
1,3-Butadiynamides the Ethynylogous Ynamides: Synthesis, Properties and Applications in Heterocyclic Chemistry

Abstract
:1. Introduction
2. Synthesis of 1,3-Butadiynamides
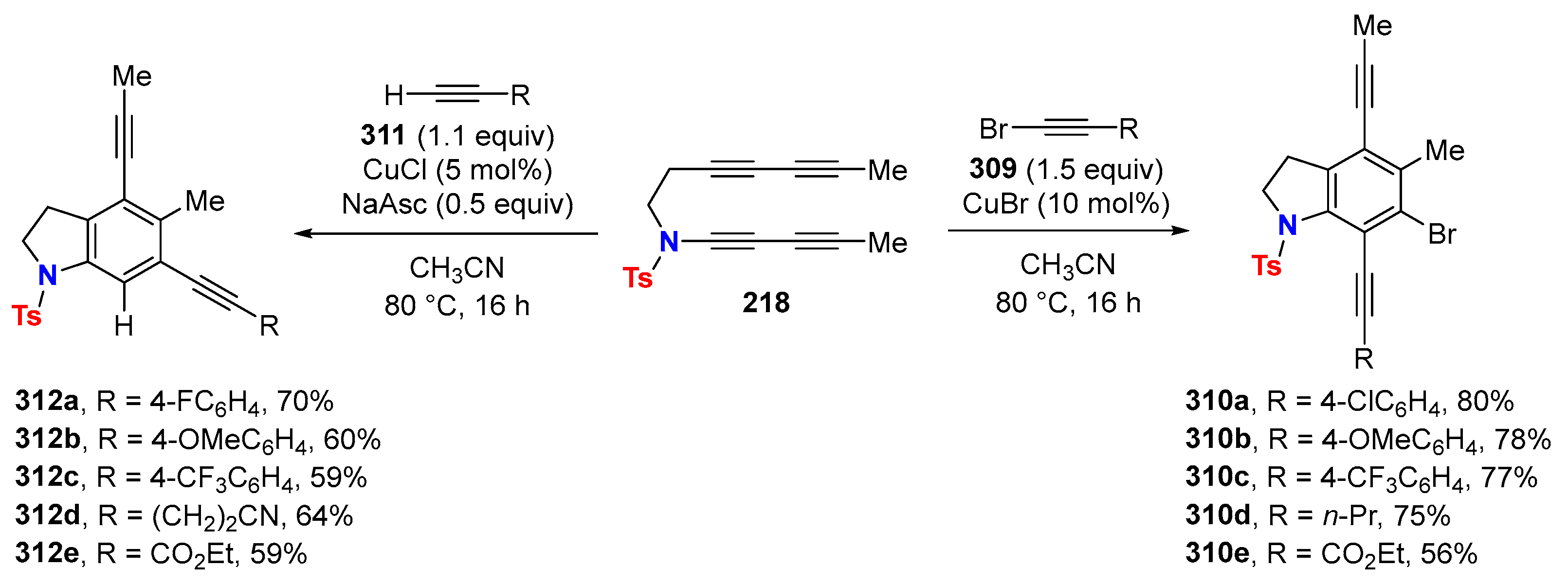
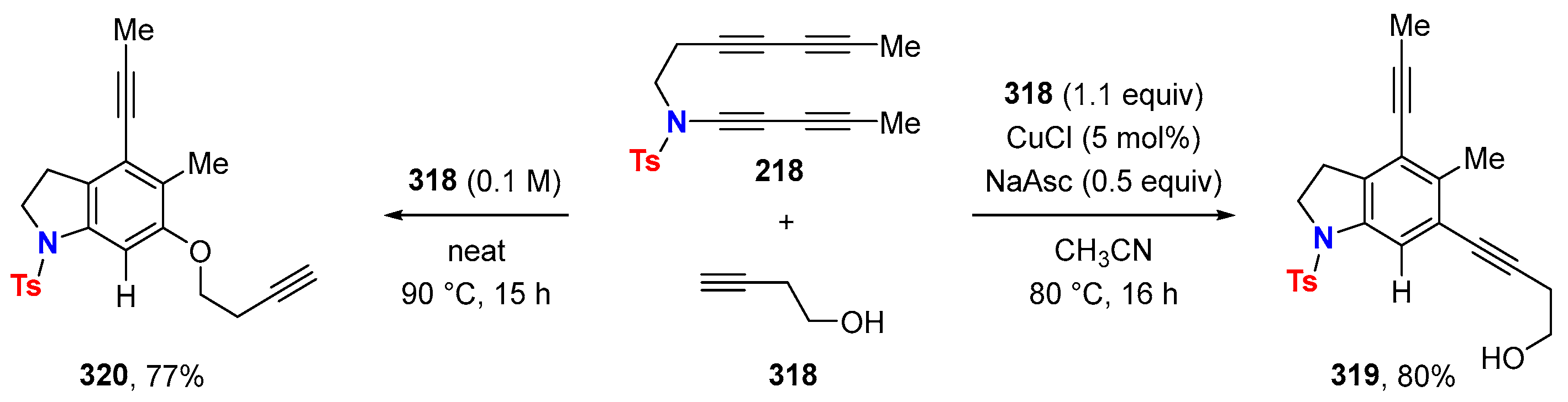
2.1. Disconnection [a]: Cu-Catalyzed Cross-Coupling Reactions
