrogen atom of the HX molecule, where the HX molecule acted as the proton donor. The magnitude of this interaction is comparable to the interaction in the water dimer [
43,
44]. The hydrogen bonds in Type 2 structures were weaker because the noble-gas molecule was the proton donor. It has been shown [
14] that complexation has a shortening effect on
, making it a less effective proton donor. Following on from the energetically unfavourable orientation, the interaction appeared weaker. However, the values of the interaction energies within the entire Group 2 were of comparable magnitude to one other, and not so diverse as for the structures in Group 1.
Conversely, the interaction origin for Type 1 structures was different. As can be seen in
Figure 1a, the interaction arose from the close vicinity of the halogen atoms in both interacting molecules, and thus, the interaction energy was different for each interacting case: it systematically increased with the atomic number of the halogen atom in the HX molecule; however, for a given HX molecule, it remained remarkably constant when changing the halogen atom in the HXeY molecule. Therefore, the magnitude of the interaction energy in Type 1 structures was primarily governed by the type of HX molecule.
We now compare the energy trend within each group, separately. In the case of Group 3, the total energy slightly increased (i.e., was less stabilizing) when going from HF to HI for a given noble-gas molecule. This correlated well with the decreasing dipole moment in the sequence of HF-HCl-HBr-HI. This trend, however, was reversed in Type 1 and 2 structures. This may have been caused by the relative orientation of the dipole moments of both molecules, and is discussed in
Section 2.3, in terms of SAPT contributions to the interaction energy.
In summary, the hydrogen-bonded complexes tended to have intermolecular interactions of comparable magnitude for a given geometry, independent of the composition. Conversely, the magnitude of the interaction for the complexes interacting via halogen atoms varied, depending on the halogen hydride. Simultaneously, it must be noted that complexes containing fluorine atoms are often exceptions to these considerations. The HXeF⋯HF complex was previously computationally reported by Jankowska and Sadlej [
38], and by Mohajeri and Bitaab [
13], in the forms of the Type 3 and 2a structures. Jankowska obtained
kJ mol
for the Type 3 structure, and
kJ mol
for the Type 2a structure, employing the MP2 method. Mohajeri and Bitaab reported
kJ mol
for the Type 3 structure, with the DFT/BMK approach. Our results agree with their data, giving the value of
kJ mol
at the MP2 theory level. The results discussed are also in agreement with previous studies of the complexes of HXeBr and HXeCl molecules [
14,
20].
2.3. The SAPT Analysis of the Intermolecular Interaction Energy
We now discuss the different terms, as obtained by the SAPT method. Aside from structures containing fluorine, all the studied complexes had a repulsive sum of first-order SAPT energies, i.e., the electrostatic and exchange interactions were not sufficient to explain the existence of the found minima, which means that they were stabilized by higher-order interaction terms: namely, by induction and dispersion.
The results are presented as follows, in a group-wise manner according to the structure types indicated in
Section 2.1. The numerical results of the total SAPT energy are presented in
Table 1. Furthermore, three types of graphs are shown, to facilitate different types of comparisons. In
Figure 2,
Figure 3,
Figure 4 and
Figure 5, values for different energy terms for the complexes grouped by the HX molecule are presented. In
Figure A1,
Figure A2 and
Figure A3, the same results are arranged by the HXeY molecule (the Type 2a and Type 3a results were not rearranged, due to their limited number). In the
Appendix A, in
Figure A4,
Figure A5,
Figure A6,
Figure A7,
Figure A8,
Figure A9 and
Figure A10, are the relative values of different components of the interaction energy. Accordingly, each individual term was divided by
, prior to plotting, the total SAPT interaction energy being represented by 1: this provided insight into the nature of the interaction, abstracting it from the magnitude of the interaction as a whole, which could be understood as a qualitative “fingerprint” of the interaction.
