
Peripheralization Strategies Applied to Morphinans and Implications for Improved Treatment of Pain

Abstract
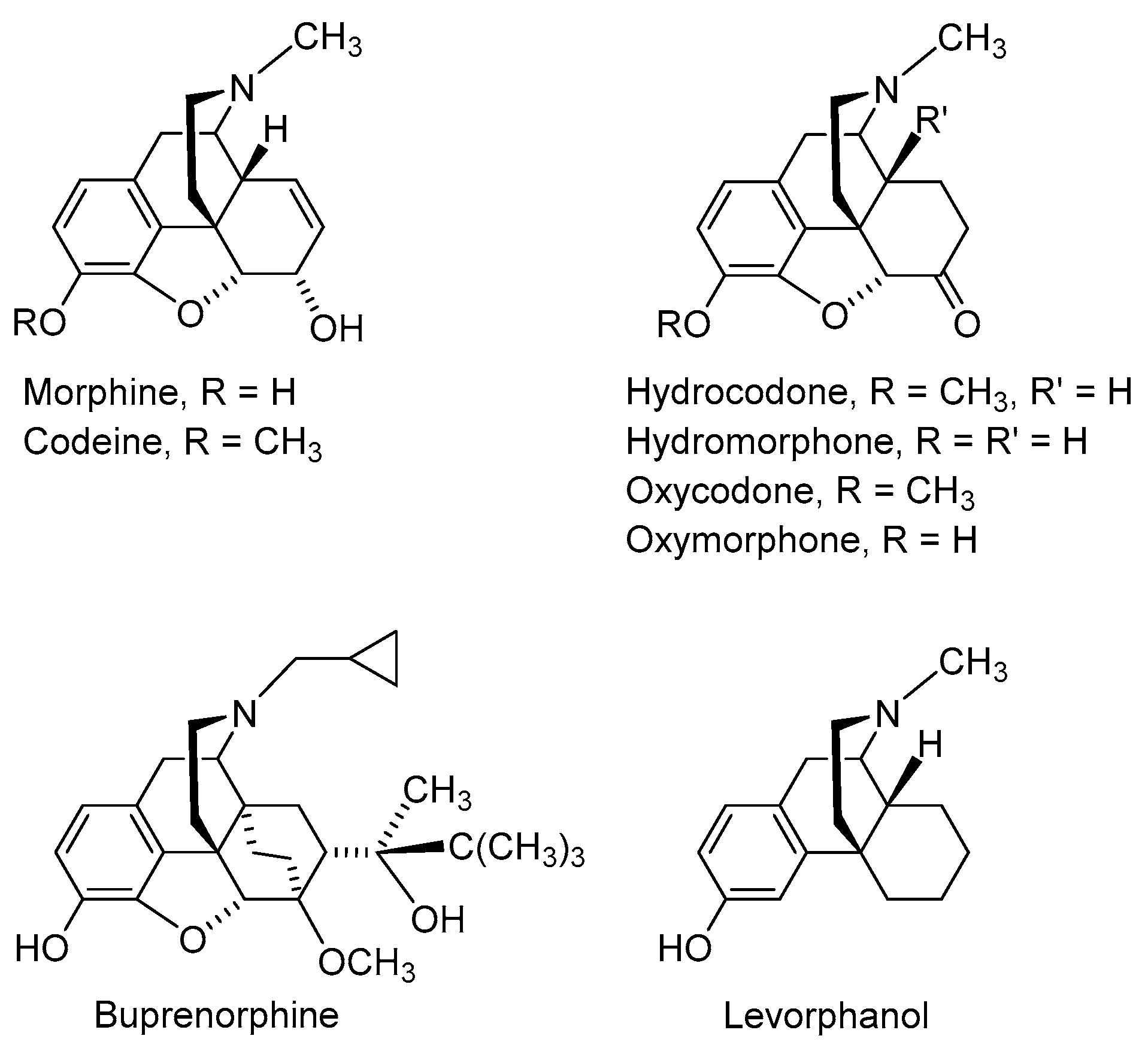
:1. Introduction
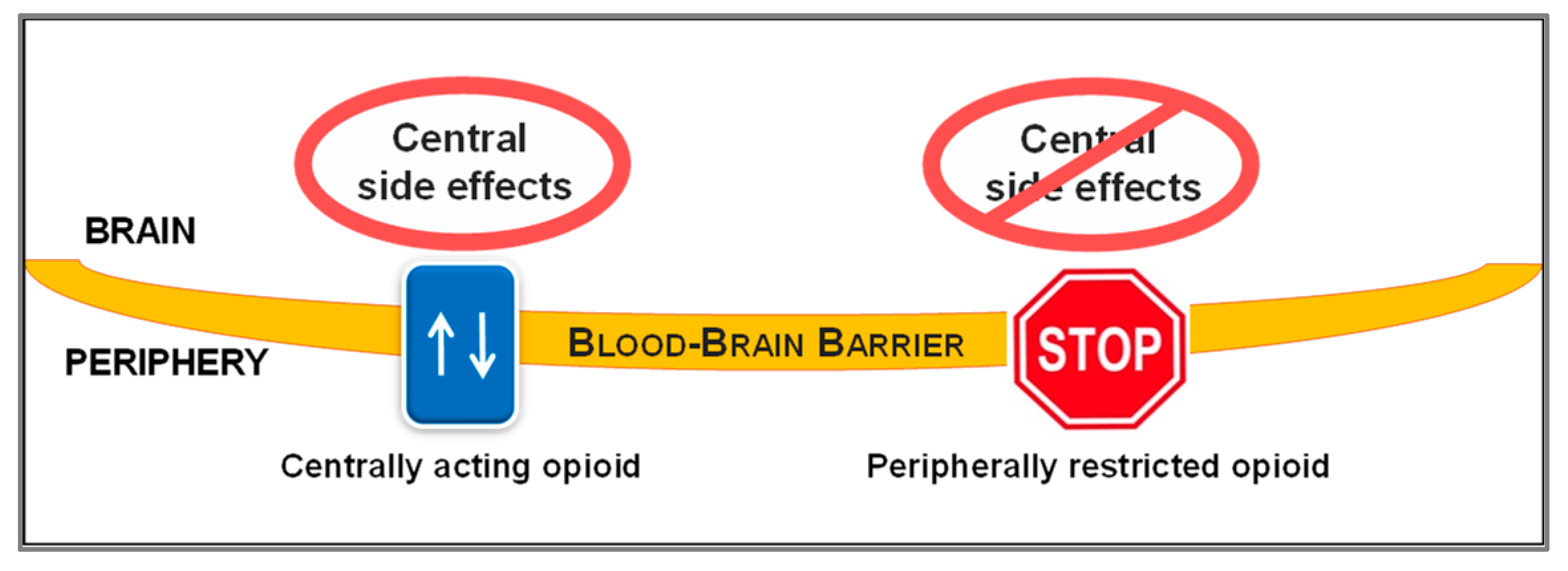
2. Peripheralization Strategies Applied to Morphinans
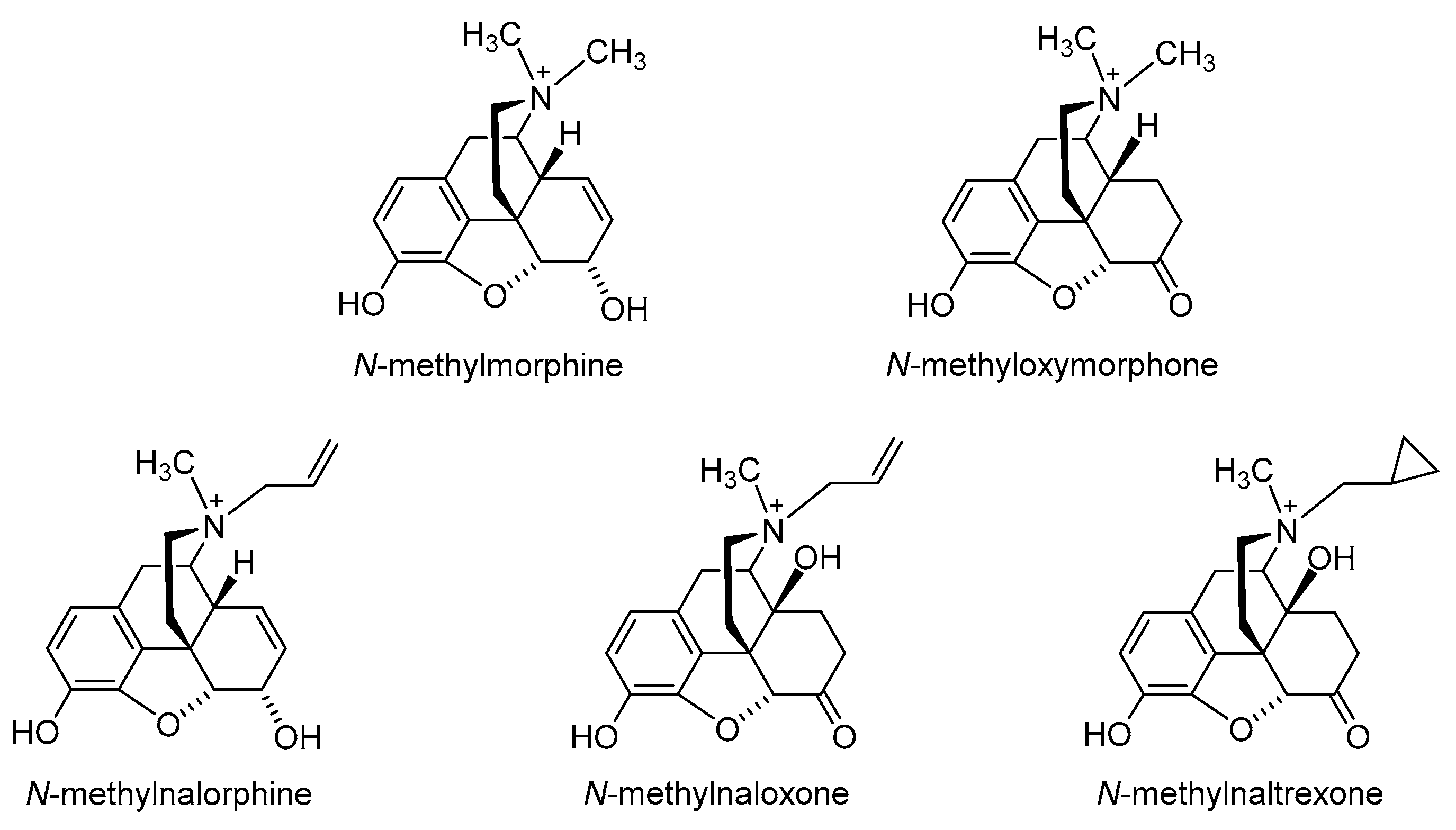
2.1. Quaternization of the Morphinan Nitrogen
2.2. Introduction of Hydrophilic Substituents at Position 6
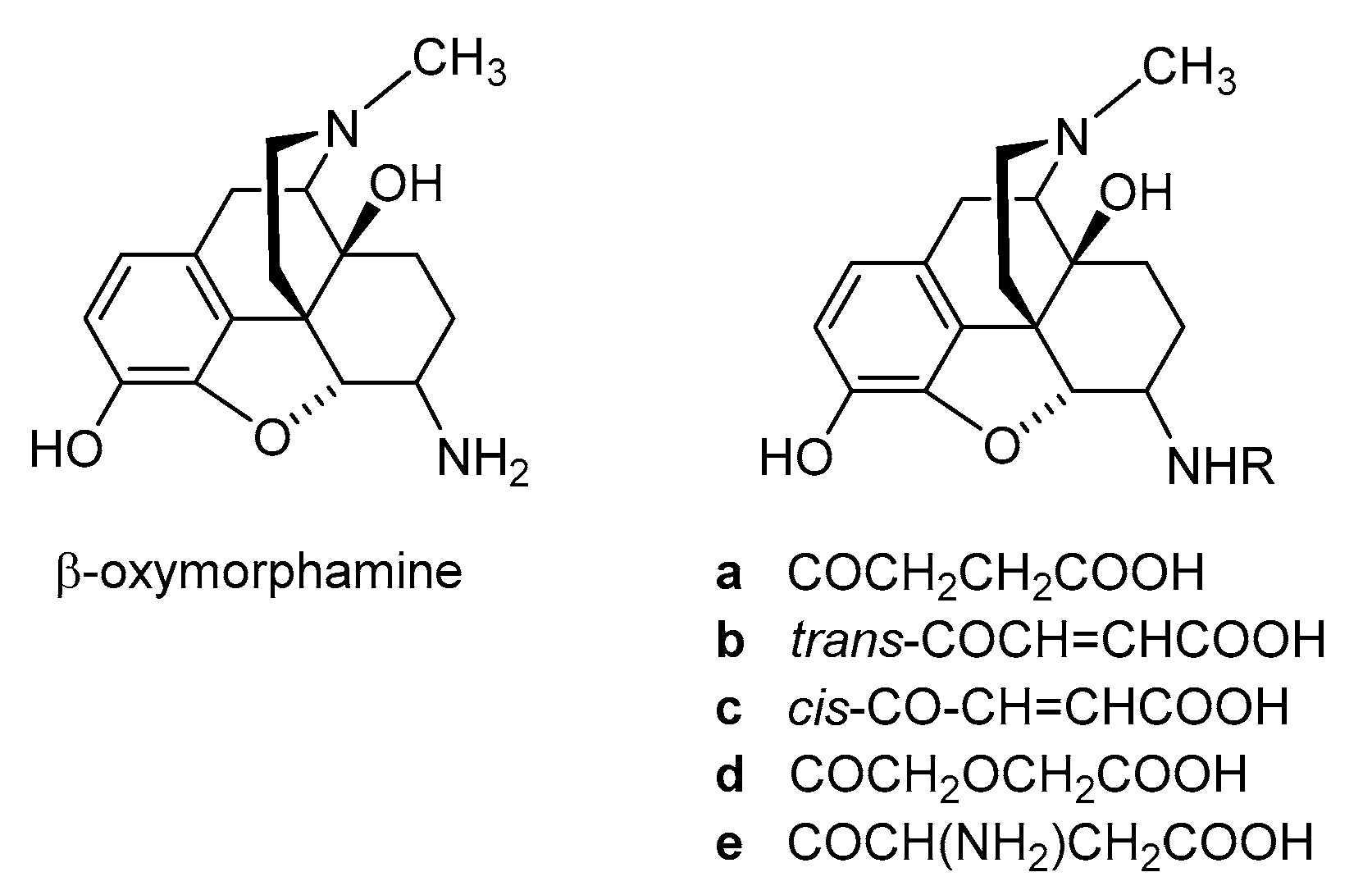
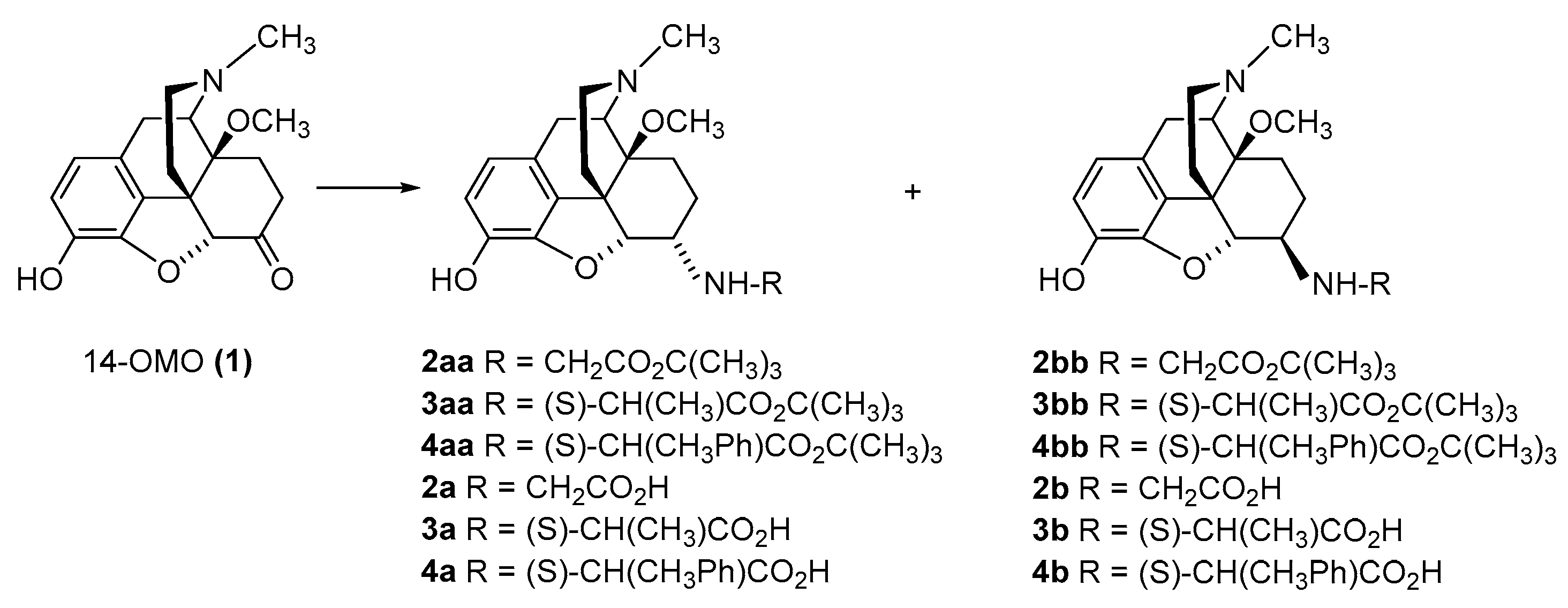
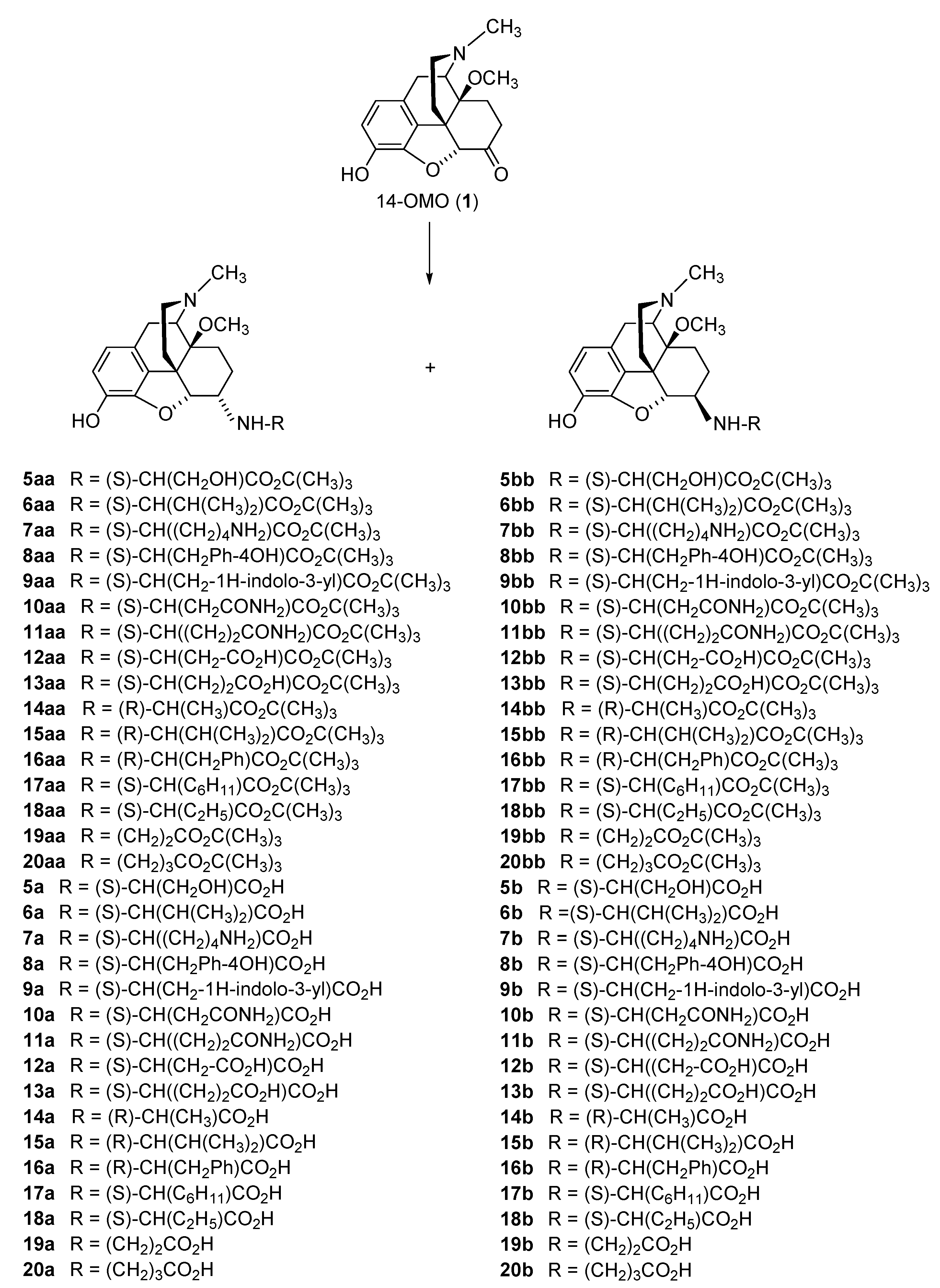
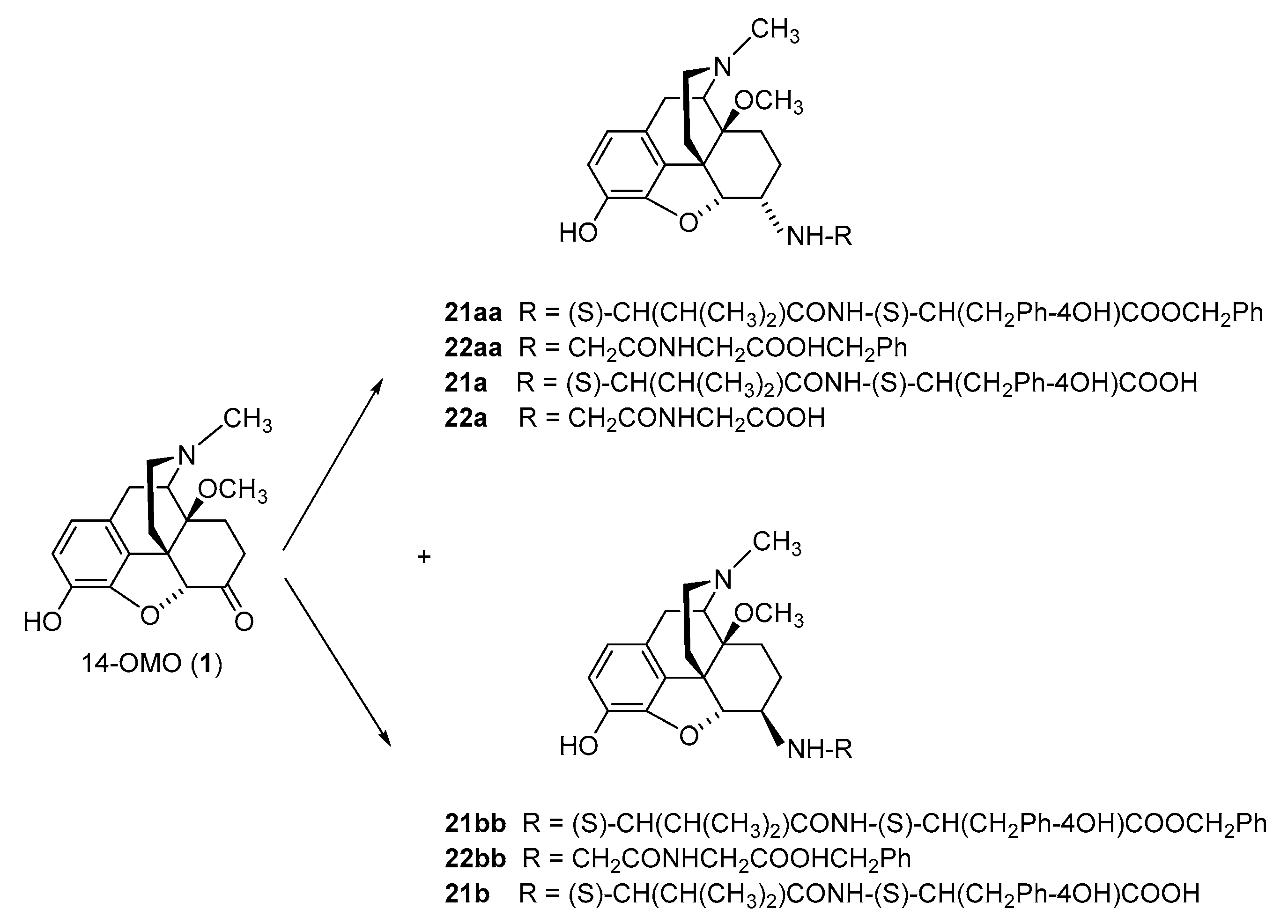
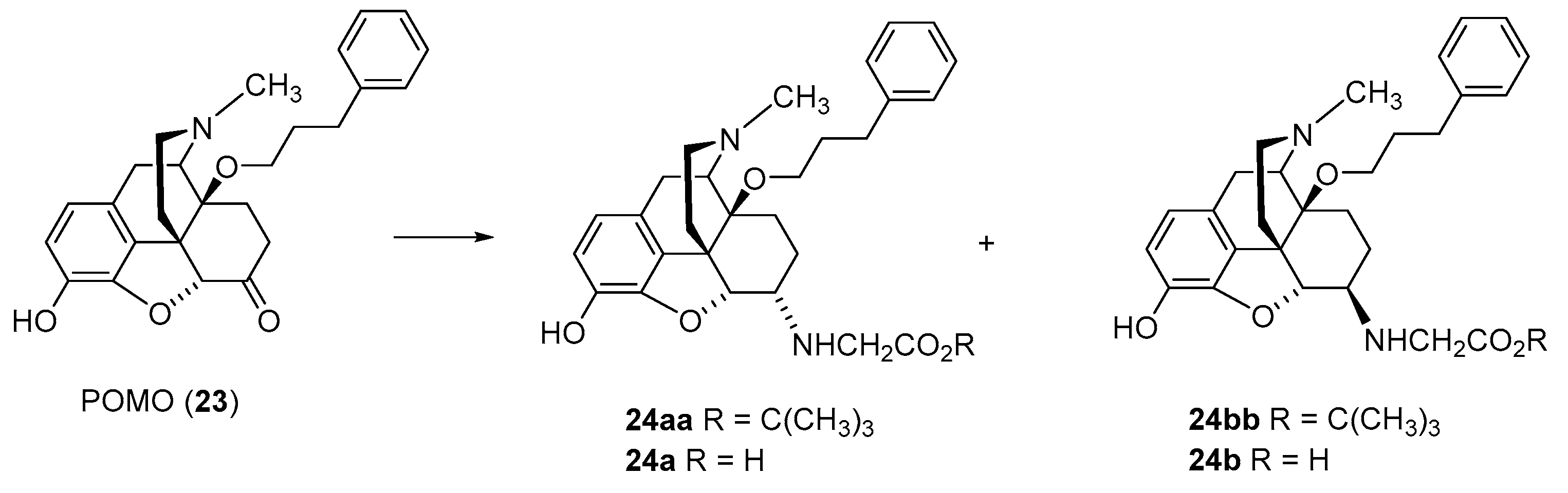
2.2.1. 6-Amino-acid-substituted 14-Alkoxymorphinans
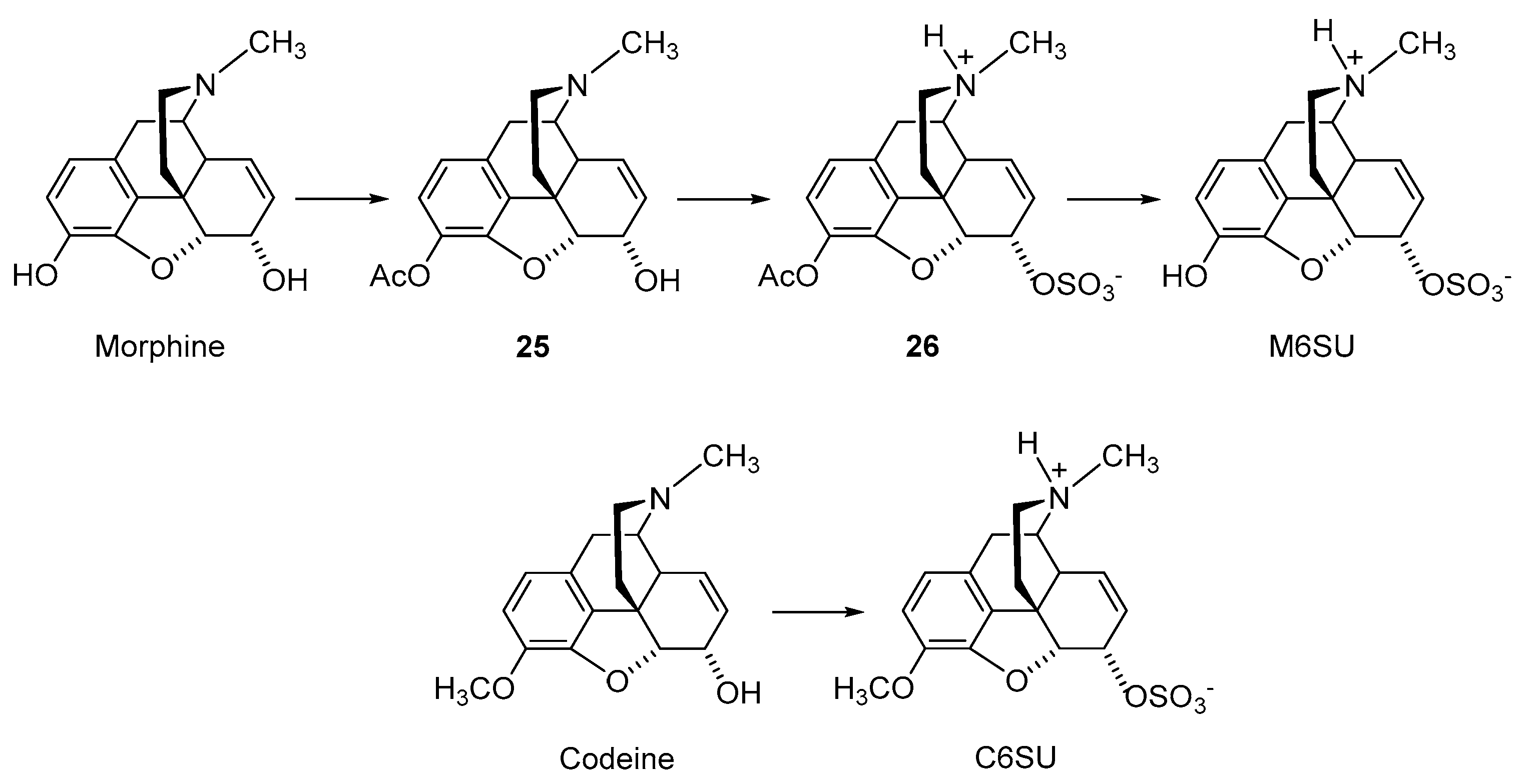
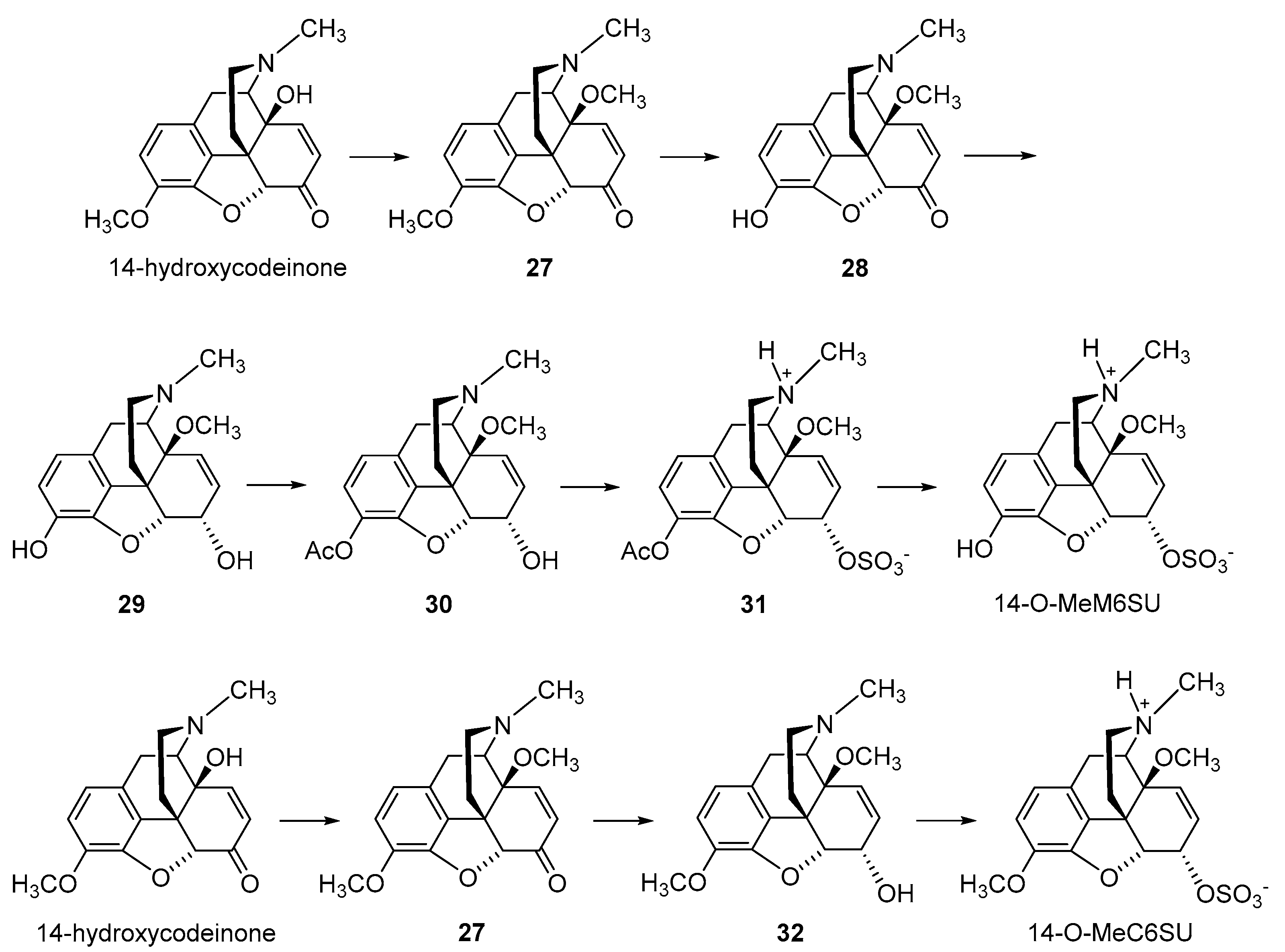
2.2.2. 6-O-Sulfate Esters of Morphine and Codeine
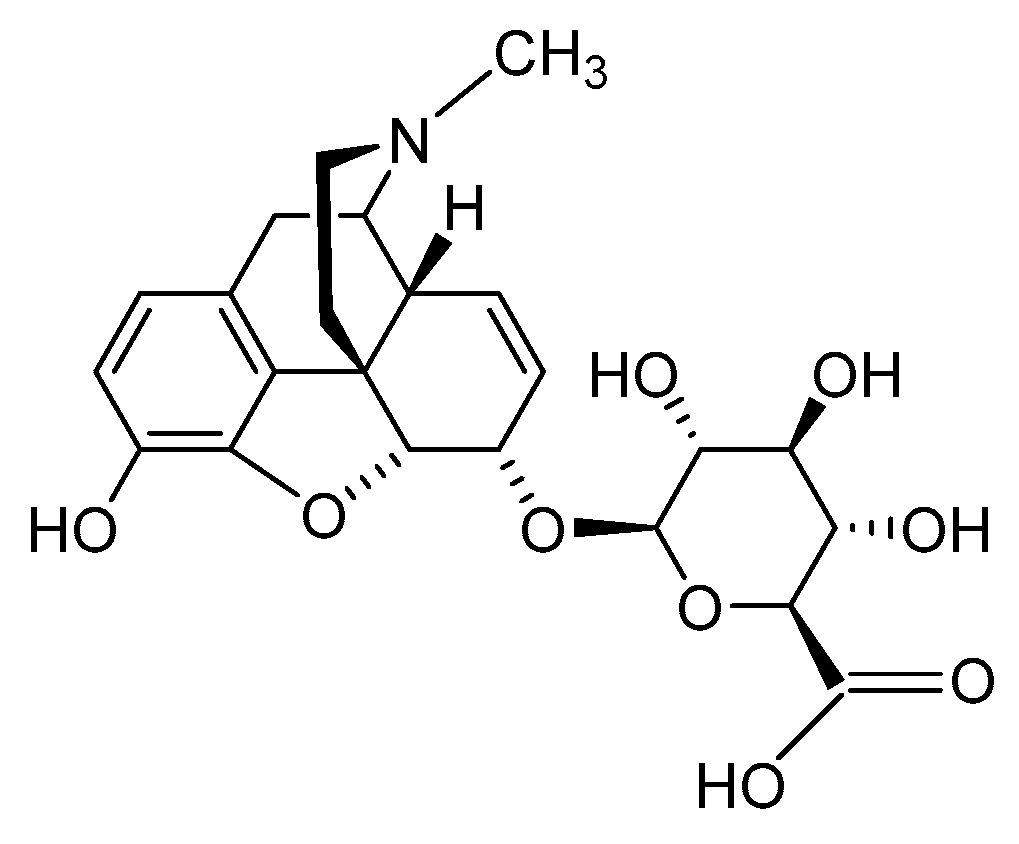
2.2.3. Morphine-6-glucuronide
2.3. Nanocarrier-Based Approaches of Drug Delivery
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Corder, G.; Castro, D.C.; Bruchas, M.R.; Scherrer, G. Endogenous and exogenous opioids in pain. Annu. Rev. Neurosci. 2018, 41, 453–473. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.K.; Smith, C.M.; Rahmatullah, M.; Nissapatorn, V.; Wilairatana, P.; Spetea, M.; Gueven, N.; Dietis, N. Opioid analgesia and opioid-induced adverse effects: A review. Pharmaceuticals 2021, 14, 1091. [Google Scholar] [CrossRef] [PubMed]
- Volkow, N.; Benveniste, H.; McLellan, T.A. Use and misuse of opioids in chronic pain. Annu. Rev. Med. 2018, 69, 451–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobczak, Ł.; Goryński, K. Pharmacological aspects of over-the-counter opioid drugs misuse. Molecules 2020, 25, 3905. [Google Scholar] [CrossRef]
- National Institute on Drug Abuse (NIDA). Available online: https://nida.nih.gov/research-topics/trends-statistics/overdose-death-rates (accessed on 28 April 2023).
- Spetea, M. Opioid receptors and their ligands in the musculoskeletal system and relevance for pain control. Curr. Pharm. Des. 2013, 19, 7382–7390. [Google Scholar] [CrossRef]
- Sein, C. Opioid receptors. Annu. Rev. Med. 2016, 67, 433–451. [Google Scholar]
- Che, T.; Roth, B.L. Structural insights accelerate the discovery of opioid alternatives. Annu. Rev. Biochem. 2021, 90, 739–761. [Google Scholar] [CrossRef]
- Spetea, M.; Asim, M.F.; Wolber, G.; Schmidhammer, H. The opioid receptor and ligands acting at the µ opioid receptor, as therapeutics and potential therapeutics. Curr. Pharm. Des. 2013, 19, 7415–7434. [Google Scholar] [CrossRef]
- Busserolles, J.; Lolignier, S.; Kerckhove, N.; Bertin, C.; Authier, N.; Eschalier, A. Replacement of current opioid drugs focusing on MOR-related strategies. Pharmacol. Ther. 2020, 210, 107519. [Google Scholar] [CrossRef]
- Darcq, E.; Kieffer, B.L. Opioid receptors: Drivers to addiction? Nat. Rev. Neurosci. 2018, 19, 499–514. [Google Scholar] [CrossRef]
- Imam, M.Z.; Kuo, A.; Ghassabian, S.; Smith, M.T. Progress in understanding mechanisms of opioid-induced gastrointestinal adverse effects and respiratory depression. Neuropharmacology 2018, 131, 238–255. [Google Scholar] [CrossRef]
- Fürst, S.; Hosztafi, S. The chemical and pharmacological importance of morphine analogues. Acta Physiol. Hung. 2008, 95, 3–44. [Google Scholar] [CrossRef] [PubMed]
- Pasternak, G.W.; Pan, Y.X. Mu opioids and their receptors: Evolution of a concept. Pharmacol. Rev. 2013, 65, 1257–1317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spetea, M.; Schmidhammer, H. Recent advances in the development of 14-alkoxy substituted morphinans as potent and safer opioid analgesics. Curr. Med. Chem. 2012, 19, 2442–2457. [Google Scholar] [CrossRef]
- Lewis, J.W.; Husbands, S.M. 14-Amino-4,5-epoxymorphinan derivatives and their pharmacological actions. Top. Curr. Chem. 2011, 299, 93–119. [Google Scholar] [PubMed]
- Stavitskaya, L.; Coop, A. Most recent developments and modifications of 14-alkylamino and 14-alkoxy-4,5-epoxymorphinan derivatives. Mini. Rev. Med. Chem. 2011, 11, 1002–1008. [Google Scholar] [CrossRef] [Green Version]
- Schmidhammer, H.; Spetea, M.; Windisch, P.; Schütz, J.; Riba, P.; Al-Khrasani, M.; Fürst, S. Functionalization of the carbonyl group in position 6 of morphinan-6-ones. Development of novel 6-amino and 6-guanidino substituted 14-alkoxymorphinans. Curr. Pharm. Des. 2013, 19, 7391–7399. [Google Scholar] [CrossRef]
- Devereaux, A.L.; Mercer, S.L.; Cunningham, C.W. DARK classics in chemical neuroscience: Morphine. ACS Chem. Neurosci. 2018, 9, 2395–2407. [Google Scholar] [CrossRef]