2.2. Disconnection [b]: Amide N-Alkynylations
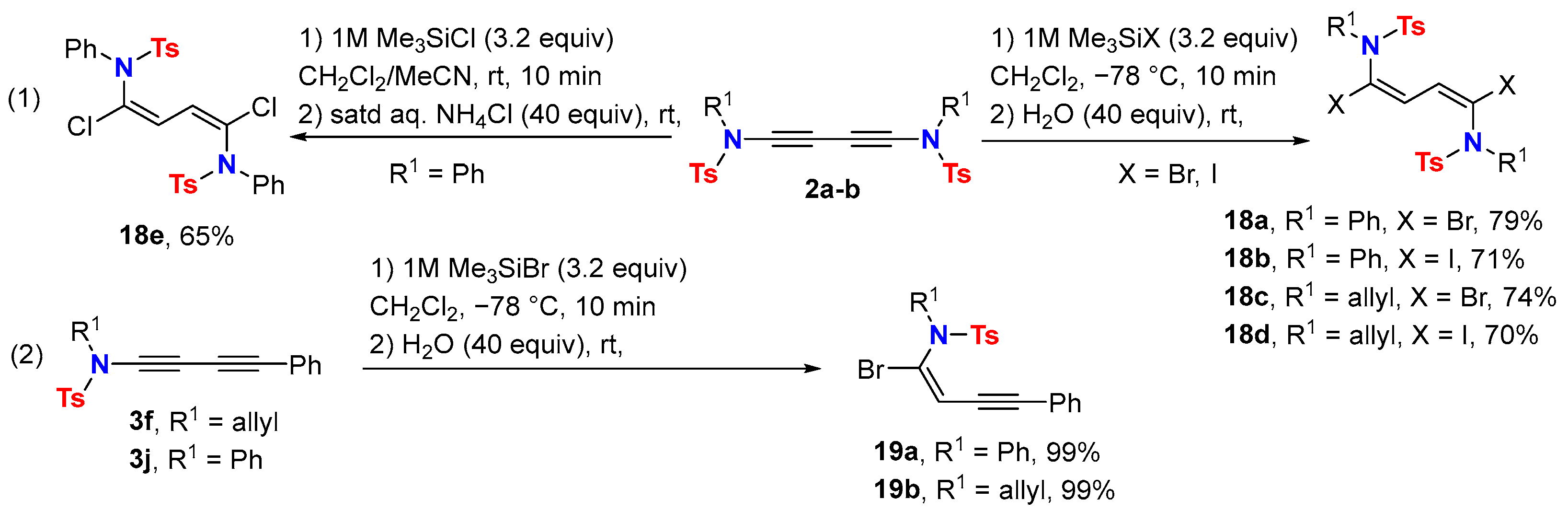
2.3. Disconnection [c]: Functional Group Interconversion (FGI) at the C-Terminus
3. Molecular Structure and Electronic Properties
4. Addition, Cycloaddition and Oxidation Reactions
4.1. Addition Reactions
4.2. Cycloaddition Reactions
4.3. Oxidation Reactions
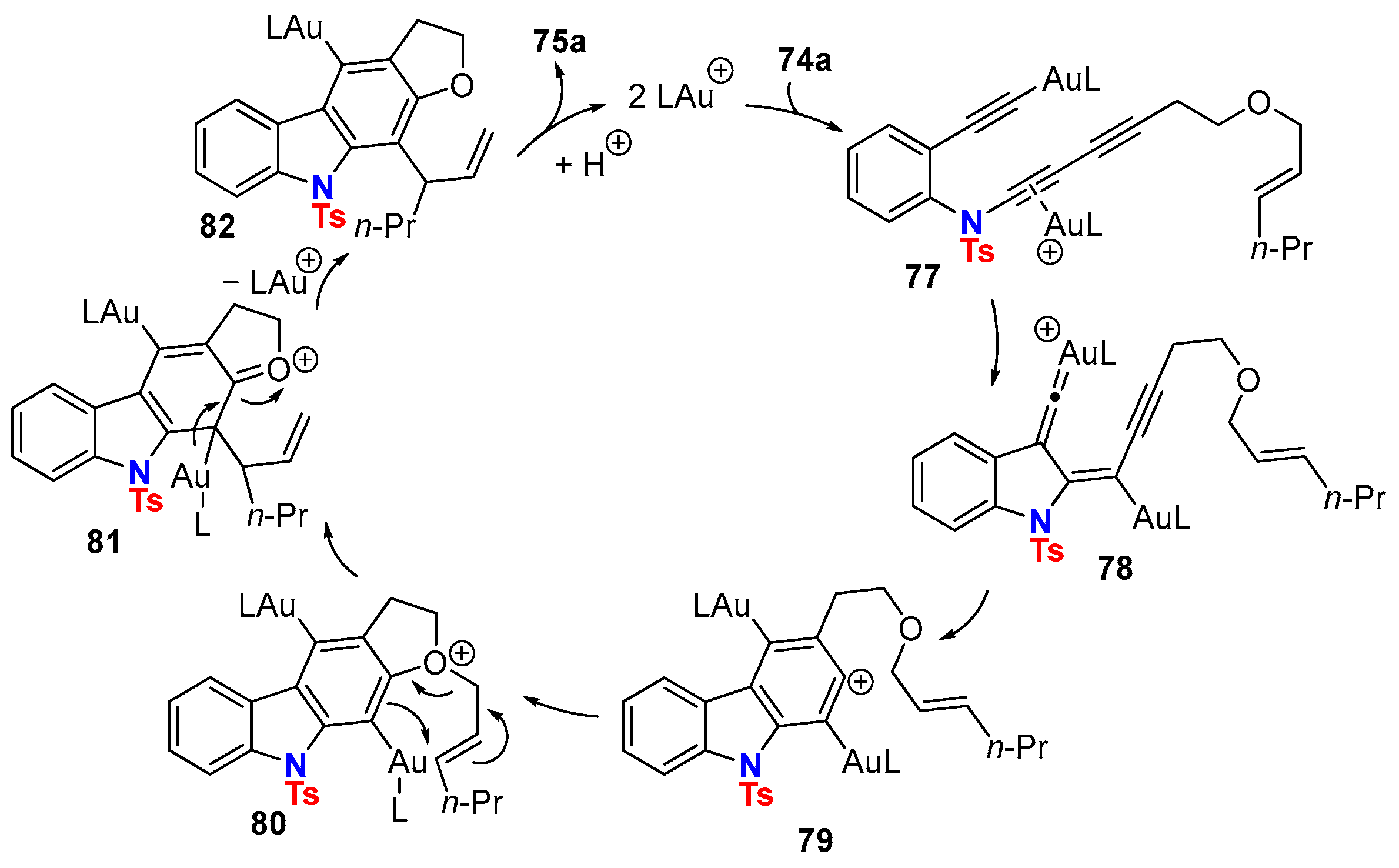
5. Metal-Catalyzed Cascade-Type Cyclization and Annulation Reactions