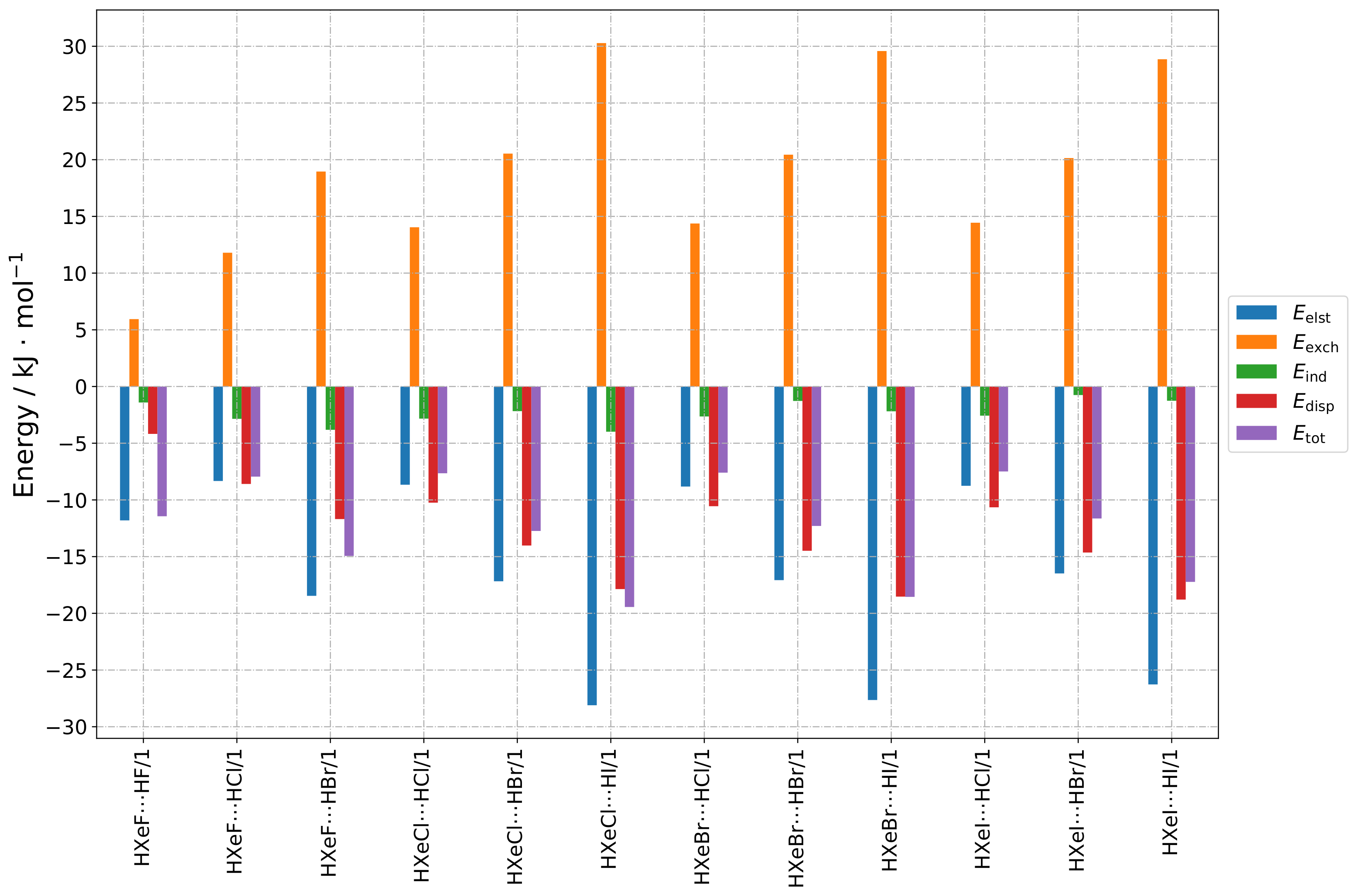
The SAPT contributions to the total interaction energy of the Group 1 structures are presented in
Figure 2 and
Figure A1. In this type of geometry, the HX molecule faces the halogen atom of HXeY, and the Xe–Y⋯X angle is acute (see
Figure 1a). The main sources of stabilization are the electrostatic and dispersive terms, while induction plays a minor role. Dispersion was clearly the dominant second-order contribution to the interaction energy. For a given HX molecule, the individual contributions remained remarkably constant. For a given HXeY molecule in this structure type, the electrostatic, exchange, and dispersion terms increased significantly when changing the mass of HX, which produced the increasing total interaction energy. This was in line with our supermolecular results (see
Section 2.2).