- Spetea, M.; Schmidhammer, H. Recent chemical and pharmacological developments on 14-oxygenated-N-methylmorphinan-6-ones. Molecules 2021, 26, 5677. [Google Scholar] [CrossRef]
- Kalso, E.; Smith, L.; McQuay, H.J.; Moore, R.A. No pain, no gain: Clinical excellence and scientific rigour—Lessons learned from IA morphine. Pain 2002, 98, 269–275. [Google Scholar] [CrossRef]
- Stein, C.; Schäfer, M.; Machelska, H. Attaching pain at its source: New perspectives on opioids. Nat. Med. 2003, 9, 1003–1008. [Google Scholar] [CrossRef]
- Kapitzke, D.; Vetter, I.; Cabot, P.J. Endogenous opioid analgesia in peripheral tissues and the clinical implications for pain control. Ther. Clin. Risk Manag. 2005, 1, 279–297. [Google Scholar]
- Martinez, V.; Abalo, R. Peripherally acting opioid analgesics and peripherally-induced analgesia. Behav. Pharmacol. 2020, 31, 136–158. [Google Scholar] [CrossRef]
- Stein, C.; Comisel, K.; Haimaeri, E.; Yassouridis, A.; Lehrberger, K. Analgesic effects of intraarticular morphine after arthroscopic knee surgery. N. Engl. J. Med. 1991, 325, 1123–1126. [Google Scholar] [CrossRef]
- Likar, R.; Kapral, S.; Steinkellner, H.; Stein, C.; Schäfer, M. Dose-dependency of intra-articular morphine analgesia. Br. J. Anaesth. 1999, 83, 241–244. [Google Scholar] [CrossRef] [Green Version]
- Iorio, M.A.; Frigeni, V. Narcotic agonist/antagonist properties of quaternary diastereoisomers derived from oxymorphone and naloxone. Eur. J. Med. Chem. 1984, 19, 301–303. [Google Scholar]
- Brown, D.R.; Goldberg, L.I. The use of quaternary narcotic antagonists in opiate research. Neuropharmacology 1985, 24, 181–191. [Google Scholar] [CrossRef]
- Foster, R.S.; Jenden, D.J.; Lomax, P. A comparison of the pharmacologic effects of morphine and N-methyl morphine. J. Pharmacol. Exp. Ther. 1967, 157, 185–195. [Google Scholar] [PubMed]
- Smith, T.W.; Buchan, P.; Parsons, D.N.; Wilkinson, S. Peripheral antinociceptive effects of N-methyl morphine. Life Sci. 1982, 31, 1205–1208. [Google Scholar] [CrossRef] [PubMed]
- Oluyomi, A.O.; Hart, S.L.; Smith, T.W. Differential antinociceptive effects of morphine and methylmorphine in the formalin test. Pain 1992, 49, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Kosterlitz, H.W.; Waterfield, A.A. In vitro models in the study of structure-activity relationships of narcotic analgesics. Annu. Rev. Pharmacol. 1975, 15, 29–47. [Google Scholar] [CrossRef]
- Botros, S.; Lipkowski, A.; Larson, D.; Stark, P.; Takemori, A.; Portoghese, P.S. Opioid agonist and antagonist activities of peripherally selective derivatives of naltrexamine and oxymorphamine. J. Med. Chem. 1989, 32, 2068–2071. [Google Scholar] [CrossRef]
- Larson, D.L.; Hua, M.; Takemori, A.K.; Portoghese, P.S. Possible contribution of a glutathione conjugate to the long-duration action of funaltrexamine. J. Med. Chem. 1993, 36, 3669–3673. [Google Scholar] [CrossRef]
- Schütz, J.; Brandt, W.; Spetea, M.; Wurst, K.; Wunder, G.; Schmidhammer, H. Synthesis of 6-amino acid substituted derivatives of the highly potent analgesic 14-O-methyloxymorphone. Helv. Chim. Acta 2003, 86, 2142–2148. [Google Scholar] [CrossRef]
- Spetea, M.; Rief, S.B.; Haddou, T.B.; Fink, M.; Kristeva, E.; Mittendorfer, H.; Haas, S.; Hummer, N.; Follia, V.; Guerrieri, E.; et al. Synthesis, biological, and structural explorations of new zwitterionic derivatives of 14-O-methyloxymorphone, as potent μ/δ opioid agonists and peripherally selective antinociceptives. J. Med. Chem. 2019, 62, 641–653. [Google Scholar] [CrossRef] [Green Version]
- Spetea, M.; Windisch, P.; Guo, Y.; Bileviciute-Ljungar, I.; Schütz, J.; Asim, M.F.; Berzetei-Gurske, I.P.; Riba, P.; Kiraly, K.; Fürst, S.; et al. Synthesis and pharmacological activities of 6-glycine substituted 14-phenylpropoxymorphinans, a novel class of opioids with high opioid receptor affinities and antinociceptive potencies. J. Med. Chem. 2011, 54, 980–988. [Google Scholar] [CrossRef] [PubMed]
- Spetea, M.; Friedmann, T.; Riba, P.; Schütz, J.; Wunder, G.; Langer, T.; Schmidhammer, H.; Fürst, S. In vitro opioid activity profiles of 6-amino acid substituted derivatives of 14-O-methyloxymorphone. Eur. J. Pharmacol. 2004, 483, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Lattanzi, R.; Rief, S.; Schmidhammer, H.; Negri, L.; Spetea, M. In vitro and in vivo pharmacological activities of 14-O-phenylpropyloxymorphone, a potent mixed mu/delta/kappa-opioid receptor agonist with reduced constipation in mice. Front. Pharmacol. 2018, 9, 1002. [Google Scholar] [CrossRef] [Green Version]
