
Novel Tryptanthrin Derivatives with Selectivity as c–Jun N–Terminal Kinase (JNK) 3 Inhibitors

 ,
,  , ,
, , 
Abstract
:1. Introduction
2. Results and Discussion
2.1. Synthesis of Tryptanthrin Derivatives
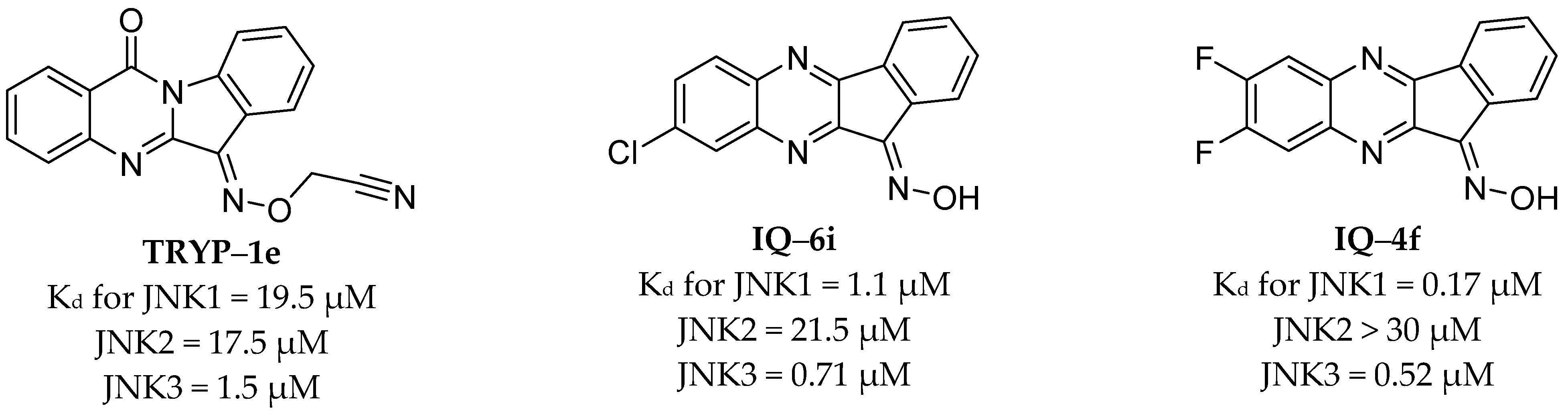
2.2. Binding Affinity of the Compounds for JNK1-3
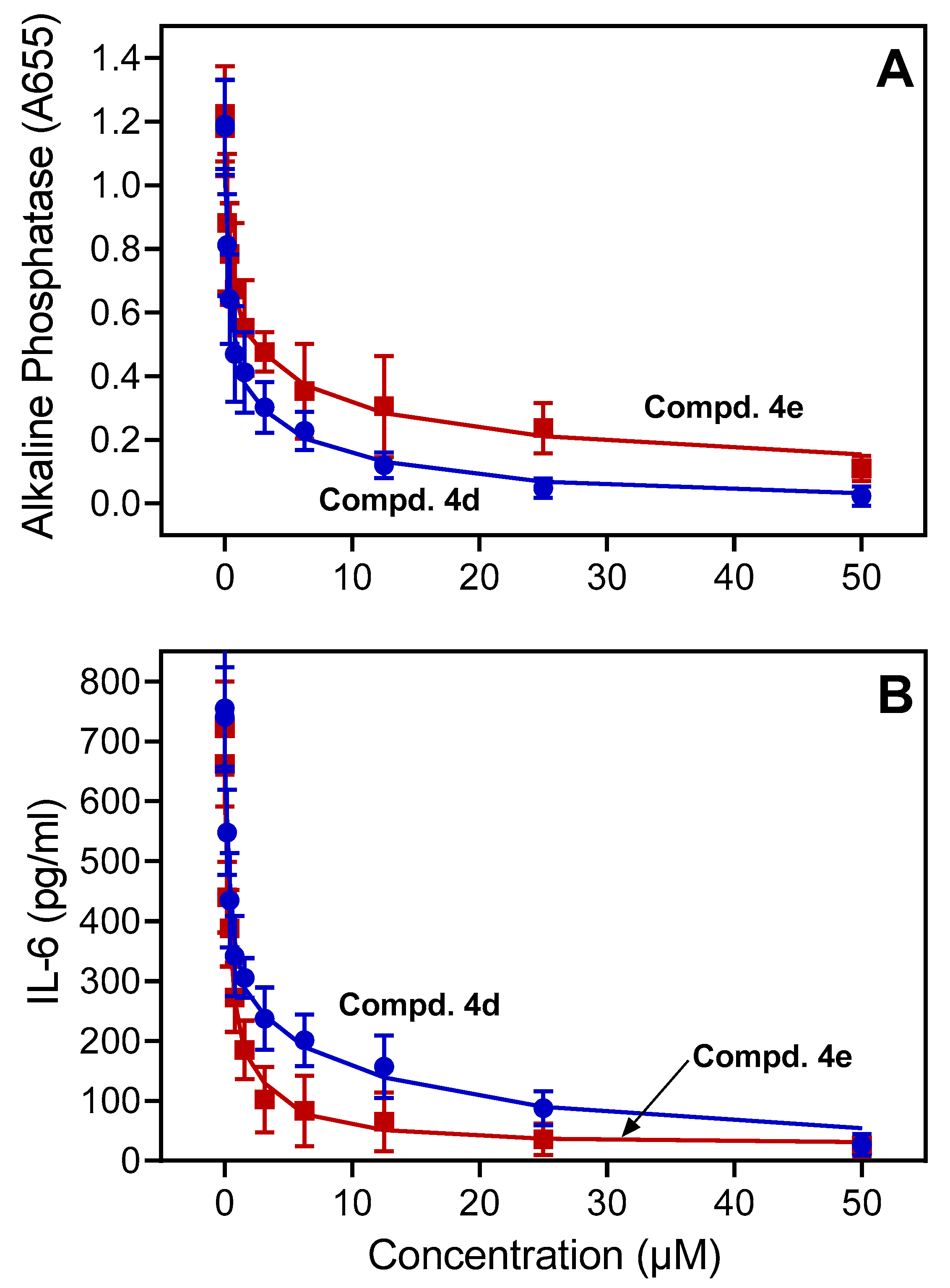
2.3. Activity of Compounds in Monocytic Cells
2.4. Molecular Modeling
3. Experimental Section
3.1. Chemistry
3.1.1. General Procedure for Synthesis of Ketones 3a–3n
3.1.2. General Procedure for Synthesis of Ketones 3o–3u
3.1.3. General Procedure for Synthesis of Compounds 4a–u
3.1.4. General Procedure for Synthesis of Compounds 5a,b
3.1.5. General Procedure for Synthesis of Compounds 5c,d
3.1.6. Preparation of 6-(((Benzo[d][1,3]dioxol-5-carbonyl)oxy)imino)indolo[2,1-b]quinazolin-12(6H)-one (5e)
3.2. Biological Analysis
3.2.1. Kinase Kd Determination
3.2.2. Cell Culture
3.2.3. Analysis of AP-1/NF-κB Activation
3.2.4. Cytotoxicity Assay
3.2.5. Analysis of c-Jun Phosphorylation
3.3. Molecular Docking
3.4. DFT Calculations
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Kyriakis, J.M.; Banerjee, P.; Nikolakaki, E.; Dai, T.; Rubie, E.A.; Ahmad, M.F.; Avruch, J.; Woodgett, J.R. The stress-activated protein kinase subfamily of c-Jun kinases. Nature 1994, 369, 156–160. [Google Scholar] [CrossRef] [PubMed]
- Abdelrahman, K.S.; Hassan, H.A.; Abdel-Aziz, S.A.; Marzouk, A.A.; Narumi, A.; Konno, H.; Abdel-Aziz, M. JNK signaling as a target for anticancer therapy. Pharmacol. Rep. 2021, 73, 405–434. [Google Scholar] [CrossRef] [PubMed]
- Johnson, G.L.; Lapadat, R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 2002, 298, 1911–1912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowes, V.L.; Ip, N.Y.; Wong, Y.H. Integration of signals from receptor tyrosine kinases and G protein-coupled receptors. Neurosignals 2002, 11, 5–19. [Google Scholar] [CrossRef]
- Hammouda, M.B.; Ford, A.E.; Liu, Y.; Zhang, J.Y. The JNK Signaling Pathway in Inflammatory Skin Disorders and Cancer. Cells 2020, 9, 857. [Google Scholar] [CrossRef] [Green Version]
- Ip, Y.T.; Davis, R.J. Signal transduction by the c-Jun N-terminal kinase (JNK)—From inflammation to development. Curr. Opin. Cell Biol. 1998, 10, 205–219. [Google Scholar] [CrossRef]
- Javadov, S.; Jang, S.; Agostini, B. Crosstalk between mitogen-activated protein kinases and mitochondria in cardiac diseases: Therapeutic perspectives. Pharmacol. Ther. 2014, 144, 202–225. [Google Scholar] [CrossRef] [Green Version]
- Nijboer, C.H.; van der Kooij, M.A.; van Bel, F.; Ohl, F.; Heijnen, C.J.; Kavelaars, A. Inhibition of the JNK/AP-1 pathway reduces neuronal death and improves behavioral outcome after neonatal hypoxic-ischemic brain injury. Brain Behav. Immun. 2010, 24, 812–821. [Google Scholar] [CrossRef]
- Geng, C.; Wei, J.; Wu, C. Mammalian STE20-like kinase 1 knockdown attenuates tnfalpha-mediated neurodegenerative disease by repressing the JNK pathway and mitochondrial stress. Neurochem. Res. 2019, 44, 1653–1664. [Google Scholar] [CrossRef]