The relative contributions to the total interaction energy provided a qualitative picture of the interaction energy composition, i.e., they formed a fingerprint of the interaction type. The relative contributions are depicted in
Figure A4 and
Figure A10: one can see that the fingerprint remained generally the same for all these structures, independent of the composition.
The low induction contribution may come as a surprise, because the noble-gas molecules had large dipole moments; however, one may note that the dipole moments of both interacting molecules pointed towards similar directions, i.e., no large dipole moment induction should have been expected. Thus, there were no co-operative forces to induce additional partial charges.
This was in stark contrast to what was observed for energy partitioning for Type 3 structures. These structures generally had the lowest total interaction energy, which made them potentially the most stable. Their SAPT energies are presented in
Figure 4 and
Figure A3. For these geometries, the hydrogen atom from HX faced the halogen atom from HXeY, and the Xe–Y⋯H angle was acute (
Figure 1d). These structures depicted a hydrogen bond HXeY⋯HX interaction; however, as they were strongly bent, a larger interaction between entire constituent molecules was also expected. This is evidenced by the bar graphs in
Figure 4. One can see that the first-order energies provided little stabilizing effect, or were destabilizing, while the second-order induction and dispersion provided crucial attractive contributions to the total interaction. This shows that the Type 3 complexes were induction–dispersion-stabilised, whereas induction played the leading role in most of the structures. The role of dispersion increased with the atomic number of the halogen atom. In general terms, the magnitude of the induction and dispersion interactions was comparable for all Type 3 structures, especially for a given HXeY molecule, with the exception of those containing F. It can be concluded that the observed weakening of the total interaction, when going from HF to HI, was caused by the increasing electrostatic and exchange terms. The same trend could be observed for a fixed HX.
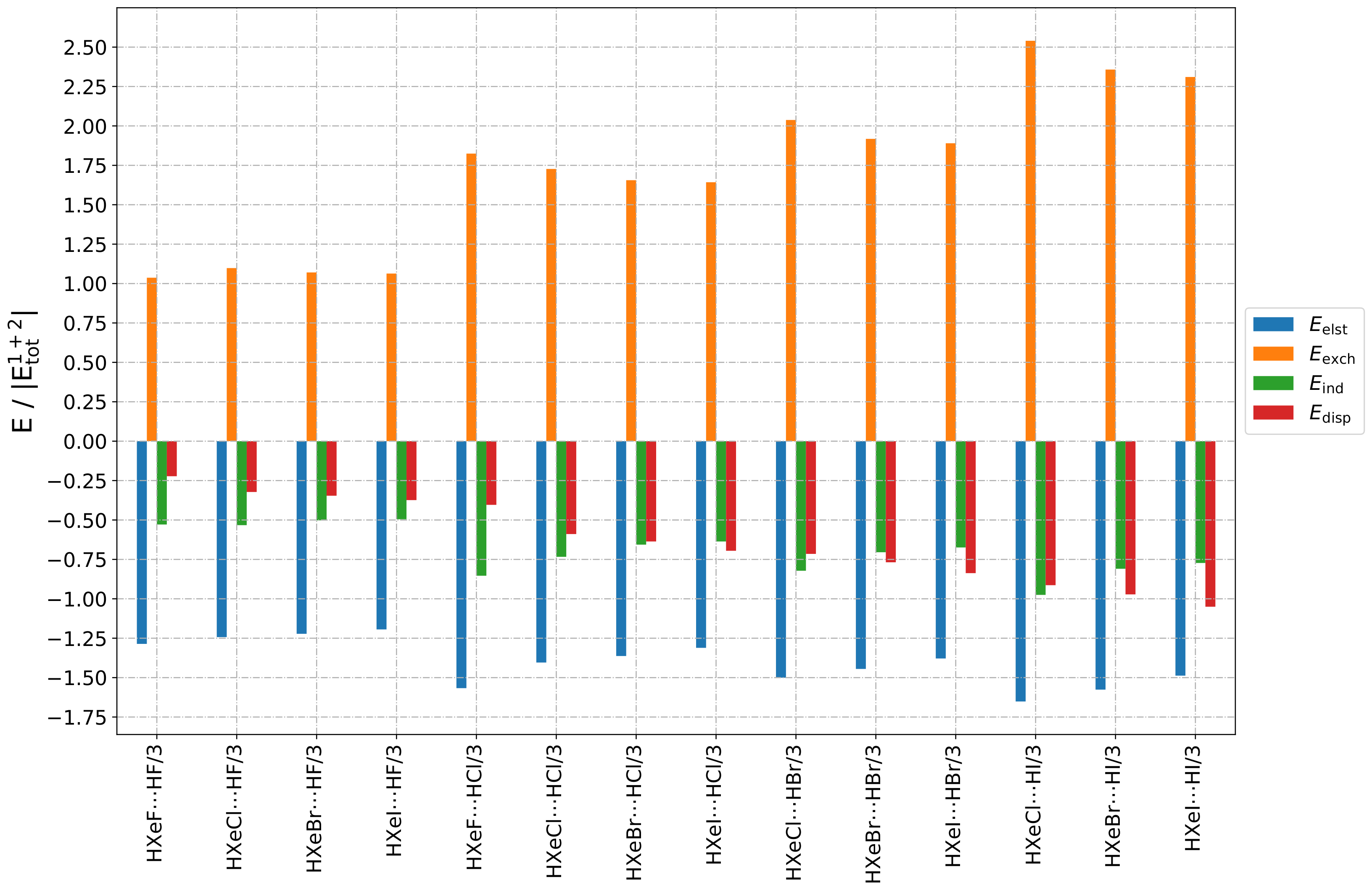
The relative contributions of the individual terms to the total interaction (see
Figure A6) were quite similar for all Type 3 structures, but there was a slight variation in dispersion-to-induction ratio, as noted above: the ratio changed from ca.