- Fürst, S.; Riba, P.; Friedmann, T.; Tímar, J.; Al-Khrasani, M.; Obara, I.; Makuch, W.; Spetea, M.; Schütz, J.; Przewlocki, R.; et al. Peripheral versus central antinociceptive actions of 6-amino acid-substituted derivatives of 14-O-methyloxymorphone in acute and inflammatory pain in the rat. J. Pharmacol. Exp. Ther. 2005, 312, 609–618. [Google Scholar] [CrossRef] [Green Version]
- Bileviciute-Ljungar, I.; Spetea, M.; Guo, Y.; Schütz, J.; Windisch, P.; Schmidhammer, H. Peripherally mediated antinociception of the mu-opioid receptor agonist 2-(4,5alpha-epoxy-3-hydroxy-14beta-methoxy-17-methylmorphinan-6beta-yl)aminoacetic acid (HS-731) after subcutaneous and oral administration in rats with carrageenan-induced hindpaw inflammation. J. Pharmacol. Exp. Ther. 2006, 317, 220–227. [Google Scholar]
- Al-Khrasani, M.; Spetea, M.; Friedmann, T.; Riba, P.; Király, K.; Schmidhammer, H.; Fürst, S. DAMGO and 6β-glycine substituted 14-O-methyloxymorphone but not morphine show peripheral, preemptive antinociception after systemic administration in a mouse visceral pain model and high intrinsic efficacy in the isolated rat vas deferens. Brain Res. Bull. 2007, 74, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Baillie, L.D.; Schmidhammer, H.; Mulligan, S.J. Peripheral μ-opioid receptor mediated inhibition of calcium signaling and action potential-evoked calcium fluorescent transients in primary afferent CGRP nociceptive terminals. Neuropharmacology 2015, 93, 267–273. [Google Scholar] [CrossRef]
- Obara, I.; Makuch, W.; Spetea, M.; Schütz, J.; Schmidhammer, H.; Przewlocki, R.; Przewlocka, B. Local peripheral antinociceptive effects of 14-O-methyloxymorphone derivatives in inflammatory and neuropathic pain in the rat. Eur. J. Pharmacol. 2007, 558, 60–67. [Google Scholar] [CrossRef]
- Puls, K.; Schmidhammer, H.; Wolber, G.; Spetea, M. Mechanistic characterization of the pharmacological profile of HS-731, a peripherally acting opioid analgesic, at the µ-, δ-, κ-opioid and nociceptin receptors. Molecules 2022, 27, 919. [Google Scholar] [CrossRef] [PubMed]
- Fürst, S.; Zádori, Z.S.; Zádor, F.; Király, K.; Balogh, M.; László, S.B.; Hutka, B.; Mohammadzadeh, A.; Calabrese, C.; Galambos, A.R.; et al. On the role of peripheral sensory and gut mu opioid receptors: Peripheral analgesia and tolerance. Molecules 2020, 25, 2473. [Google Scholar] [CrossRef] [PubMed]
- Herz, A.; Teschemacher, H.J. Activities and sites of antinociceptive action of morphine-like analgesics and kinetics of distribution following intravenous, intracerebral and intraventricular application. In Advances in Drug Research; Harper, N., Simmonds, A.B., Eds.; Academic Press Limited: London, UK, 1971; Volume 6, pp. 79–118. [Google Scholar]
- Frances, B.; Gout, R.; Monsarrat, B.; Cros, J.; Zajac, J.M. Further evidence that morphine-6β-glucuronide is a more potent opioid agonist than morphine. J. Pharmacol. Exp. Ther. 1992, 262, 2531. [Google Scholar]
- Tukey, R.H.; Strassburg, C.P. Human UDP-glucuronosyltransferases: Metabolism, expression, and disease. Annu. Rev. Pharmacol. Toxicol. 2000, 40, 581–616. [Google Scholar] [CrossRef]
- Mori, M.; Oguri, K.; Yoshimura, H.; Shimomura, K.; Kamata, O.; Ueki, S. Chemical synthesis and analgesic effect of morphine ethereal sulfates. Life Sci. 1972, 11, 525–533. [Google Scholar] [CrossRef]
- Váradi, A.; Gergely, A.; Béni, S.; Jankovics, P.; Noszál, B.; Hosztafi, S. Sulfate esters of morphine derivatives: Synthesis and characterization. Eur. J. Pharm. Sci. 2011, 42, 65–72. [Google Scholar] [CrossRef]
- Zuckerman, A.; Bolan, E.; de Paulis, T.; Schmidt, D.; Spector, S.; Pasternak, G.W. Pharmacological characterization of morphine-6-sulfate and codeine-6-sulfate. Brain Res. 1999, 842, 1–5. [Google Scholar] [CrossRef]
- Zádor, F.; Mohammadzadeh, A.; Balogh, M.; Zádori, Z.S.; Király, K.; Barsi, S.; Galambos, A.R.; László, S.B.; Hutka, B.; Váradi, A.; et al. Comparisons of in vivo and in vitro opioid effects of newly synthesized 14-methoxycodeine-6-O-sulfate and codeine-6-O-sulfate. Molecules 2020, 25, 1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Preechagoon, D.; Brereton, I.; Staatz, C.; Prankerd, R. Ester prodrugs of a potent analgesic, morphine-6-sulfate: Syntheses, spectroscopic and physicochemical properties. Int. J. Pharm. 1998, 163, 177–190. [Google Scholar] [CrossRef]
- Crooks, P.A.; Kottayil, S.G.; Al-Ghananeem, A.M.; Byrn, S.R.; Butterfield, A.D. Opiate receptor binding properties of morphine-, dihydromorphine-, and codeine 6-O-sulfate ester congeners. Bioorg. Med. Chem. Lett. 2006, 16, 4291–4295. [Google Scholar] [CrossRef]
- Kaspersen, F.M.; Van Boeckel, C.A.A. A review of the methods of chemical synthesis of sulphate and glucuronide conjugates. Xenobiotica 1987, 17, 1451–1471. [Google Scholar] [CrossRef]