- Johnson, G.L.; Nakamura, K. The c-jun kinase/stress-activated pathway: Regulation, function and role in human disease. Biochim. Biophys. Acta 2007, 1773, 1341–1348. [Google Scholar] [CrossRef] [Green Version]
- Waetzig, V.; Herdegen, T. Context-specific inhibition of JNKs: Overcoming the dilemma of protection and damage. Trends Pharmacol. Sci. 2005, 26, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Guma, M.; Ronacher, L.M.; Firestein, G.S.; Karin, M.; Corr, M. JNK-1 deficiency limits macrophage-mediated antigen-induced arthritis. Arthritis Rheum. 2011, 63, 1603–1612. [Google Scholar] [CrossRef]
- Bogoyevitch, M.A.; Boehm, I.; Oakley, A.; Ketterman, A.J.; Barr, R.K. Targeting the JNK MAPK cascade for inhibition: Basic science and therapeutic potential. Biochim. Biophys. Acta 2004, 1697, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.Y.; Zhang, Q.G. Agents targeting c-Jun N-terminal kinase pathway as potential neuroprotectants. Expert Opin. Investig. Drugs 2005, 14, 1373–1383. [Google Scholar] [CrossRef] [PubMed]
- Ge, H.X.; Zou, F.M.; Li, Y.; Liu, A.M.; Tu, M. JNK pathway in osteoarthritis: Pathological and therapeutic aspects. J. Recept. Sig. Transd. 2017, 37, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Solinas, G.; Becattini, B. JNK at the crossroad of obesity, insulin resistance, and cell stress response. Mol. Metab. 2017, 6, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Singh, U.K.; Kini, S.G.; Garg, V.; Agrawal, S.; Tomar, P.K.; Pathak, P.; Chaudhary, A.; Gupta, P.; Malik, A. JNK pathway signaling: A novel and smarter therapeutic targets for various biological diseases. Future Med. Chem. 2015, 7, 2065–2086. [Google Scholar] [CrossRef]
- Wu, Q.H.; Wu, W.D.; Jacevic, V.; Franca, T.C.C.; Wang, X.; Kuca, K. Selective inhibitors for JNK signalling: A potential targeted therapy in cancer. J. Enzym. Inhib. Med. Chem. 2020, 35, 574–583. [Google Scholar] [CrossRef] [Green Version]
- Plotnikov, M.B.; Aliev, O.I.; Shamanaev, A.Y.; Sidekhmenova, A.V.; Anishchenko, A.M.; Fomina, T.I.; Rydchenko, V.S.; Khlebnikov, A.I.; Anfinogenova, Y.J.; Schepetkin, I.A.; et al. Antihypertensive activity of a new c-Jun N-terminal kinase inhibitor in spontaneously hypertensive rats. Hypertens. Res. 2020, 43, 1068–1078. [Google Scholar] [CrossRef]
- Plotnikov, M.B.; Chernysheva, G.A.; Aliev, O.I.; Smol’iakova, V.I.; Fomina, T.I.; Osipenko, A.N.; Rydchenko, V.S.; Anfinogenova, Y.J.; Khlebnikov, A.I.; Schepetkin, I.A.; et al. Protective effects of a new c-Jun N-terminal kinase inhibitor in the model of global cerebral ischemia in rats. Molecules 2019, 24, 1722. [Google Scholar] [CrossRef] [Green Version]
- Plotnikov, M.B.; Chernysheva, G.A.; Smolyakova, V.I.; Aliev, O.I.; Trofimova, E.S.; Sherstoboev, E.Y.; Osipenko, A.N.; Khlebnikov, A.I.; Anfinogenova, Y.J.; Schepetkin, I.A.; et al. Neuroprotective effects of a novel inhibitor of c-Jun N-terminal kinase in the rat model of transient focal cerebral ischemia. Cells 2020, 9, 1860. [Google Scholar] [CrossRef]