to
. Summarising these trends, it appears that the share of
, when compared to
, was highest for the HXeI⋯HI structure, and lowest for HXeF⋯HF. Additionally, for Type 3 structures, both the induction and dispersion interactions were the largest, compared to the other groups of the studied complexes.
In the same way as the small induction terms could be rationalized by the orientation of the interacting molecules for the Type 1 structures, the large induction interaction for the Type 3 structures could be explained by the dipole moments being oriented in an advantageous manner relative to one other, i.e., they were close to antiparallel orientation.
The SAPT energy contributions for the Type 2 structures are presented in
Figure 3 and
Figure A2: of all the considered types of structures, they had the highest total energy in general and, therefore, they were potentially the least stable complexes. The halogen atom of HX faced the hydrogen atom of HXeY, while the angle H-X⋯H was close to 90 degrees (see
Figure 1b). The structures were stabilised by electrostatic, induction, and dispersion energies, with the contributions of all three being important; however, as the electrostatic term was again quenched by the exchange interaction, it was straightforward that the stabilisation of these complexes arose mostly from the second-order corrections, i.e., dispersion and induction. Unlike the Type 3 structures, the Type 2 structures were clearly dominated by the dispersion contribution. Both induction and dispersion fell in order from HF to HI for the structures with a given HXeY. Similarly, both energy contributions fell in order from HXeF to HXeI for the structures with a given HX; however, this fall was generally smoother. There was an exception in the trends for structures with HXeF, in which the relative contribution of induction (see
Figure A9) and dispersion rose slightly in order from HF to HI. On the relative graphs, one can observe that the overall fingerprint of the interaction remained similar for all the Type 2 structures.
These energetic characteristics of the Type 1, 2, and 3 structures were in agreement with a previous report for HXeY⋯HX for X, Y = Cl and Br (see Table 4 in Ref. [
14]); however, direct comparison is difficult, because of the different methodology employed: namely, the Morokuma analysis. The present results show that trends in energy decomposition apply to different compositions of the complexes, and depend mostly on the geometry.