- Al-Horani, R.A.; Desai, U.R. Chemical sulfation of small molecules—Advances and challenges. Tetrahedron 2010, 66, 2907–2918. [Google Scholar] [CrossRef] [Green Version]
- Lacko, E.; Varadi, A.; Rapavi, R.; Zador, F.; Riba, P.; Benyhe, S.; Borsodi, A.; Hosztafi, S.; Timar, J.; Noszal, B.; et al. A novel µ-opioid receptor ligand with high in vitro and in vivo agonist efficacy. Curr. Med. Chem. 2012, 54, 4699–4707. [Google Scholar] [CrossRef]
- Brown, C.E.; Roerig, S.C.; Burger, V.T. Analgesic potencies of morphine 3- and 6-sulfates, after intracerebroventricular administration in mice: Relationship to structural characteristics defined by mass spectrometry and nuclear magnetic resonance. J. Pharm. Sci. 1985, 74, 821–824. [Google Scholar] [CrossRef]
- Holtman, J.R.; Crooks, P.A.; Johnson-Hardy, J.; Wala, E.P. Antinociceptive effects and toxicity of morphine-6-O-sulfate sodium salt in rat models of pain. Eur. J. Pharmacol. 2010, 648, 87–94. [Google Scholar] [CrossRef]
- Al-Khrasani, M.; Lackó, E.; Riba, P.; Király, K.; Sobor, M.; Timár, J.; Mousa, S.; Schäfer, M.; Fürst, S. The central versus peripheral antinociceptive effects of µ-opioid receptor agonists in the new model of rat visceral pain. Brain Res. Bull. 2012, 87, 238–243. [Google Scholar] [CrossRef]
- Lackó, E.; Riba, P.; Giricz, Z.; Váradi, A.; Cornic, L.; Balogh, M.; Király, K.; Cseko, K.; Mousa, S.A.; Hosztafi, S.; et al. New morphine analogs produce peripheral antinociception within a certain dose range of their systemic administration. J. Pharmacol. Exp. Ther. 2016, 359, 171–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadlapalli, J.S.K.; Ford, B.M.; Ketkar, A.; Wan, A.; Penthala, N.R.; Eoff, R.L.; Prather, P.L.; Dobretsov, M.; Crooks, P.A. Antinociceptive effects of the 6-O-sulfate ester of morphine in normal and diabetic rats: Comparative role of mu- and delta-opioid receptors. Pharmacol. Res. 2016, 13, 335–347. [Google Scholar] [CrossRef] [Green Version]
- Yadlapalli, J.S.K.; Jai Shankar, K.; Dogra, N.; Walbaum, A.W.; Wessinger, W.D.; Prather, P.L.; Crooks, P.A.; Dobretsov, M. Evaluation of analgesia, tolerance, and the mechanism of action of morphine-6-O-sulfate across multiple pain modalities in Sprague-Dawley Rats. Anesth. Analg. 2017, 125, 1021–1031. [Google Scholar] [CrossRef] [PubMed]
- Balogh, M.; Zádori, Z.S.; Lázár, B.; Karádi, D.; László, S.; Mousa, S.A.; Hosztafi, S.; Zádor, F.; Riba, P.; Schäfer, M.; et al. The peripheral versus central antinociception of a novel opioid agonist: Acute inflammatory pain in rats. Neurochem. Res. 2018, 43, 1250–1257. [Google Scholar] [CrossRef] [PubMed]
- Balogh, M.; Zádor, F.; Zádori, Z.S.; Shaqura, M.; Király, K.; Mohammadzadeh, A.; Varga, B.; Lázár, B.; Mousa, S.A.; Hosztafi, S.; et al. Efficacy-based perspective to overcome reduced opioid analgesia of advanced painful diabetic neuropathy in rats. Front. Pharmacol. 2019, 10, 347. [Google Scholar] [CrossRef] [Green Version]
- Khalefa, B.I.; Mousa, S.A.; Shaqura, M.; Lackó, E.; Hosztafi, S.; Riba, P.; Schäfer, M.; Ferdinandy, P.; Fürst, S.; Al-Khrasani, M. Peripheral antinociceptive efficacy and potency of a novel opioid compound 14-O-MeM6SU in comparison to known peptide and non-peptide opioid agonists in a rat model of inflammatory pain. Eur.J. Pharmacol. 2013, 713, 54–57. [Google Scholar] [CrossRef] [PubMed]
- Rosow, C.E. Anesthetic drug interaction: An overview. J. Clin. Anesthn. 1997, 9, 27S–32S. [Google Scholar] [CrossRef] [PubMed]
- Yadlapalli, J.S.K.; Albayati, Z.A.F.; Penthala, N.R.; Hendrickson, H.P.; Crooks, H.A. Stability studies of potent opioid analgesic, morphine-6-O-sulfate in various buffers and biological matrices by HPLC-DAD analysis. Biomed. Chromatogr. 2017, 31, e3957. [Google Scholar] [CrossRef]
- Kiraly, K.; Caputi, F.F.; Hanuska, A.; Kató, E.; Balogh, M.; Köles, L.; Palmisano, M.; Riba, P.; Hosztafi, S.; Romualdi, P.; et al. A new potent analgesic agent with reduced liability to produce morphine tolerance. Brain Res. Bull. 2015, 117, 32–38. [Google Scholar] [CrossRef]
- Paul, D.; Standifer, K.M.; Inturrisi, C.E.; Pasternak, G.W. Pharmacological characterization of morphine-6 beta-glucuronide, a very potent morphine metabolite. J. Pharmacol. Exp. Ther. 1989, 251, 477–483. [Google Scholar]
- Osborne, R.; Joel, S.; Trew, D.; Slevin, M. Morphine and metabolite behavior after different routes of morphine administration: Demonstration of the importance of the active metabolite morphine-6-glucuronide. Clin. Pharmacol. Ther. 1990, 47, 12–19. [Google Scholar] [CrossRef]
