
Three-Step Synthesis of the Antiepileptic Drug Candidate Pynegabine

Abstract
:1. Introduction
2. Results and Discussions
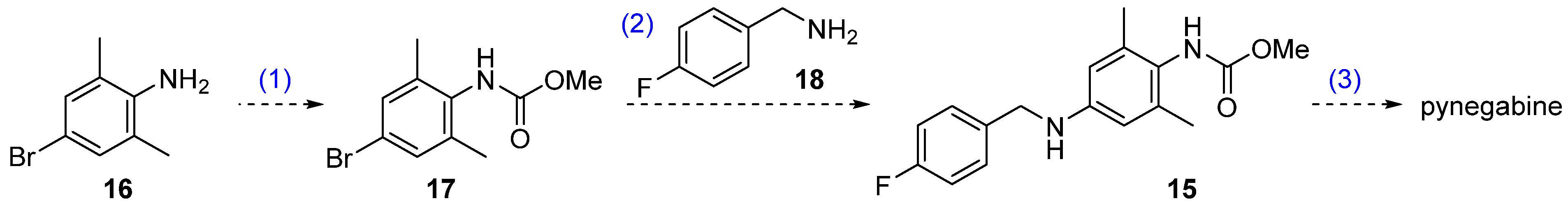
2.1. Methoxycarbonylation of Aniline 16
2.2. Buchwald–Hartwig Cross Coupling of the Compounds 17 and 18
2.3. Propargylation of Compound 15
3. Materials and Methods
3.1. General Information
3.2. Synthesis of Methyl (4-Bromo-2,6-dimethylphenyl)carbamate (17)
3.3. Synthesis of Methyl (4-(4-Fluorobenzylamino)-2,6-dimethylphenyl)carbamate (15)
3.4. Synthesis of Pynegabine
4. Conclusions
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- World Health Organization. Epilepsy, 9 February 2023. Available online: https://www.who.int/en/news-room/fact-sheets/detail/epilepsy (accessed on 27 May 2023).
- Elkommos, S.; Mula, M. Current and future pharmacotherapy options for drug-resistant epilepsy. Expert Opin. Pharmacother. 2022, 23, 2023–2034. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Li, K.; Chu, Y.; Li, C.; Zhang, T.; Liu, P.; Sun, T.; Jiang, C. ROS-removing nano-medicine for navigating inflammatory microenvironment to enhance anti-epileptic therapy. Acta Pharm. Sin. B 2023, 13, 1246–1261. [Google Scholar] [CrossRef] [PubMed]
- Stafstrom, C.E.; Grippon, S.; Kirkpatrick, P. Ezogabine (retigabine). Nat. Rev. Drug Discov. 2011, 10, 729–730. [Google Scholar] [CrossRef]
- Clark, S.; Antell, A.; Kaufman, K. New antiepileptic medication linked to blue discoloration of the skin and eyes. Ther. Adv. Drug Saf. 2015, 6, 15–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- GlaxoSmithKline: Trobalt®/Potiga® Discontinuation—Important Reminder. Available online: https://www.ilae.org/files/dmfile/GSK_RetigabineTrobalt-Reminder.pdf (accessed on 27 May 2023).