The Type 2a structures were similar to the Type 2 structures: the HX halogen atom faced the hydrogen atom of HXeY, but with the difference that the structure was linear, i.e., the angle H–X⋯H was close to 180 degrees (see
Figure 5). Only two structures of this kind were found to converge, and both these cases contained HF: for these structures, the relative share of electrostatic energy was much higher than the share of dispersion and induction energies. Compared to this, the structure with HXeCl had a lower total interaction energy, along with all other energy components. The destabilisation arose from greater exchange energy; moreover, the share of the induction energy compared to the dispersion energy was much higher for the structure with HXeCl (see
Figure A7).
Analogically, the Type 3a structures were similar to the Type 3 structures, the only difference being that the Xe–Y⋯H angle was obtuse. Only two such structures, containing HF, were successfully converged. Again, the share of electrostatic energy was much higher than the share of dispersion and induction energies. Here, the structure with HXeCl had higher total energy, which made it potentially less stable. The induction and electrostatic energies were clearly higher in the structure with HXeF, whereas the dispersion energy was lower. The destabilisation coming from the exchange energy was also lower. The share of induction compared to dispersion was significantly lower for the structure containing the HXeCl molecule.
2.4. Vibrational Spectroscopy
In this section, the focus is on features of the computed anharmonic vibrational spectra that are particularly interesting from the experimental point of view, i.e., the predicted wavenumbers of the stretching vibrational mode of Xe–H,
, and the stretching vibrational mode of H–X,
. The point of this discussion is to analyse the influence of the complexation on each property. The results of
are collected in
Table 2,
Table 3,
Table 4,
Table 5 and
Table 6, and the wavenumbers for
are collected in
Table 7,
Table 8,
Table 9,
Table 10 and
Table 11. The analysis was based on experimental values regarding the complexation effect, from existing literature resources, and the effect accounted for by the computational MP2 and B3LYP methods. Where we were unable to obtain a numerical value of a frequency, or where the literature data on a particular species were not available, we marked these cases with a hash character (#) in the tables.
Especially, Xe–H vibrational frequencies for isolated HNgY molecules were overestimated drastically on the levels used here. It has been previously suggested that using more sophisticated correlation methods to compute the harmonic spectra (such as CCSD(T)), and to effectively correct that with lower-level computed anharmonicity (such as MP2), can result in a reasonably good correlation between computations and experimental matrix isolation findings [
45,
46]. The importance of the anharmonicity of the HXeY molecules was noted by Runeberg et al. [
47] on HXeH, indicating that the multireference PES, when stretching the molecular bonds, was connected to the mismatch of vibrational fundamental modes between heavy ab initio calculations and experimental matrix isolation data; therefore, the abovementioned “CCSD(T) harmonic + MP2 anharmonic approach” could be considered a cost-effective way to predict IR features, compared to expensive and extensive molecular computations, in order to support and identify experimental vibrational features of systems involving noble-gas species, such as HXeY, studied here.
2.4.1. Xe–H Stretching Mode
As seen in
Table 2,
Table 3,
Table 4,
Table 5 and
Table 6, 32 of the 41 structures studied exhibited a blueshift of the Xe–H stretching mode: in accordance with previous studies [
14,
15,
31,
33,
35,
36], this shows that this behaviour was typical for the investigated compounds, even when the noble-gas molecule was not the proton donor. Two methodological points should be noted: firstly, the DFT/B3LYP/aug-cc-pVTZ(-PP) model seemed not to perform well, because it predicted redshifts for some of the structures, e.g., HXeCl⋯HCl/1, while the blueshifts seemed to be experimentally confirmed; secondly, although our monomer wavenumber values were overestimated by about 200 cm
, compared to the reported experimental results, the blueshifted values were under a minor influence of anharmonicity. For example, Lignell et al. [
14] studied the HXeY⋯HX complexes for Y, X = Cl and Br, and reported the experimental range observed for the blueshifted
for HXeCl⋯HCl to be 30.1–115.5 cm
; their harmonic computational results for the same range were 10–117 cm
, which agreed with our MP2 anharmonic results: 8–109 cm
(see
Table 3). The results for HXeCl⋯HBr and HXeBr⋯HBr and HXeBr⋯HCl compared equally well (see
Table 4).