- Tegeder, I.; Meier, S.; Burian, M.; Schmidt, H.; Geisslinger, G.; Lötsch, J. Peripheral opioid analgesia in experimental human pain models. Brain 2003, 126, 1092–1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanna, M.H.; Elliott, K.M.; Fung, M. Randomized, double-blind study of the analgesic efficacy of morphine-6-glucuronide versus morphine sulfate for postoperative pain in major surgery. Anesthesiology 2005, 102, 815–821. [Google Scholar] [CrossRef]
- Binning, A.R.; Przesmycki, K.; Sowinski, P.; Morrison, L.M.M.; Smith, T.W.; Marcus, P.; Lees, J.P.; Dahan, A. A randomised controlled trial on the efficacy and side-effect profile (nausea/vomiting/sedation) of morphine-6-glucuronide versus morphine for post-operative pain relief after major abdominal surgery. Eur. J. Pain 2011, 15, 402–408. [Google Scholar] [CrossRef]
- Sverrisdóttir, E.; Lund, T.M.; Olesen, A.E.; Drewes, A.M.; Christrup, L.L.; Kreilgaard, M. A review of morphine and morphine-6-glucuronide’s pharmacokinetic-pharmacodynamic relationships in experimental and clinical pain. Eur. J. Pharm. Sci. 2015, 74, 45–62. [Google Scholar] [CrossRef]
- Kilpatrick, G.J.; Smith, T.W. Morphine-6-glucuronide: Actions and mechanisms. Med. Res. Rev. 2005, 25, 521–544. [Google Scholar] [CrossRef]
- Lötsch, J.; Geisslinger, G. Morphine-6-glucuronide: An analgesic of the future? Clin. Pharmacokinet. 2001, 40, 485–499. [Google Scholar] [CrossRef]
- Bickel, U.; Schumacher, O.P.; Kang, Y.S.; Voigt, K. Poor permeability of morphine 3-glucuronide and morphine 6-glucuronide through the blood barrier in the rat. J. Pharmacol. Exp. Ther. 1996, 278, 107–113. [Google Scholar] [PubMed]
- Huwyler, J.; Drewe, J.; Klusemann, C.; Fricker, G. Evidence for P-glycoprotein-modulated penetration of morphine-6-glucuronide into brain capillary endothelium. Br. J. Pharmacol. 1996, 118, 1879–1885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sattari, M.; Routledge, P.; Mashayekhi, S. The influence of active transport systems on morphine -6-glucuronide transport in MDCKII and MDCK-PGP cells. Daru 2011, 19, 412–416. [Google Scholar] [PubMed]
- Bourasset, F.; Cisternino, S.; Temsamani, J.; Scherrmann, J.-M. Evidence for an active transport of morphine-6-β-d-glucuronide but not P-glycoprotein-mediated at the blood–brain barrier. J. Neurochem. 2003, 86, 1564–1567. [Google Scholar] [CrossRef] [PubMed]
- Cashman, J.R.; MacDougall, J.M. Dynamic medicinal chemistry in the elaboration of morphine-6-glucuronide analogs. Curr. Top. Med. Chem. 2005, 5, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Klimas, R.; Mikus, G. Morphine-6-glucuronide is responsible for the analgesic effect after morphine administration: A quantitative review of morphine, morphine-6-glucuronide, and morphine-3-glucuronide. Br. J. Anaesth. 2014, 113, 935–944. [Google Scholar] [CrossRef] [Green Version]
- Koning, G.A.; Schiffelers, R.M.; Storm, G. Endothelial cells at inflammatory sites as target for therapeutic intervention. Endothelium 2002, 9, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Fleige, E.; Quadir, M.A.; Haag, R. Stimuli-responsive polymeric nanocarriers for the controlled transport of active compounds: Concepts and applications. Adv. Drug Deliv. Rev. 2012, 64, 866–884. [Google Scholar] [CrossRef] [PubMed]
- Kraft, J.C.; Freeling, J.P.; Wang, Z.; Ho, R.J. Emerging research and clinical development trends of liposome and lipid nanoparticle drug delivery systems. J. Pharm. Sci. 2014, 103, 29–52. [Google Scholar] [CrossRef] [Green Version]
- Nehoff, H.; Parayath, N.N.; Domanovitch, L.; Taurin, S.; Greish, K. Nanomedicine for drug targeting: Strategies beyond the enhanced permeability and retention effect. Int. J. Nanomed. 2014, 22, 2539–2555. [Google Scholar]
- Gurunathan, S.; Kang, M.H.; Qasim, M.; Kim, J.H. Nanoparticle-mediated combination therapy: Two-in-one approach for cancer. Int. J. Mol. Sci. 2018, 19, 3264. [Google Scholar] [CrossRef] [Green Version]
- Moradkhani, M.R.; Karimi, A.; Negahdari, B. Nanotechnology application for pain therapy. Artif. Cells Nanomed. Biotechnol. 2018, 46, 368–373. [Google Scholar] [CrossRef] [Green Version]
- Chakravarthy, K.V.; Boehm, F.J.; Christo, P.J. Nanotechnology: A promising new paradigm for the control of pain. Pain Med. 2018, 19, 232–243. [Google Scholar] [CrossRef] [Green Version]
- González-Rodríguez, S.; Quadir, M.A.; Gupta, S.; Walker, K.A.; Zhang, X.; Spahn, V. Polyglycerol-opioid conjugate produces analgesia devoid of side effects. Elife 2017, 6, e27081. [Google Scholar] [CrossRef]