- Cho, H.; Hah, J.M. A Perspective on the development of c-Jun N-terminal kinase inhibitors as therapeutics for Alzheimer’s Disease: Investigating Structure through Docking Studies. Biomedicines 2021, 9, 1431. [Google Scholar] [CrossRef] [PubMed]
- Gehringer, M.; Muth, F.; Koch, P.; Laufer, S.A. c-Jun N-terminal kinase inhibitors: A patent review (2010–2014). Expert Opin. Ther. Pat. 2015, 25, 849–872. [Google Scholar] [CrossRef] [PubMed]
- Rehfeldt, S.C.H.; Majolo, F.; Goettert, M.I.; Laufer, S. c-Jun N-terminal kinase inhibitors as potential leads for new therapeutics for Alzheimer’s Diseases. Int. J. Mol. Sci. 2020, 21, 9677. [Google Scholar] [CrossRef] [PubMed]
- Ha, J.; Kang, E.; Seo, J.; Cho, S. Phosphorylation Dynamics of JNK signaling: Effects of dual-specificity phosphatases (DUSPs) on the JNK pathway. Int. J. Mol. Sci. 2019, 20, 6157. [Google Scholar] [CrossRef] [Green Version]
- Bode, A.M.; Dong, Z. The functional contrariety of JNK. Mol. Carcinog. 2007, 46, 591–598. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Barrett, T.; Whitmarsh, A.J.; Cavanagh, J.; Sluss, H.K.; Derijard, B.; Davis, R.J. Selective interaction of JNK protein kinase isoforms with transcription factors. EMBO J. 1996, 15, 2760–2770. [Google Scholar] [CrossRef] [Green Version]
- Abdelli, S.; Bonny, C. JNK3 Maintains expression of the insulin receptor substrate 2 (IRS2) in insulin-secreting cells: Functional consequences for insulin signaling. PLoS ONE 2012, 7, 35997. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Zhang, P.X.; Zheng, X.Y.; Ye, C.T.; Li, M.Y.; Bian, P.Y.; Fan, C.; Zhang, Y. miR-155 regulates pro- and anti-inflammatory cytokine expression in human monocytes during chronic hepatitis C virus infection. Ann. Transl. Med. 2021, 9, 1618. [Google Scholar] [CrossRef]
- Gorogh, T.; Beress, L.; Quabius, E.S.; Ambrosch, P.; Hoffmann, M. Head and neck cancer cells and xenografts are very sensitive to palytoxin: Decrease of c-jun n-terminale kinase-3 expression enhances palytoxin toxicity. Mol. Cancer 2013, 12, 12. [Google Scholar] [CrossRef] [Green Version]
- Lapouge, G.; Millon, R.; Muller, D.; Abecassis, J.; Eber, M.; Bergerat, J.P.; Klein-Soyer, C. Cisplatin-induced genes as potential markers for thyroid cancer. Cell. Mol. Life Sci. 2005, 62, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Pi, X.; Wu, Y.; Ferguson, J.E., 3rd; Portbury, A.L.; Patterson, C. SDF-1α stimulates JNK3 activity via eNOS-dependent nitrosylation of MKP7 to enhance endothelial migration. Proc. Natl. Acad. Sci. USA 2009, 106, 5675–5680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lattin, J.E.; Greenwood, K.P.; Daly, N.L.; Kelly, G.; Zidar, D.A.; Clark, R.J.; Thomas, W.G.; Kellie, S.; Craik, D.J.; Hume, D.A.; et al. Beta-arrestin 2 is required for complement C1q expression in macrophages and constrains factor-independent survival. Mol. Immunol. 2009, 47, 340–347. [Google Scholar] [CrossRef]