- Groseclose, M.R.; Castellino, S. An investigation into retigabine (ezogabine) associated dyspigmentation in rat eyes by MALDI imaging mass spectrometry. Chem. Res. Toxicol. 2019, 32, 294–303. [Google Scholar] [CrossRef]
- Bock, C.; Link, A. How to replace the lost keys? Strategies toward safer KV7 channel openers. Future Med. Chem. 2019, 11, 337–355. [Google Scholar] [CrossRef]
- Borgini, M.; Mondal, P.; Liu, R.; Wipf, P. Chemical modulation of Kv7 potassium channels. RSC Med. Chem. 2021, 12, 483–537. [Google Scholar] [CrossRef]
- Hernandez, C.C.; Tarfa, R.A.; Limcaoco, J.M.I.; Liu, R.; Mondal, P.; Hill, C.; Duncan, R.K.; Tzounopoulos, T.; Stephenson, C.R.J.; O’Meara, M.J.; et al. Development of an automated screen for Kv7.2 potassium channels and discovery of a new agonist chemotype. Bioorg. Med. Chem. Lett. 2022, 71, 128841. [Google Scholar] [CrossRef]
- Liu, S.; Guo, P.; Wang, K.; Zhang, S.; Li, Y.; Shen, J.; Mei, L.; Ye, Y.; Zhang, Q.; Yang, H. General pharmacological activation mechanism of K+ channels bypassing channel gates. J. Med. Chem. 2022, 65, 10285–10299. [Google Scholar] [CrossRef]
- Musella, S.; Carotenuto, L.; Iraci, N.; Baroli, G.; Ciaglia, T.; Nappi, P.; Basilicata, M.G.; Salviati, E.; Barrese, V.; Vestuto, V.; et al. Beyond retigabine: Design, synthesis, and pharmacological characterization of a potent and chemically stable neuronal Kv7 channel activator with anticonvulsant activity. J. Med. Chem. 2022, 65, 11340–11364. [Google Scholar] [CrossRef]
- Wang, T.; Krauss, G.L. XEN1101: A novel potassium channel modulator for the potential treatment of focal epilepsy in adults. Touchrev. Neurol. 2022, 18, 2–4. [Google Scholar] [CrossRef]
- Wurm, K.W.; Bartz, F.-M.; Schulig, L.; Bodtke, A.; Bednarski, P.J.; Link, A. Modifications of the triaminoaryl metabophore of flupirtine and retigabine aimed at avoiding quinone diimine formation. ACS Omega 2022, 7, 7989–8012. [Google Scholar] [CrossRef]
- Wurm, K.W.; Bartz, F.-M.; Schulig, L.; Bodtke, A.; Bednarski, P.J.; Link, A. Carba analogues of flupirtine and retigabine with improved oxidation resistance and reduced risk of quinoid metabolite formation. ChemMedChem 2022, 17, e202200262. [Google Scholar] [CrossRef]
- Wurm, K.W.; Bartz, F.-M.; Schulig, L.; Bodtke, A.; Bednarski, P.J.; Link, A. Replacing the oxidation-sensitive triaminoaryl chemotype of problematic KV7 channel openers: Exploration of a nicotinamide scaffold. Arch. Pharm. 2023, 356, e2200473. [Google Scholar] [CrossRef]
- Nan, F.-J.; Li, M.; Gao, Z.-B.; Zhang, Y.-M.; Hu, H.-N.; Xu, H.-Y.; Liu, H.-N.; Pi, X.-P. Novel KCNQ Potassium Channel Agonist and Preparation Method and Application Thereof. CN201410175315, 29 June 2018. [Google Scholar]
- Zhang, Y.-M.; Xu, H.-Y.; Hu, H.-N.; Tian, F.-Y.; Chen, F.; Liu, H.-N.; Zhan, L.; Pi, X.-P.; Liu, J.; Gao, Z.-B.; et al. Discovery of HN37 as a potent and chemically stable antiepileptic drug candidate. J. Med. Chem. 2021, 64, 5816–5837. [Google Scholar] [CrossRef]
- Rudén, C.; Hansson, S.O. How accurate are the European Union’s classifications of chemical substances. Toxicol. Lett. 2003, 144, 159–172. [Google Scholar] [CrossRef] [PubMed]
- Hachoose, R.C. How to handle hydrogen in process plants. Chem. Eng. 2006, 113, 54–59. [Google Scholar]