5.1. Intramolecular Processes
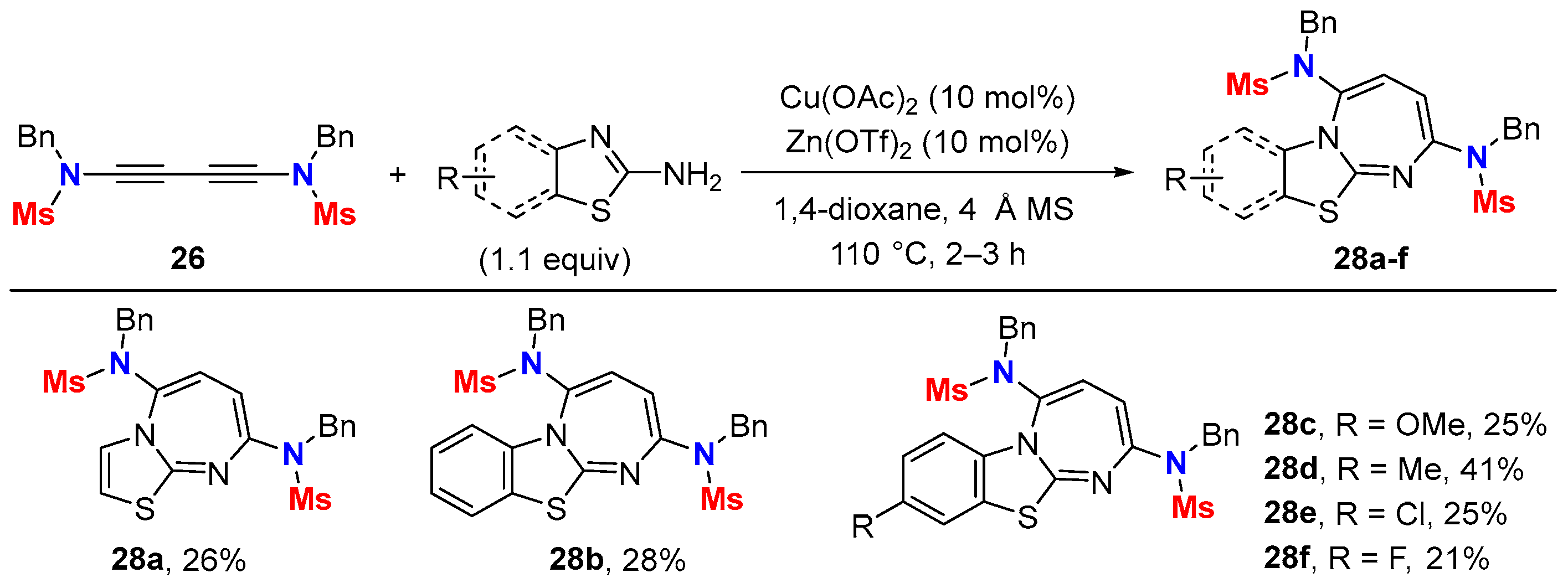
5.2. Intermolecular Processes
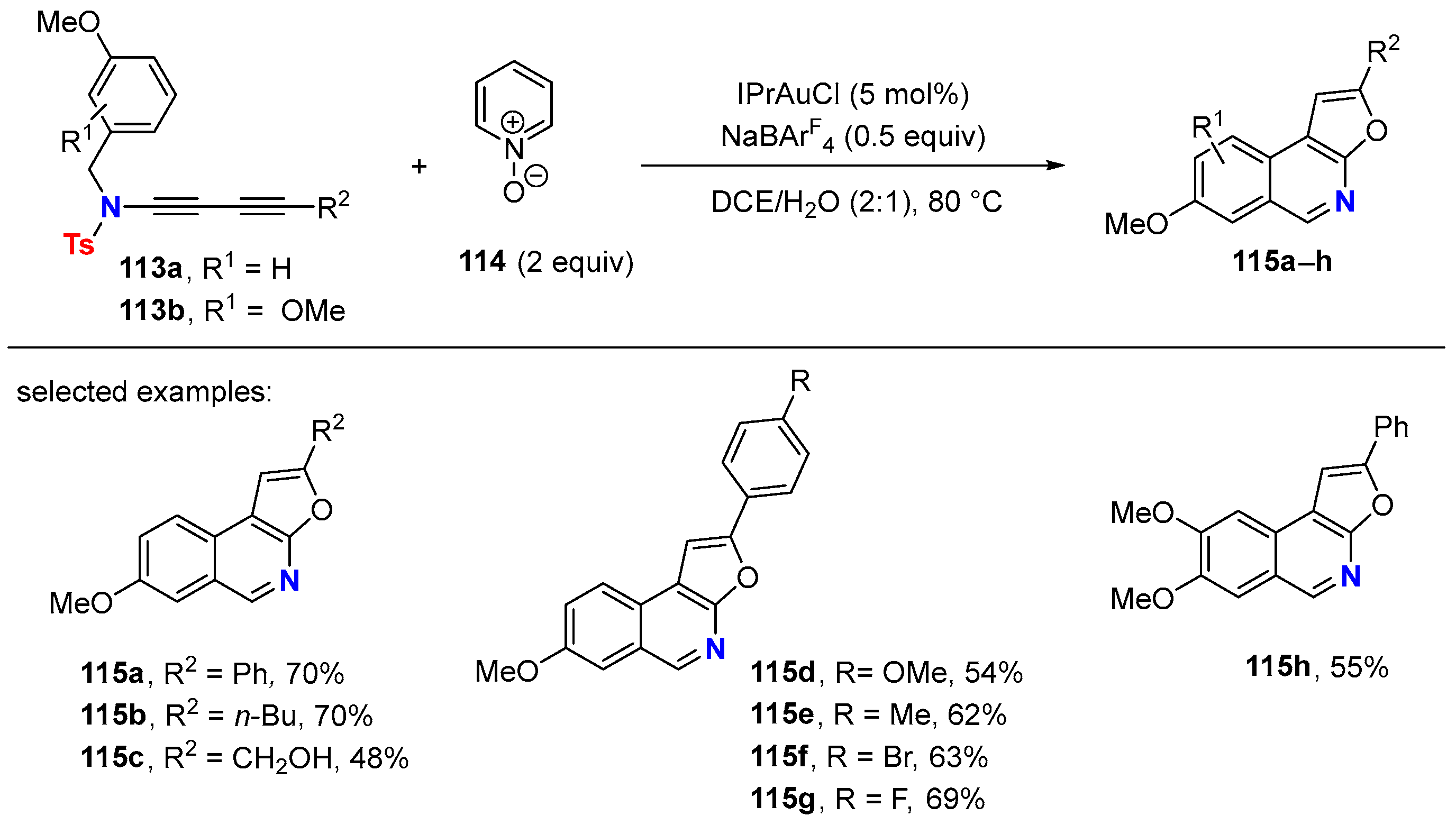