- Ebelt, N.D.; Cantrell, M.A.; van den Berg, C.L. c-Jun N-terminal kinases mediate a wide range of targets in the metastatic cascade. Genes Cancer 2013, 4, 378–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Xiang, X.; Xia, M.; Su, J.; Wu, Y.; Shen, L.; Xu, Y.; Sun, L. Inhibition of JNK3 promotes apoptosis induced by BH3 mimetic S1 in chemoresistant human ovarian cancer cells. Anat. Rec. 2015, 298, 386–395. [Google Scholar] [CrossRef] [PubMed]
- Yan, F.; Wang, X.M.; Liu, Z.C.; Pan, C.; Yuan, S.B.; Ma, Q.M. JNK1, JNK2, and JNK3 are involved in P-glycoprotein-mediated multidrug resistance of hepatocellular carcinoma cells. Hepatobiliary Pancreat. Dis. Int. 2010, 9, 287–295. [Google Scholar]
- Butterfield, L.; Zentrich, E.; Beekman, A.; Heasley, L.E. Stress- and cell type-dependent regulation of transfected c-Jun N-terminal kinase and mitogen-activated protein kinase kinase isoforms. Biochem. J. 1999, 338, 681–686. [Google Scholar] [CrossRef]
- Sun, R.; Xiang, T.X.; Tang, J.; Peng, W.Y.; Luo, J.; Li, L.L.; Qiu, Z.; Tan, Y.Q.; Ye, L.; Zhang, M.; et al. 19q13 KRAB zinc-finger protein ZNF471 activates MAPK10/JNK3 signaling but is frequently silenced by promoter CpG methylation in esophageal cancer. Theranostics 2020, 10, 2243–2259. [Google Scholar] [CrossRef]
- Finch, A.; Davis, W.; Carter, W.G.; Saklatvala, J. Analysis of mitogen-activated protein kinase pathways used by interleukin 1 in tissues in vivo: Activation of hepatic c-Jun N-terminal kinases 1 and 2, and mitogen-activated protein kinase kinases 4 and 7. Biochem. J. 2001, 353, 275–281. [Google Scholar] [CrossRef]
- Gorogh, T.; Berwig, J.; Scola, N.; Lippert, B.M. Differential regulation of MAPK (JNK 3) gene expression in human head and neck squamous cell carcinomas. Onkologie 2004, 27, 353–357. [Google Scholar] [CrossRef]
- Zang, Y.C.; Zhu, J.; Li, Q.; Tu, J.; Li, X.Q.; Hu, R.K.; Yang, D.R. miR-137-3p modulates the progression of prostate cancer by regulating the JNK3/EZH2 axis. OncoTargets Ther. 2020, 13, 7921–7932. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Chen, S.; Lu, C. Amyloid precursor protein promotes the migration and invasion of breast cancer cells by regulating the MAPK signaling pathway. Int. J. Mol. Med. 2020, 45, 162–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.R.; Pei, P.; Shea, F.F.; Bissonnette, C.; Nieto, K.; Din, C.; Liu, Y.Y.; Schwendeman, S.P.; Lin, Y.X.; Richard, S.; et al. Fenretinide combines perturbation of signaling kinases, cell-extracellular matrix interactions and matrix metalloproteinase activation to inhibit invasion in oral squamous cell carcinoma cells. Carcinogenesis 2022, 22, 851–864. [Google Scholar] [CrossRef] [PubMed]
- Rehfeldt, S.C.H.; Laufer, S.; Goettert, M.I. A highly selective in vitro JNK3 inhibitor, FMU200, restores mitochondrial membrane potential and reduces oxidative stress and apoptosis in SH-SY5Y cells. Int. J. Mol. Sci. 2021, 22, 3701. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.T.; Zhu, G.W.; Liu, X.A.; Gao, T.F.; Fang, F.; Dou, X.D.; Li, Y.Y.; Zheng, R.Q.; Jin, H.W.; Zhang, L.R.; et al. The structure-based optimization of 3-substituted indolin-2-one derivatives as potent and isoform-selective c-Jun N-terminal kinase 3 (JNK3) inhibitors and biological evaluation. Eur. J. Med. Chem. 2023, 250, 115167. [Google Scholar] [CrossRef]