- Martin, C.; Oyen, E.; Mangelschots, J.; Bibian, M.; Ben Haddou, T.; Andrade, J.; Gardiner, J.; Van Mele, B.; Madder, A.; Hoogenboom, R.; et al. Injectable peptide hydrogels for controlled-release of opioids. MedChemComm 2016, 7, 542–549. [Google Scholar] [CrossRef]
- Martin, C.; Oyen, E.; Van Wanseele, Y.; Haddou, T.B.; Schmidhammer, H.; Andrade, J.; Waddington, L.; Van Eeckhaut, A.; Van Mele, B.; Gardiner, J.; et al. Injectable peptide-based hydrogel formulations for the extended in vivo release of opioids. Mater. Today Chem. 2017, 3, 49–59. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Agonist Potencies IC50 × 108 (M) a | Antinociceptive Potencies ED50 b |
|---|---|---|
| a | 14.4 | 0.27 nmol/mouse, i.c.v. 9.8 µmol/kg, i.v. 30.3 µmol/kg, p.o. |
| b | 13.0 | - c |
| c | 3.9 | 0.17 nmol/mouse, i.c.v. 11 µmol/kg, i.v. ~20 µmol/kg, p.o. |
| d | 2.7 | 0.25 nmol/mouse, i.c.v. <20 µmol/kg, i.v. >25 µmol/kg, p.o. |
| e | 3.1 | 0.44 nmol/mouse, i.c.v. <0 µmol/kg, i.v. |

| Compound | R | Opioid Receptor Binding a | Agonist Activity b | clogD7.4 c | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MOR Ki (nM) | DOR Ki (nM) | KOR Ki (nM) | Ki Ratio MOR/DOR/KOR | MOR EC50 (nM) | MOR % stim. | DOR EC50 (nM) | DOR % stim. | KOR EC50 (nM) | KOR % stim. | |||
| 14-OMO (1) | 0.10 | 4.80 | 10.2 | 1/48/102 | 3.83 | 97 | 37.3 | 106 | 116 | 77 | 0.48 | |
| HS-730 (2a) | α-Gly | 0.89 | 15.4 | 43.2 | 1/7/49 | 1.16 | 99 | 9.61 | 103 | 399 | 87 | −3.35 |
| HS-731 (2b) | β-Gly | 0.83 | 7.86 | 44.8 | 1/9.5/54 | 3.78 | 98 | 7.92 | 103 | 361 | 82 | −3.35 |
| HS-935 (3a) | α-L-Ala | 0.77 | 26.9 | 142 | 1/35/184 | 1.34 | 97 | 9.55 | 93 | 214 | 51 | −2.81 |
| HS-936 (3b) | β-L-Ala | 1.90 | 7.71 | 63.7 | 1/4.1/34 | 6.24 | 87 | 5.20 | 104 | 392 | 64 | −2.81 |
| HS-937 (4a) | α-L-Phe | 0.95 | 3.67 | 28.5 | 1/3.9/30 | 0.38 | 93 | 0.39 | 102 | 219 | 39 | −1.13 |
| HS-938 (4b) | β-L-Phe | 2.58 | 1.03 | 151 | 1/0.4/59 | 6.76 | 99 | 0.48 | 94 | 1172 | 81 | −1.13 |
| 5a | α-L-Ser | 2.21 | 5.32 | 196 | 1/2.4/89 | 1.60 | 87 | 13.9 | 101 | 1213 | 44 | −3.89 |
| 5b | β-L-Ser | 2.14 | 5.29 | 152 | 1/2.5/71 | 3.56 | 101 | 6.98 | 98 | 201 | 88 | −3.89 |
| 6a | α-L-Val | 3.16 | 3.91 | 325 | 1/1.2/103 | 10.5 | 95 | 33.8 | 91 | 462 | 51 | −1.94 |
| 6b | β-L-Val | 3.04 | 3.52 | 305 | 1/1.2/100 | 11.7 | 84 | 5.73 | 96 | 1117 | 68 | −1.94 |
| 7a | α-L-Lys | 0.19 | 1.27 | 12.6 | 1/6.7/66 | 2.25 | 9 | 152 | 106 | 118 | 79 | −5.57 |
| 7b | β-L-Lys | 0.53 | 3.34 | 33.7 | 1/6.3/64 | 6.85 | 90 | 45.1 | 93 | 525 | 62 | −5.57 |
| 8a | α-L-Tyr | 0.83 | 2.18 | 39.5 | 1/2.6/48 | 1.87 | 92 | 1.76 | 88 | 100 | 62 | −1.41 |
| 8b | β-L-Tyr | 3.20 | 3.89 | 186 | 1/1.2/58 | 24.7 | 93 | 6.23 | 95 | 774 | 60 | −1.41 |
| 9a | α-L-Trp | 0.36 | 1.02 | 25.1 | 1/2.8/70 | 0.51 | 93 | 2.52 | 102 | 70.1 | 61 | −1.03 |
| 9b | β-L-Trp | 0.65 | 1.19 | 8.66 | 1/1.8/13 | 1.64 | 101 | 2.18 | 96 | 181 | 87 | −1.03 |
| 10a | α-L-Asn | 1.17 | 3.37 | 74.0 | 1/2.9/63 | 0.83 | 99 | 9.78 | 106 | 81.7 | 67 | −4.29 |
| 10b | β-L-Asn | 1.26 | 2.25 | 103 | 1/1.8/82 | 2.04 | 96 | 3.18 | 88 | 923 | 71 | −4.29 |
| 11a | α-L-Gln | 3.24 | 5.13 | 351 | 1/1.6/108 | 2.27 | 90 | 7.80 | 104 | 185 | 70 | −4.04 |
| 11b | β-L-Gln | 2.48 | 4.87 | 290 | 1/2.0/117 | 9.54 | 98 | 3.96 | 103 | 1410 | 63 | −4.04 |
| 12a | α-L-Asp | 1.36 | 14.6 | 50.2 | 1/11/37 | 4.10 | 90 | 10.1 | 97 | 2991 | 83 | −5.64 |
| 12b | β-L-Asp | 3.42 | 22.6 | 351 | 1/6.6/103 | 1.45 | 74 | 11.8 | 101 | 753 | 49 | −5.64 |
| 13a | α-L-Glu | 1.45 | 9.03 | 87.2 | 1/6.2/60 | 3.11 | 105 | 10.8 | 98 | 1167 | 68 | −5.39 |
| 13b | β-L-Glu | 11.6 | 7.64 | 1252 | 1/0.7/108 | 12.7 | 98 | 4.60 | 101 | 2233 | 76 | −5.39 |
| 14a | α-D-Ala | 0.69 | 10.4 | 71.5 | 1/15/104 | 1.44 | 100 | 24.3 | 106 | 254 | 67 | −2.81 |
| 14b | β-D-Ala | 1.48 | 11.3 | 142 | 1/7.6/96 | 15.4 | 102 | 5.46 | 106 | 1001 | 86 | −2.81 |
| 15a | α-D-Val | 1.70 | 1.93 | 202 | 1/1.1/119 | 4.51 | 105 | 1.12 | 93 | 2218 | 96 | −1.94 |
| 15b | β-D-Val | 1.02 | 1.68 | 159 | 1/1.6/156 | 2.38 | 101 | 1.30 | 99 | 1278 | 98 | −1.94 |
| 16a | α-D-Phe | 0.61 | 3.69 | 76.4 | 1/6.0/125 | 0.77 | 96 | 8.36 | 95 | 215 | 80 | −1.13 |
| 16b | β-D-Phe | 1.28 | 1.19 | 139 | 1/0.9/109 | 0.68 | 78 | 1.71 | 96 | 611 | 92 | −1.13 |
| 17a | α-L-Chg | 1.23 | 14.3 | 177 | 1/12/144 | 2.88 | 86 | 20.5 | 101 | 250 | 52 | −1.26 |
| 17b | β-L-Chg | 1.66 | 1.30 | 118 | 1/0.8/71 | 5.33 | 86 | 3.57 | 96 | 282 | 59 | −1.26 |
| 18a | α-L-Abu | 0.76 | 37.5 | 144 | 1/49/189 | 5.12 | 88 | 98.2 | 104 | 942 | 61 | −2.34 |
| 18b | β-L-Abu | 1.83 | 1.30 | 201 | 1/0.7/110 | 5.47 | 83 | 2.22 | 100 | 572 | 72 | −2.34 |
| 19a | α-β-Ala | 1.30 | 60.0 | 182 | 1/46/140 | 3.52 | 99 | 96.4 | 97 | 186 | 78 | −3.18 |