- Heravi, M.M.; Zadsirjan, V.; Malmir, M.; Mohammadi, L. Buchwald–Hartwig reaction: An update. Monatsh. Chem. 2021, 152, 1127–1171. [Google Scholar] [CrossRef]
- Knölker, H.-J.; Braxmeier, T. Isocyanates—Part 3. Synthesis of carbamates by DMAP-catalyzed reaction of amines with di-tert-butyldicarbonate and alcohols. Tetrahedron Lett. 1996, 37, 5861–5864. [Google Scholar] [CrossRef]
- Bak, I.G.; Chae, C.-G.; Lee, J.-S. Synthetic control of helical polyisocyanates by living anionic polymerization toward peptide mimicry. Macromolecules 2022, 55, 1923–1945. [Google Scholar] [CrossRef]
- Maiti, D.; Fors, B.P.; Henderson, J.L.; Nakamura, Y.; Buchwald, S.L. Palladium-catalyzed coupling of functionalized primary and secondary amines with aryl and heteroaryl halides: Two ligands suffice in most cases. Chem. Sci. 2011, 2, 57–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singer, R.A. BippyPhos: A highly versatile ligand for Pd-catalyzed C-N, C-O and C-C couplings. Isr. J. Chem. 2020, 60, 294–302. [Google Scholar] [CrossRef]
- Stewart, W.E.; Siddall, T.H., III. Nuclear magnetic resonance studies of amides. Chem. Rev. 1970, 70, 517–551. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | 16 (g) | Boc2O (eq) | DMAP (eq) | MeCN (mL) | MeOH (mL) | Time 1 (h) | Yield 2 (%) |
| 1 | 5.0 | 1.4 | 1.0 | 100 | 5 | 20 | 63 |
| 2 | 5.0 | 1.4 | 0.1 | 100 | 5 | 20 | 66 |
| 3 | 20.0 | 1.4 | 0.1 | 200 | 50 | 8 | 65 |
| 4 3 | 2.0 | 1.4 | 0.1 | 0 | 20 | 80 | <20 4 |
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | 18 (eq) | Ligand | Base | Solvent | Temp (°C) | Time (h) | Yield (%) |
| 1 | 1.1 | BrettPhos | Cs2CO3 | CPME | 95 | 16 | 66 1 |
| 2 | 1.1 | BrettPhos | Cs2CO3 | PhMe | 95 | 16 | 25 1 |
| 3 | 1.1 | BrettPhos | Cs2CO3 | tAmOH | 95 | 16 | 22 1 |
| 4 | 1.1 | BrettPhos | Cs2CO3 | DMAc | 95 | 16 | 18 1 |
| 5 | 1.1 | BrettPhos | tBuONa | THF | 40 | 16 | 86 1 |
| 6 | 1.1 | XPhos | tBuONa | THF | 40 | 16 | 86 1 |
| 7 | 1.1 | tBu-XPhos | tBuONa | THF | 40 | 16 | 89 1 |
| 8 | 1.1 | Me4tBu-XPhos | tBuONa | THF | 40 | 16 | 96 1 |
| 9 | 1.1 | JohnPhos | tBuONa | THF | 40 | 16 | 83 1 |
| 10 | 1.1 | DavePhos | tBuONa | THF | 40 | 16 | 80 1 |
| 11 | 1.1 | CPhos | tBuONa | THF | 40 | 16 | 86 1 |
| 12 | 1.1 | BippyPhos | tBuONa | THF | 40 | 16 | 98 1 |
| 13 4 | 1.1 | BippyPhos | tBuONa | THF | 40 | 23 | 73 2 |
| 14 4 | 0.95 | BippyPhos | tBuONa | THF | 40 | 23 | 63 3 |
| 15 4 | 0.95 | BippyPhos | tBuONa | THF | 50 | 5 | 86 3 |
| 16 5 | 0.95 | BippyPhos | tBuONa | THF | 50 | 3 | 72 3 |
| 17 6 | 0.95 | BippyPhos | tBuONa | THF | 50 | 2 | 78 3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, Y.-J.; Gong, Y.-L.; Lu, S.-C.; Zhang, S.-P.; Xu, S. Three-Step Synthesis of the Antiepileptic Drug Candidate Pynegabine. Molecules 2023, 28, 4888. https://doi.org/10.3390/molecules28134888
Sun Y-J, Gong Y-L, Lu S-C, Zhang S-P, Xu S. Three-Step Synthesis of the Antiepileptic Drug Candidate Pynegabine. Molecules. 2023; 28(13):4888. https://doi.org/10.3390/molecules28134888
Chicago/Turabian StyleSun, Yi-Jing, Ya-Ling Gong, Shi-Chao Lu, Shi-Peng Zhang, and Shu Xu. 2023. "Three-Step Synthesis of the Antiepileptic Drug Candidate Pynegabine" Molecules 28, no. 13: 4888. https://doi.org/10.3390/molecules28134888