- Shuai, W.; Bu, F.Q.; Zhu, Y.M.; Wu, Y.Y.; Xiao, H.; Pan, X.L.; Zhang, J.F.; Sun, Q.; Wang, G.; Ouyang, L. Discovery of novel indazole chemotypes as isoform-selective JNK3 inhibitors for the treatment of Parkinson’s Disease. J. Med. Chem. 2023, 66, 1273–1300. [Google Scholar] [CrossRef]
- Jun, J.; Baek, J.; Kang, D.; Moon, H.; Kim, H.; Cho, H.; Hah, J.M. Novel 1,4,5,6-tetrahydrocyclopenta[d]imidazole-5-carboxamide-based JNK3 inhibitors: Design, synthesis, molecular docking, and therapeutic potential in neurodegenerative diseases. Eur. J. Med. Chem. 2023, 245, 114917. [Google Scholar] [CrossRef]
- Kaur, R.; Manjal, S.K.; Rawal, R.K.; Kumar, K. Recent synthetic and medicinal perspectives of tryptanthrin. Bioorganic Med. Chem. 2017, 25, 4533–4552. [Google Scholar] [CrossRef]
- Schepetkin, I.A.; Kovrizhina, A.R.; Stankevich, K.S.; Khlebnikov, A.I.; Kirpotina, L.N.; Quinn, M.T.; Cook, M.J. Design, synthesis and biological evaluation of novel O-substituted tryptanthrin oxime derivatives as c-Jun N-terminal kinase inhibitors. Front. Pharmacol. 2022, 13, 958687. [Google Scholar] [CrossRef]
- Schepetkin, I.A.; Khlebnikov, A.I.; Potapov, A.S.; Kovrizhina, A.R.; Matveevskaya, V.V.; Belyanin, M.L.; Atochin, D.N.; Zanoza, S.O.; Gaidarzhy, N.M.; Lyakhov, S.A.; et al. Synthesis, biological evaluation, and molecular modeling of 11H-indeno[1,2-b]quinoxalin-11-one derivatives and tryptanthrin-6-oxime as c-Jun N-terminal kinase inhibitors. Eur. J. Med. Chem. 2019, 161, 179–191. [Google Scholar] [CrossRef]
- Kirpotina, L.N.; Schepetkin, I.A.; Hammaker, D.; Kuhs, A.; Khlebnikov, A.I.; Quinn, M.T. Therapeutic effects of tryptanthrin and tryptanthrin-6-oxime in models of rheumatoid arthritis. Front. Pharmacol. 2020, 11, 1145. [Google Scholar] [CrossRef] [PubMed]
- Nie, Z.L.; Xia, X.L.; Zhao, Y.; Zhang, S.; Zhang, Y.W.; Wang, J.H. JNK selective inhibitor, IQ-1S, protects the mice against lipopolysaccharides-induced sepsis. Bioorgan. Med. Chem. 2021, 30, 115945. [Google Scholar] [CrossRef] [PubMed]
- Schepetkin, I.A.; Kirpotina, L.N.; Khlebnikov, A.I.; Hanks, T.S.; Kochetkova, I.; Pascual, D.W.; Jutila, M.A.; Quinn, M.T. Identification and characterization of a novel class of c-Jun N-terminal kinase inhibitors. Mol. Pharmacol. 2012, 81, 832–845. [Google Scholar] [CrossRef] [Green Version]
- Liakhov, S.A.; Schepetkin, I.A.; Karpenko, O.S.; Duma, H.I.; Haidarzhy, N.M.; Kirpotina, L.N.; Kovrizhina, A.R.; Khlebnikov, A.I.; Bagryanskaya, I.Y.; Quinn, M.T. Novel c-Jun N-Terminal Kinase (JNK) Inhibitors with an 11H-Indeno[1,2-b]quinoxalin-11-one Scaffold. Molecules 2021, 26, 5688. [Google Scholar] [CrossRef]
- Jia, F.C.; Zhou, Z.W.; Xu, C.; Wu, Y.D.; Wu, A.X. Divergent synthesis of quinazolin-4(3H)-ones and tryptanthrins enabled by a tert-butyl hydroperoxide/K3PO4-promoted oxidative cyclization of isatins at room temperature. Org. Lett. 2016, 18, 2942–2945. [Google Scholar] [CrossRef]