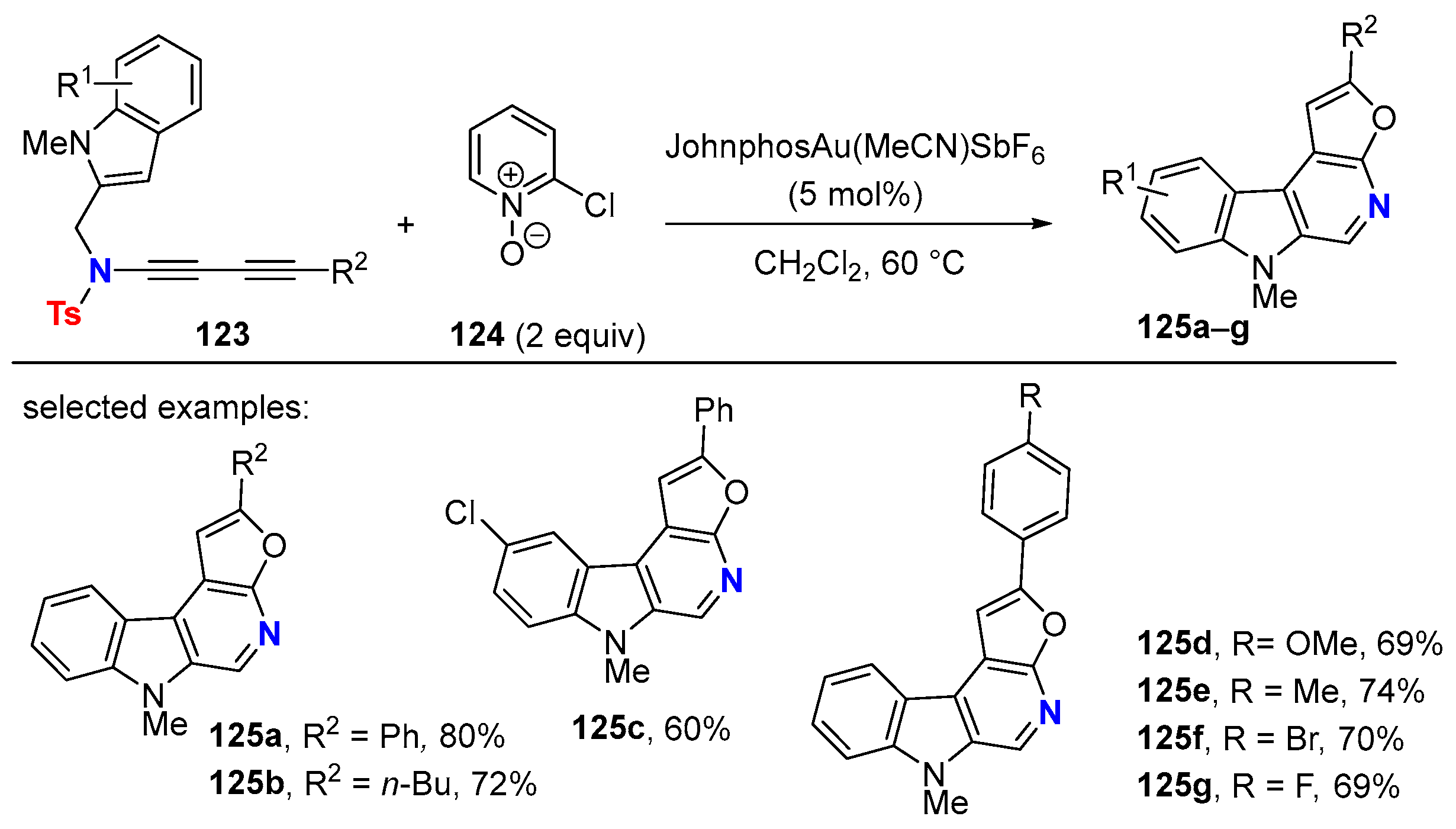
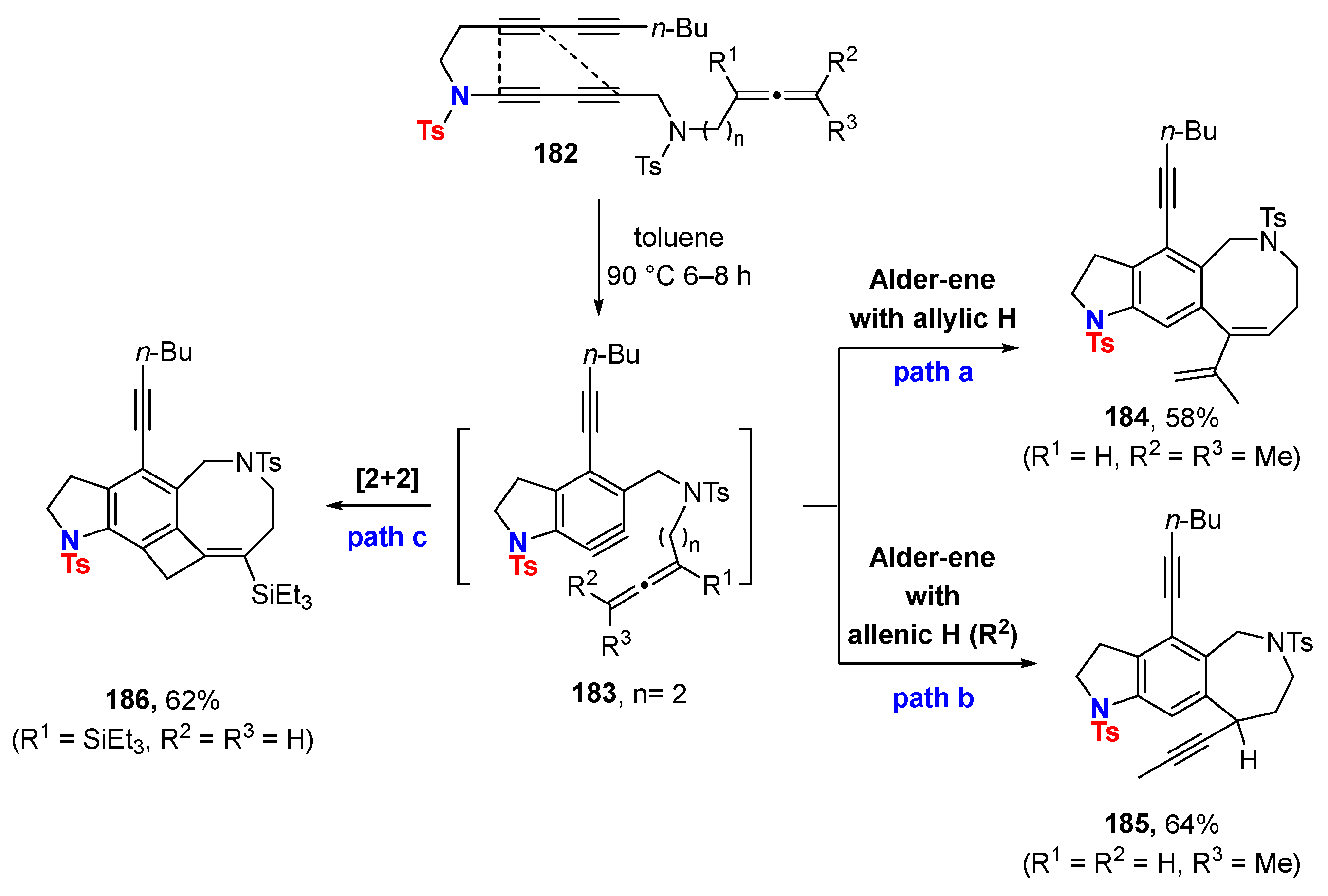
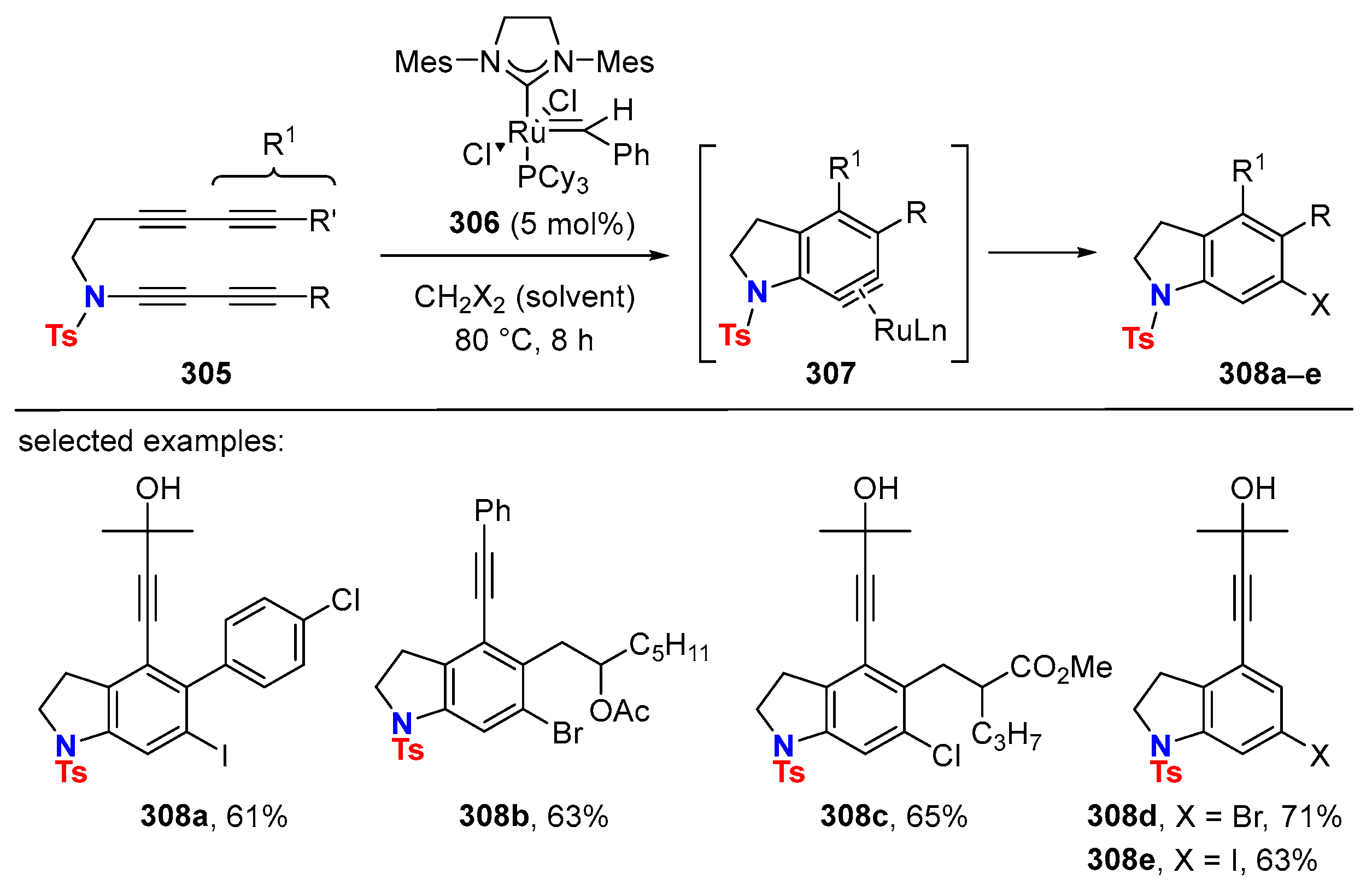
6. HDDA Cascade Reactions