| 19b | β-β-Ala | 1.04 | 13.9 | 71.4 | 1/13/69 | 5.74 | 78 | 20.1 | 99 | 622 | 67 | −3.18 |
| 20a | α-GABA | 0.77 | 12.5 | 45.6 | 1/16/59 | 2.88 | 103 | 34.4 | 103 | 2034 | 104 | −2.93 |
| 20b | β-GABA | 1.41 | 6.61 | 147 | 1/4.7/104 | 12.3 | 86 | 6.06 | 103 | 3396 | 74 | −2.93 |
| 21a | α-L-Val-L-Tyr | 0.82 | 1.19 | 69.0 | 1/1.5/84 | 0.89 | 84 | 1.16 | 88 | 330 | 50 | −1.02 |
| 21b | β-L-Val-L-Tyr | 0.44 | 1.38 | 390 | 1/3.1/886 | 0.16 | 73 | 1.56 | 89 | 1884 | 63 | −1.02 |
| 22a | β-Gly-Gly | 4.62 | 7.52 | 203 | 1/1.6/44 | 4.39 | 85 | 2.84 | 102 | 885 | 75 | −4.27 |

| Compound | R | Opioid Receptor Binding, Ki (nM) a | clogD7.4 b | |||
|---|---|---|---|---|---|---|
| MOR | DOR | KOR | Ki Ratio MOR/DOR/KOR | |||
| POMO (23) | 0.073 | 0.13 | 0.30 | 1/1.8/4.1 | 2.89 | |
| 24a | α-Gly | 0.19 | 0.22 | 0.73 | 1/1.2/3.8 | −0.85 |
| 24b | β-Gly | 0.16 | 0.19 | 0.81 | 1/1.2/5.1 | −0.85 |
| Compound | Amino Acid Substitution at Position 6 | Radiant Heat Tail-Flick Test (Rat) ED50 (nmol/kg, s.c.) | Writhing Assay (Mouse) ED50 (µg/kg, s.c.) | Formalin Test (Rat) ED50 (nmol/kg, s.c.) | |
|---|---|---|---|---|---|
| Phase I | Phase II | ||||
| Morphine | 6053 | 437 | 1613 | 1472 | |
| Fentanyl | 38.6 | ||||
| 14-OMO (1) | 14.9 | 3.26 | |||
| HS-730 (2a) | α-Gly | 58.5 | 35.7 | 72 | 110 |
| HS-731 (2b) | β-Gly | 29.0 | 27.5 | 125 | 204 |
| HS-935 (3a) | α-L-Ala | 68.9 | 16.0 | ||
| HS-936 (3b) | β-L-Ala | 53.4 | 86.3 | ||
| HS-937 (4a) | α-L-Phe | 315 | 31.1 | 171 | 292 |
| HS-938 (4b) | β-L-Phe | >3600 | 579 | 79 | 107 |
| 5a | α-L-Ser | 32.1 | |||
| 6b | β-L-Val | 117 | |||
| 7a | α-L-Lys | 20.6 | |||
| 8a | α-L-Tyr | 14.6 | |||
| 9b | β-L-Trp | 92.7 | |||
| 10a | α-L-Asn | 15.2 | |||
| 12a | α-L-Asp | 38.2 | |||
| 13a | α-L-Glu | 36.1 | |||
| 15b | β-D-Val | 14.0 | |||
| 16a | α-D-Phe | 18.1 | |||
| 16b | β-D-Phe | 250 | |||
| 17a | α-L-Chg | 20.6 | |||
| 18a | α-L-Abu | 17.5 | |||
| 19a | α-β-Ala | 31.2 | |||
| 20a | α-GABA | 41.9 | |||
| 21a | β-L-Val-L-Tyr | 178 | |||
| 22a | β-Gly-Gly | 104 | |||
| 24a | α-Gly | 81.1 | |||
| 24b | β-Gly | 130 | |||
| Pain Model | Route | ED50 | Reference |
|---|---|---|---|
| Acute nociception Radiant heat tail-flick test (rat) | i.c.v. s.c. | 0.030 nmol/rat 29.0 nmol/kg | [40] [40] |
| Trigeminal nociception Eye wiping test (mouse) | i.p. | 50 µg/kg a | [43] |
| Visceral pain | |||
| Acetic-acid-induced writhing assay (mouse) | i.c.v. s.c. | 0.49 pmol/mouse 51 nmol/kg | [42] [42] |
| s.c. | 27.5 µg/kg | [36] | |
| Inflammatory pain Formalin test (rat) Carrageenan-induced thermal and mechanical hyperalgesia (rat) | i.pl. s.c. s.c. p.o. | Phase I: 0.2 nmol; Phase II: 0.4 nmol Phase I: 125 nmol/kg; Phase II: 204 nmol/kg 20 µg/kg a 10 mg/kg a | [44] [40] [41] [41] |
| Neuropathic pain, sciatic nerve ligation—Mechanical hyperalgesia (rat) | i.pl. | 441 nmol | [44] |

| Compound | R, Amino Acid Substitution at Position 6 | Radiant Heat Tail-Flick Test (Rat), ED50 | Ratio ED50 (nmol/kg, s.c.)/ ED50 (nmol/rat, i.c.v.). | |
|---|---|---|---|---|
| s.c. (nmol/kg) | i.c.v. (nmol/rat) | |||
| Morphine | 6053 | 35.1 | 172 | |
| Fentanyl | 38.6 | 1.66 | 23 | |
| 14-OMO (1) | 14.9 | 0.172 | 87 | |
| HS-730 (2a) | α-Gly | 58.5 | 0.031 | 1887 |
| HS-731 (2b) | β-Gly | 29.0 | 0.030 | 967 |
| HS-935 (3a) | α-L-Ala | 68.9 | 0.121 | 569 |
| HS-936 (3b) | β-L-Ala | 53.4 | 0.082 | 651 |
| HS-937 (4a) | α-L-Phe | 315 | 0.063 | 5000 |
| HS-938 (4b) | β-L-Phe | >3600 | 0.776 | >4600 |
| Compound | Opioid Receptor Binding, Ki (nM) a | |||
|---|---|---|---|---|
| MOR | DOR | KOR | Ki Ratio MOR/DOR/KOR | |
| Morphine | 4.37 | 2951 | 113 | 1/675/26 |
| Codeine | 737 | - b | - | - |
| M6SU | 11.5 | 525 | 275 | 1/46/24 |
| C6SU | 96.9 | 968 b | - | 1/10/- |
| 14-O-MeM6SU | 1.12 | 10.2 | 295 | 1/9/263 |
| 14-O-MeC6SU | 3.37 | 346 | 246 | 1/103/73 |
| Compound | MVD Bioassay | [35S]GTPγS Binding Assay | ||
|---|---|---|---|---|
| EC50 (nM) | Tissue | EC50 (nM) | Emax (%) | |
| Morphine | 347 | Rat brain Guinea-pig brain | 250 462 | 129 119 |
| Codeine | >1000 | Rat brain Guinea-pig brain | - a - | 110 104 |
| M6SU | 103 | Rat brain Guinea-pig brain | 105 - | 133 - |
| C6SU | >1000 | Rat brain Guinea-pig brain | >10,000 - | 121 102 |
| 14-O-MeM6SU | 4.38 | Rat brain Guinea-pig brain | 19.1 - | 201 - |
| 14-O-MeC6SU | 238 | Rat brain Guinea-pig brain | 301 >1000 | 128 130 |
| Compound | Pain Model (Species) | ED50 (Systemic Administration) | ED50 (Central Administration) | Reference |
|---|---|---|---|---|
| Morphine | hot-plate test (mouse) | 4.5 mg/kg, s.c. | [50] | |
| hot water tail-flick test (rat) | 3.41 mg/kg, i.p. | [64] | ||