- Tucker, A.M.; Grundt, P. The chemistry of tryptanthrin and its derivatives. Arkivoc 2012, 1, 546–569. [Google Scholar] [CrossRef] [Green Version]
- Trofimov, B.A.; Schmidt, E.Y. Reactions of acetylenes in superbasic media. Recent advances. Russ. Chem. Rev. 2014, 83, 600–619. [Google Scholar] [CrossRef]
- Karaman, M.W.; Herrgard, S.; Treiber, D.K.; Gallant, P.; Atteridge, C.E.; Campbell, B.T.; Chan, K.W.; Ciceri, P.; Davis, M.I.; Edeen, P.T.; et al. A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2008, 26, 127–132. [Google Scholar] [CrossRef]
- Ansideri, F.; Dammann, M.; Boeckler, F.M.; Koch, P. Fluorescence polarization-based competition binding assay for c-Jun N-terminal kinases 1 and 2. Anal. Biochem. 2017, 532, 26–28. [Google Scholar] [CrossRef]
- Guha, M.; Mackman, N. LPS induction of gene expression in human monocytes. Cell. Signal. 2001, 13, 85–94. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Toll-like receptors; their physiological role and signal transduction system. Int. Immunopharmacol. 2001, 1, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Dugave, C.; Demange, L. Cis-trans isomerization of organic molecules and biomolecules: Implications and applications. Chem. Rev. 2003, 103, 2475–2532. [Google Scholar] [CrossRef] [PubMed]
- Blanco, F.; Alkorta, I.; Elguero, J. Barriers about double carbon-nitrogen bond in imine derivatives (aldimines, oximes, hydrazones, azines). Croat. Chem. Acta 2009, 82, 173–183. [Google Scholar]
- Scapin, G.; Patel, S.B.; Lisnock, J.; Becker, J.W.; LoGrasso, P.V. The structure of JNK3 in complex with small molecule inhibitors: Structural basis for potency and selectivity. Chem. Biol. 2003, 10, 705–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef] [Green Version]
- Weiss, K.; Warren, C.H.; Wettermark, G. Cis-trans isomerization about the carbon-nitrogen double bond. Structures of the isomers of N-benzylideneaniline. J. Am. Chem. Soc. 1971, 93, 4658–4663. [Google Scholar] [CrossRef]
- Lakeev, A.P.; Frelikh, G.A.; Yanovskaya, E.A.; Kovrizhina, A.R.; Udut, V.V. Quantification of a promising JNK inhibitor and nitrovasodilator IQ-1 and its major metabolite in rat plasma by LC-MS/MS. Bioanalysis 2022, 14, 1423–1441. [Google Scholar] [CrossRef]
- Krivogorsky, B.; Nelson, A.C.; Douglas, K.A.; Grundt, P. Tryptanthrin derivatives as Toxoplasma gondii inhibitors-structure-activity-relationship of the 6-position. Bioorganic Med. Chem. Lett. 2013, 23, 1032–1035. [Google Scholar] [CrossRef]
- Thomsen, R.; Christensen, M.H. MolDock: A new technique for high-accuracy molecular docking. J. Med. Chem. 2006, 49, 3315–3321. [Google Scholar] [CrossRef]
- Neese, F.; Wennmohs, F.; Becker, U.; Riplinger, C. The ORCA quantum chemistry program package. J. Chem. Phys. 2020, 152, 224108. [Google Scholar] [CrossRef]