The blueshifts were the largest for Type 3 structures, which can be traced back to their large interaction energies, i.e., stronger interaction and larger perturbation of electronic structures upon complexation, as discussed above. Their values ranged from 106 to 154 cm. The largest blueshifts were observed for complexes containing HF. The shifts for geometries of Types 1 and 2 of all compositions were positive, and ranged from a couple of units to 59 cm. The only exceptions to this blueshifting tendency were the complexes with HI, which—according to our anharmonic results—mostly exhibited a redshift.
The curious exception of complexes with HI is puzzling. We did not observe any drastic differences between this complex and the others, in terms of interaction energy components; however, the HXeCl⋯HI/3 complex was the least stable in the HXeCl⋯HX/3 group. HI also has the smallest dipole moment (see
Table 12). From the HXeCl⋯HI complexes, the HXeCl⋯HI/2 structure had the lowest interaction energy and the largest redshift, of −365 cm
. Zhu et al. [
36] noted that no experimental evidence for the HXeCl⋯HI complex was found, but their calculations predicted a blueshift. Our results, however, suggest that the vibrational bands corresponding to complexes with HI should be sought for wavenumbers lower, compared to the isolated noble-gas molecule. Tsuge et al. [
35] did indeed find computationally a redshifting H–Xe stretching mode, but only for the Type 2 structure of HXeI⋯HI, and for another dihydrogen-bonded structure not studied here. Perhaps in the case of HI depicting lower interaction energies, the same effect that causes the blueshift of
for other complexes does not occur. On the other hand, some part of the interaction mechanism may not been accounted for in our calculations: for example, a better description of relativistic effects—applying more sophisticated electron correlation methods or taking into account intramolecular BSSE for the HXeY molecule in the complex—could be considered [
47]. This could also give insights into blue/redshift mismatch, in case there are more fundamental halogen-bond-type interactions in the interactive triangle structures of heavy atoms in Type 1 structures [
48].
The results for complexes with HXeF provided two more redshifting cases: namely, the HXeF⋯HBr/2 and HXeF⋯HCl/3 structures; the first of these, however, had a redshift of −3 cm
, which may have been a computational artefact. The latter complex redshifted substantially, but there are no experimental data to refer to. In general, we were not successful in finding all the structures for HXeF complexes, but for those reported in
Table 2 the observed trends were similar to the other noble-gas molecules.
2.4.2. Hydrogen Halide H–X Stretching Mode
Another interesting vibrational mode is that of the HX molecule. In
Table 7,
Table 8,
Table 9,
Table 10 and
Table 11, one can see that all the
modes were redshift, and that the magnitude of the complexation effect corresponded to the intermolecular interaction energy. Consequently, the HXeCl/Br/I Type 3 structures, in which HX was the proton donor, showed the largest redshifts—approximately −300 to −500 cm
—while the Types 1 and 2 structures ranged from a few to approximately −40 cm
. In the HXeF complexes, which were not synthesised, the computational redshift of HF reached −1000 cm
.
This trend can be seen in each subset of the Type 1 structures, except for the HXeF molecule. For example, in the HXeCl⋯HCl/HBr/HI complexes, the redshift increased as the interaction energy increased. This trend was reversed for the Type 2 structures.
The obtained redshifted values for the HXeI complex were in agreement with previous experimental studies: for instance, the change registered by Zhu et al. for HXeI⋯HCl/3 (see Ref. [
36], Table V) was −337 cm
, and our anharmonic value was equal to −357 cm
. The experimental shift for the Type 3 complex with HBr was −378 cm
[
35], and our calculations provided the value of −363 cm
. Our results could be used as a corroboration of the experimental assignments from the literature: for example, Tsuge et al. suggested another
band that was redshifted by −266 cm
, but this value was quite far from the anharmonic results; similarly, the same authors observed a redshift of −167 cm
for HI in complexes with HXeI/3, but we predict a number twice as large—−347 cm
.
The computed redshifts of
for complexes with the HXeF molecule were very large: −863 cm
for HXeF⋯HF/3, and even −1001 cm
in the case of HXeF⋯HCl/3. The former value compared reasonably well to a previous study by Yen et al. [
49]. These authors obtained a redshift of −668 cm
for HXeF⋯HF/3. Jankowska and Sadlej [
38] reported a value of −728 cm
.



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}