| radiant heat tail-flick test (mouse) | 5.5 nmol/mouse, i.c.v. | [59] | ||
| radiant heat tail-flick test (mouse) | 314 ng/mouse, i.c.v. | [52] | ||
| radiant heat tail-flick test (rat) | 6221 nmol/kg, s.c. | 38.6 nmol/rat, i.c.v | [58] | |
| radiant heat tail-flick test (rat) | 2.68 mg/kg, i.p. | 3.51 µg/rat, i.t. | [60] | |
| radiant heat tail-flick test (rat) | 9.1 mg/kg, p.o. | [60] | ||
| acetic-acid-induced writhing assay (mouse) | 238.6 nmol/kg, i.p. | 2.02 nmol/mouse, i.c.v. | [61] | |
| formalin-induced inflammatory pain (rat) | Phase II: 0.259 mg/kg, i.p. | [61] | ||
| formalin-induced inflammatory pain (rat) | Phase I and II: 3884, 7769, 15,538, 31,075 nmol/kg, s.c. a | [65] | ||
| neuropathic pain, CCI (rat) hyperalgesia allodynia | 2.65 mg/kg, i.p. 1.45 mg/kg, i.p. | [60] | ||
| STZ-induced diabetic neuropathic pain— tail withdrawal test (rat) | 6.47 mg/kg, i.p. | [63] | ||
| STZ-induced diabetic neuropathic pain— paw withdrawal test (rat) | 40,000 nmol/kg, s.c. a | [66] | ||
| Codeine | radiant heat tail-flick test (rat) | 54.01 µmol/kg | [53] | |
| M6SU | hot-plate test (mouse) | 1.7 mg/kg, s.c. | [50] | |
| hot water tail-flick (rat) | 0.82 mg/kg, i.p. | [63] | ||
| radiant heat tail-flick test (mouse) | 0.19 nmol/mouse, i.c.v. | [59] | ||
| radiant heat tail-flick test (mouse) | 10.6 ng/mouse, i.c.v. | [52] | ||
| radiant heat tail-flick test (rat) | 9305 nmol/kg, s.c. | 0.356 nmol/rat, i.c.v. | [58] | |
| radiant heat tail-flick test (rat) | 0.54 mg/kg i.p. | 0.29 μg/rat, i.t. | [60] | |
| radiant heat tail-flick test (rat) | 4.97 mg/kg, p.o. | [60] | ||
| paw pressure threshold test (rat) | 2.3 mg/kg, i.p. | [64] | ||
| acetic-acid-induced writhing assay (mouse) | 1993 nmol/kg, s.c. | 9 pmol/mouse, i.c.v. | [62] | |
| formalin-induced inflammatory pain (rat) | Phase II: 0.094 mg/kg, i.p. | [60] | ||
| CFA-induced inflammatory pain—paw pressure test (rat) | 292 nmol/kg, s.c. | [62] | ||
| neuropathic pain, CCI (rat) hyperalgesia allodynia | 0.40 mg/kg, i.p. 0.19 mg/kg, i.p. | [60] | ||
| STZ-induced diabetic neuropathic pain—tail withdrawal test (rat) | 0.35 mg/kg, i.p. | [63] | ||
| C6SU | radiant heat tail-flick test (mouse) | 200 ng, i.c.v b | [52] | |
| radiant heat tail-flick test (rat) | weak effect (<20%), s.c. | [53] | ||
| CFA-induced inflammatory pain—paw pressure test (rat) | 6.6 and 13.2 µmol/kg, s.c. a | [53] | ||
| 14-O-MeM6SU | radiant heat tail-flick test (rat) | 182.4 nmol/kg, s.c. | 0.0157 nmol/rat, i.c.v. | [58] |
| acetic-acid-induced writhing assay (mouse) | 87 nmol/kg, s.c. | 1.7 nmol/mouse, i.c.v. | [62] | |
| formalin-induced inflammatory pain (rat) | Phase I: 3506 and 1012 nmol/kg, s.c. a Phase II: 253, 506 and 1012 nmol/kg, s.c. a | [65] | ||
| CFA-induced inflammatory pain—paw pressure test (rat) | 45 nmol/kg, s.c. | [62] | ||
| STZ-induced diabetic neuropathic pain—paw withdrawal test (rat) | 253, 506 and 1012 nmol/kg, s.c. a | [66] | ||
| 14-O-MeC6SU | radiant heat tail-flick test (rat) | 5.34 µmol/kg, s.c. | 0.017 µmol/animal, i.c.v. | [53] |
| CFA-induced inflammatory pain—paw pressure test (rat) | 6.1 and 12.2 µmol/kg, s.c. a | [53] |
| Compound | Pain Model (Species) | ED50 Ratio Peripheral/Central Administration | Reference |
|---|---|---|---|
| Fentanyl | radiant heat tail-flick test (rat) | 23 | [50] |
| Morphine | radiant heat tail-flick test (rat) | 172 | [40] |
| 159 | [58] | ||
| 125 | [53] | ||
| acetic-acid-induced writhing assay (rat) | 118 | [61] | |
| M6SU | radiant heat tail-flick test (rat) | 25,493 | [58] |
| acetic-acid-induced writhing assay (mouse) | 221,444 | [62] | |
| 14-O-MeM6SU | radiant heat tail-flick test (rat) | 11,615 | [58] |
| acetic-acid-induced writhing assay (mouse) | 51,177 | [62] | |
| 14-O-MeC6SU | radiant heat tail-flick test (rat) | 314 | [53] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schmidhammer, H.; Al-Khrasani, M.; Fürst, S.; Spetea, M. Peripheralization Strategies Applied to Morphinans and Implications for Improved Treatment of Pain. Molecules 2023, 28, 4761. https://doi.org/10.3390/molecules28124761
Schmidhammer H, Al-Khrasani M, Fürst S, Spetea M. Peripheralization Strategies Applied to Morphinans and Implications for Improved Treatment of Pain. Molecules. 2023; 28(12):4761. https://doi.org/10.3390/molecules28124761
Chicago/Turabian StyleSchmidhammer, Helmut, Mahmoud Al-Khrasani, Susanna Fürst, and Mariana Spetea. 2023. "Peripheralization Strategies Applied to Morphinans and Implications for Improved Treatment of Pain" Molecules 28, no. 12: 4761. https://doi.org/10.3390/molecules28124761