- Ásgeirsson, V.; Birgisson, B.O.; Bjornsson, R.; Becker, U.; Neese, F.; Riplinger, C.; Jónsson, H. Nudged Elastic Band Method for Molecular Reactions Using Energy-Weighted Springs Combined with Eigenvector Following. J. Chem. Theory Comput. 2021, 17, 4929–4945. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Brandenburg, J.G.; Bannwarth, C.; Hansen, A. Consistent structures and interactions by density functional theory with small atomic orbital basis sets. J. Chem. Phys. 2015, 143, 054107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skyner, R.E.; McDonagh, J.L.; Groom, C.R.; van Mourik, T.; Mitchell, J.B. A review of methods for the calculation of solution free energies and the modelling of systems in solution. Phys. Chem. Chem. Phys. 2015, 17, 6174–6191. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 |  | ||||
| Compd. | R1 | Compd. | R1 | Compd. | R2 |
| 1a | H | 1g | 4-CH3 | 2a | H |
| 1b | 5-NO2 | 1h | 6-CH3 | 2b | 6-Br |
| 1c | 5-Br | 1i | 5-CH3 | 2c | 6-CF3 |
| 1d | 5-Et | 1j | 6-OCH3 | 2d | 7-Br |
| 1e | 5-OCH3 | 1k | 6-Br | 2e | 7-f |
| 1f | 5-C6H5 | 1l | 4-Br | 2f | 7-NO2 |
| 2g | 6-NO2 | ||||
 | |||||||||||
| Compd. | R1 | R2 | R3 | R4 | R5 | R6 | JNK1 | JNK2 | JNK3 | Selectivity Index | |
| Kd (µM) | JNK1/JNK3 | JNK2/JNK3 | |||||||||
| TRYP-Ox | H | H | H | H | H | H | 0.15 ± 0.08 | 1.0 ± 0.14 | 0.28 ± 0.21 | 0.15 | 3.6 |
| 4a | H | NO2 | H | H | H | H | N.B. | N.B. | 0.86 ± 0.042 | >40 | >40 |
| 4b | H | Br | H | H | H | H | 3.2 ± 0.21 | 8.5 ± 3.2 | 0.41 ± 0.07 | 7.8 | 20.7 |
| 4c | H | Et | H | H | H | H | 2.6 ± 0.4 | 1.3 ± 0.01 | 0.49 ± 0.09 | 5.3 | 2.7 |
| 4d | H | OMe | H | H | H | H | N.B. | N.B. | 0.34 ± 0.03 | >100 | >100 |
| 4e | H | Ph | H | H | H | H | N.B. | N.B. | 0.34 ± 0.04 | >100 | >100 |
| 4f | H | NO2 | H | H | Br | H | N.B. | N.B. | N.B. | ||
| 4g | Me | H | H | H | H | H | 6.2 ± 1.8 | 14.0 ± 0.7 | 0.40 ± 0.04 | 15.5 | 35 |
| 4h | H | H | Me | H | H | H | 0.67 ± 0.35 | 0.57 ± 0.12 | 0.23 ± 0.04 | 2.9 | 2.5 |
| 4i | H | H | H | H | CF3 | H | N.B. | N.B. | 11.0 ± 1.4 | >3 | >3 |
| 4j | H | H | H | H | Br | H | 4.1 ± 0.6 | 7.4 ± 0.6 | 1.9 ± 0.4 | 2.2 | 3.9 |
| 4k | H | H | H | H | H | Br | N.B. | N.B. | 5.7 ± 1.8 | >6 | >6 |
| 4l | H | H | H | H | H | F | 1.1 ± 0.1 | 0.72 ± 0.22 | 0.43 ± 0.04 | 2.6 | 1.7 |
| 4m | H | H | H | H | H | NO2 | N.B. | N.B. | 6.1 ± 0.4 | >5.6 | >5.6 |
| 4n | H | H | H | H | NO2 | H | N.B. | N.B. | 8.7 ± 1.0 | >3.9 | >3.9 |
| 4o | H | Me | H | H | Me | H | 3.7 ± 0.5 | 6.9 ± 0.2 | 0.87 ± 0.16 | 4.3 | 7.9 |
| 4p | Me | H | H | Me | H | H | N.B. | N.B. | N.B. | ||
| 4q | H | OMe | H | H | OMe | H | N.B. | N.B. | N.B. | ||
| 4r | H | H | OMe | H | H | OMe | 4.6 ± 0.6 | 5.8 ± 1.6 | 3.2 ± 1.1 | 1.4 | 1.8 |
| 4s | H | H | Br | H | H | Br | N.B. | N.B. | N.B. | ||
| 4t | H | Br | H | H | Br | H | N.B. | N.B. | N.B. | ||
| 4u | Br | H | H | Br | H | H | N.B. | N.B. | N.B. | ||
 | |||||||||||
| Compd. | R | JNK1 | JNK2 | JNK3 | Selectivity Index | ||||||
| Kd (µM) | JNK1/JNK3 | JNK2/JNK3 | |||||||||
| 5a | -CH2COOEt | N.B. | N.B. | N.B. | |||||||
| 5b | -CH(COOEt)2 | 16.5 ± 0.7 | N.B. | N.B. | |||||||
| 5c | -COOCH3 | 1.7 ± 0.4 | 2.8 ± 0.5 | 0.70 ± 0.12 | 2.4 | 4.0 | |||||
| 5d | -COOCH2-C≡CH | 0.95 ± 0.03 | 1.6 ± 0.1 | 0.32 ± 0.06 | 3.0 | 5.0 | |||||
| 5e |  | 0.47 ± 0.08 | 1.5 ± 0.6 | 0.27 ± 0.01 | 1.7 | 0.9 | |||||
| Compd. | AP Production in THP–1Blue | Cytotoxicity in THP–1Blue | IL–6 Production in MonoMac–6 | Cytotoxicity in MonoMac–6 |
|---|---|---|---|---|
| IC50, μM | ||||
| TRYP–Ox | 3.8 ± 1.1 | N.T. | 3.2 ± 1.2 | N.T. |
| 4a | n.d. | 8.4 ± 0.9 | n.d. | 4.1 ± 0.7 |
| 4b | 2.1 ± 0.7 | N.T. | 15.5 ± 3.8 | N.T. |
| 4c | 0.7 ± 0.2 | N.T. | n.d. | 13.3 ± 1.3 |
| 4d | 0.8 ± 0.3 | N.T. | 0.7 ± 0.1 | N.T. |
| 4e | 1.6 ± 0.1 | N.T. | 0.17 ± 0.02 | N.T. |
| 4f | n.d. | 3.8 ± 0.9 | n.d. | 2.7 ± 0.6 |
| 4g | N.A. | N.T. | 25.2 ± 5.3 | N.T. |
| 4h | 0.9 ± 0.2 | N.T. | 0.3 ± 0.1 | N.T. |
| 4i | n.d. | 5.6 ± 1.2 | n.d. | 5.7 ± 1.2 |
| 4j | n.d. | 12.5 ± 2.3 | n.d. | 8.2 ± 1.6 |
| 4k | n.d. | 24.1 ± 2.7 | 0.41 ± 0.04 | N.T. |
| 4l | 5.0 ± 0.1 | N.T. | 0.61 ± 0.04 | N.T. |
| 4m | 8.3 ± 0.2 | N.T. | 1.9 ± 0.3 | N.T. |
| 4n | n.d. | 39.3 ± 3.1 | 9.6 ± 1.3 | N.T. |
| 4o | 8.1 ± 0.1 | N.T. | 0.8 ± 0.1 | N.T. |
| 4p | N.A. | N.T. | N.A. | N.T. |
| 4q | 4.6 ± 0.3 | N.T. | 1.3 ± 0.4 | N.T. |
| 4r | n.d. | 8.3 ± 1.3 | n.d. | 4.3 ± 0.9 |
| 4s | n.d. | 49.1 ± 8.3 | n.d. | 16.9 ± 3.4 |
| 4t | n.d. | 6.6 ± 0.3 | n.d. | 9.2 ± 2.1 |
| 4u | N.A. | N.T. | N.A. | N.T. |
| 5a | N.A. | N.T. | N.A. | N.T. |
| 5b | N.A. | N.T. | N.A. | N.T. |
| 5c | 8.1 ± 2.2 | N.T. | 12.4 ± 1.3 | N.T. |
| 5d | 4.4 ± 1.6 | N.T. | 7.0 ± 0.9 | N.T. |
| 5e | 3.5 ± 1.2 | N.T. | n.d. | 27.3 ± 1.4 |
| Compound | DS |
|---|---|
| TRYP-Ox (Z) | −62.2 |
| TRYP-Ox (E) | −34.9 |
| 4a (Z) | −71.12 |
| 4a (E) | −70.17 |
| 4b (Z) | −73.32 |
| 4b (E) | −66.26 |
| 4d (E) | −84.91 |
| 4d (Z) | * |
| 4f (Z) | −51.87 |
| 4f (E) | −27.04 |
| 4j (E) | −70.95 |
| 4j (Z) | −63.44 |
| 4k (Z) | −71.36 |
| 4k (E) | * |
| 4m (Z) | −69.18 |
| 4m (E) | −44.17 |
| 4n (Z) | −65.97 |
| 4n (E) | −38.77 |
| 4q (Z) | −34.07 |
| 4q (E) | −23.00 |
| 4s (E) | −49.47 |
| 4s (Z) | −21.43 |
| 4t (E) | −29.88 |
| 4t (Z) | −16.65 |
| 4u (E) | −45.07 |
| 4u (Z) | −21.39 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schepetkin, I.A.; Karpenko, O.S.; Kovrizhina, A.R.; Kirpotina, L.N.; Khlebnikov, A.I.; Chekal, S.I.; Radudik, A.V.; Shybinska, M.O.; Quinn, M.T. Novel Tryptanthrin Derivatives with Selectivity as c–Jun N–Terminal Kinase (JNK) 3 Inhibitors. Molecules 2023, 28, 4806. https://doi.org/10.3390/molecules28124806
Schepetkin IA, Karpenko OS, Kovrizhina AR, Kirpotina LN, Khlebnikov AI, Chekal SI, Radudik AV, Shybinska MO, Quinn MT. Novel Tryptanthrin Derivatives with Selectivity as c–Jun N–Terminal Kinase (JNK) 3 Inhibitors. Molecules. 2023; 28(12):4806. https://doi.org/10.3390/molecules28124806
Chicago/Turabian StyleSchepetkin, Igor A., Oleksander S. Karpenko, Anastasia R. Kovrizhina, Liliya N. Kirpotina, Andrei I. Khlebnikov, Stepan I. Chekal, Alevtyna V. Radudik, Maryna O. Shybinska, and Mark T. Quinn. 2023. "Novel Tryptanthrin Derivatives with Selectivity as c–Jun N–Terminal Kinase (JNK) 3 Inhibitors" Molecules 28, no. 12: 4806. https://doi.org/10.3390/molecules28124806