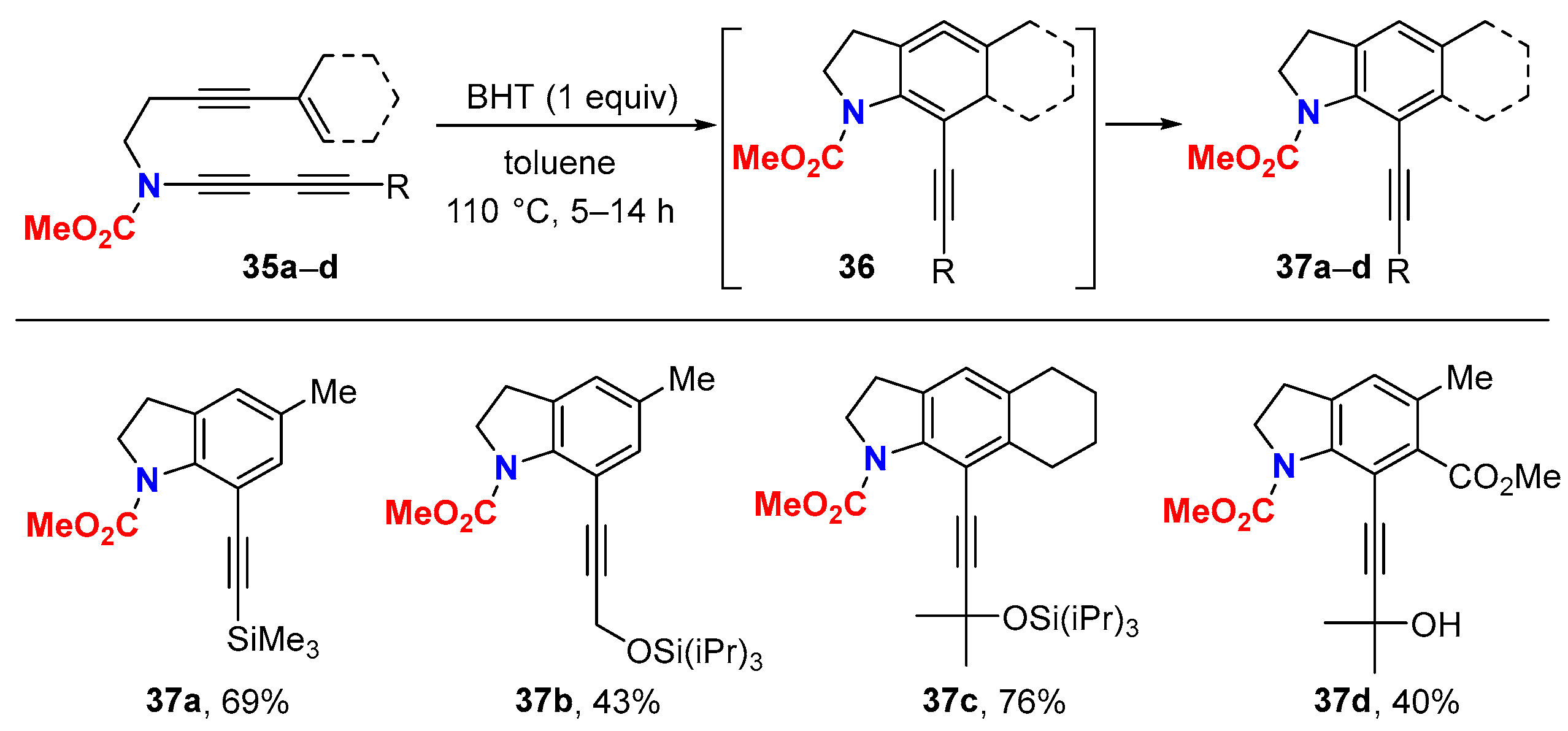
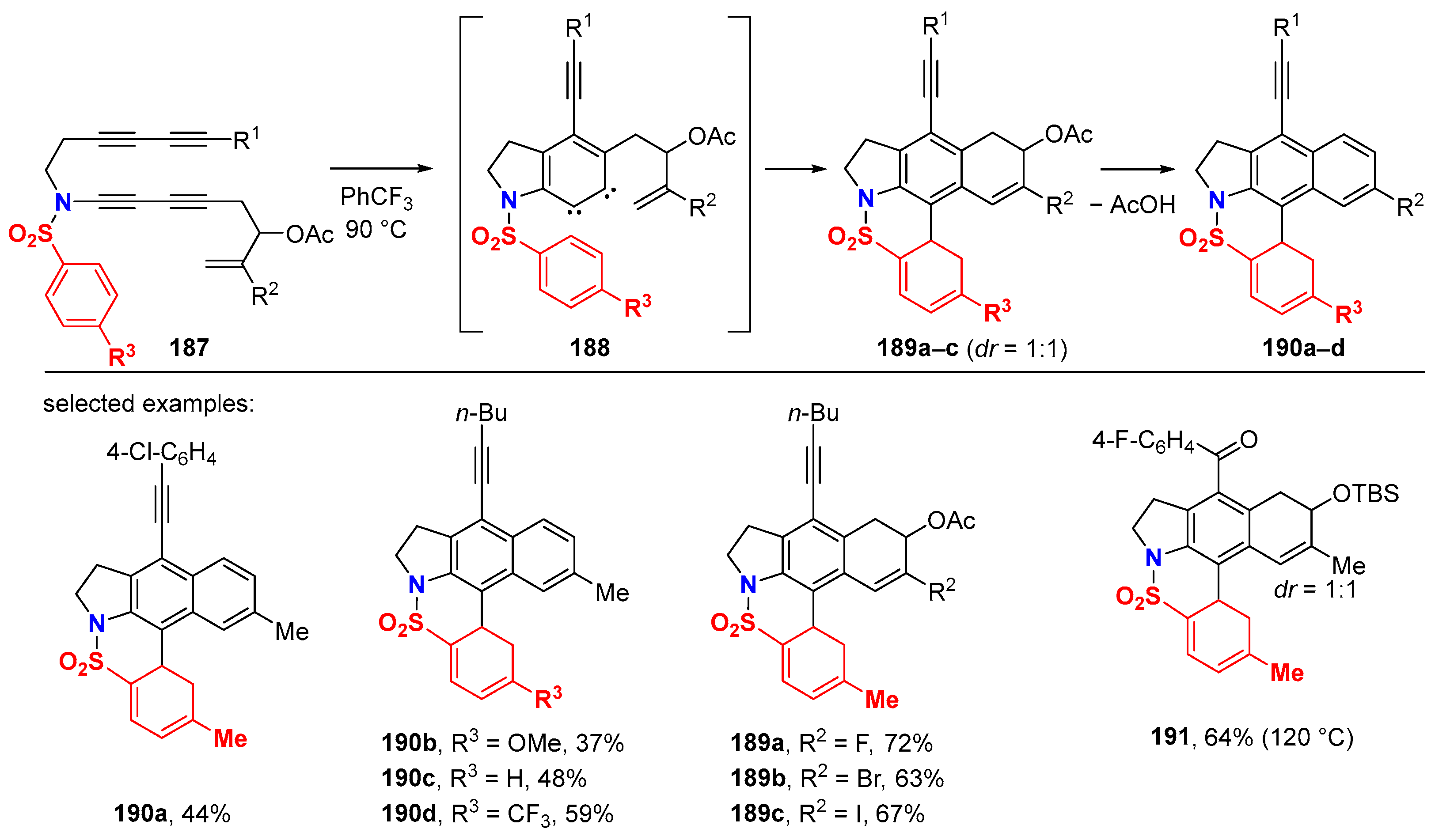
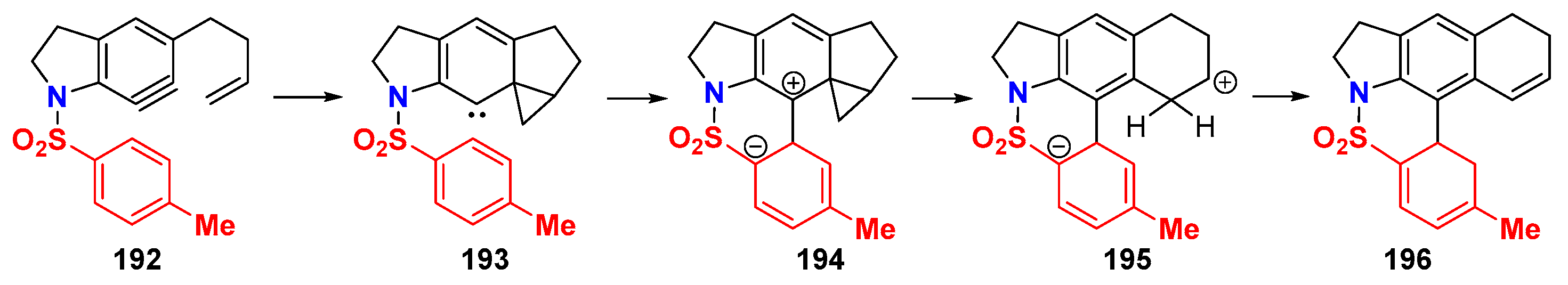
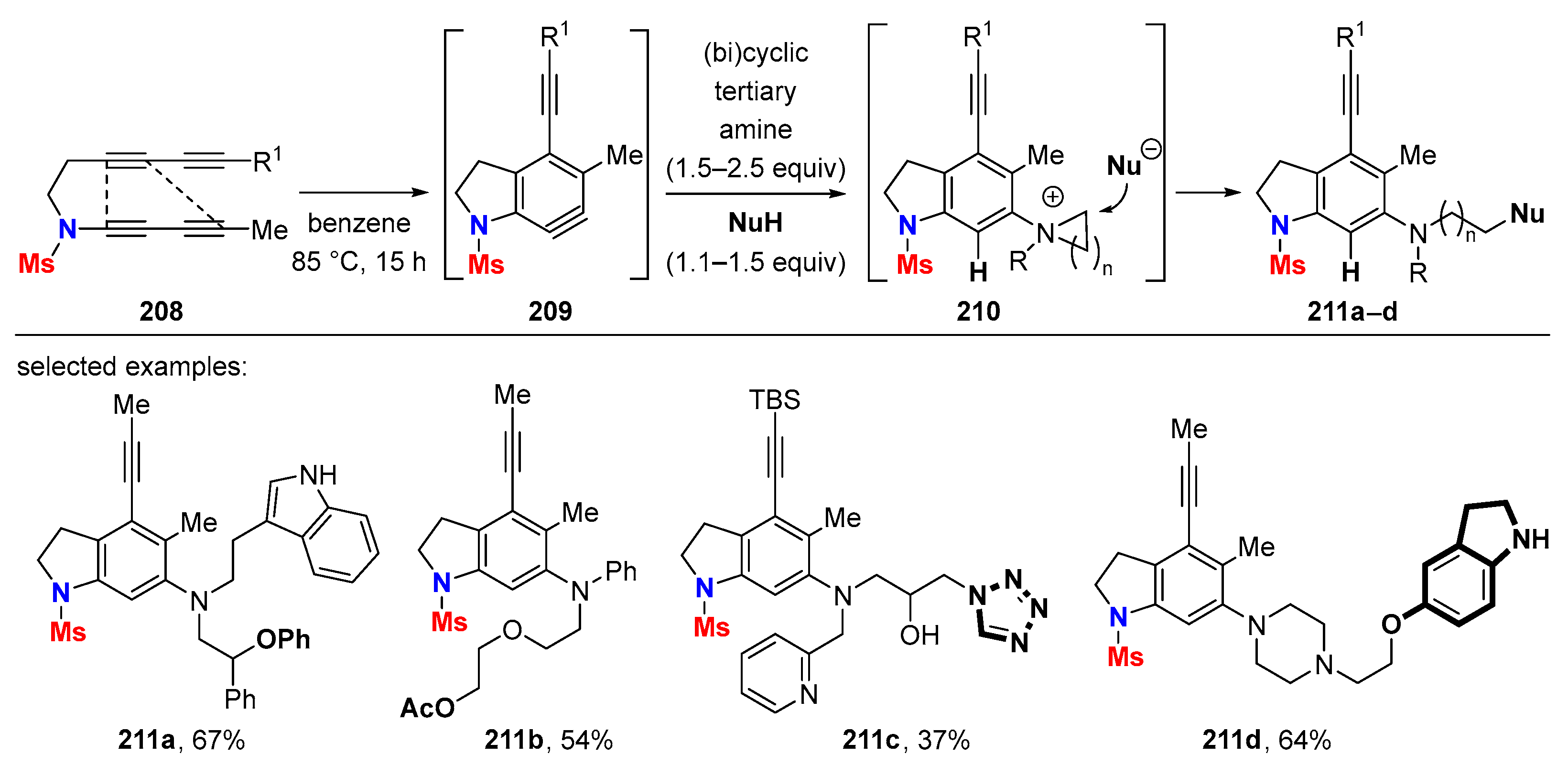
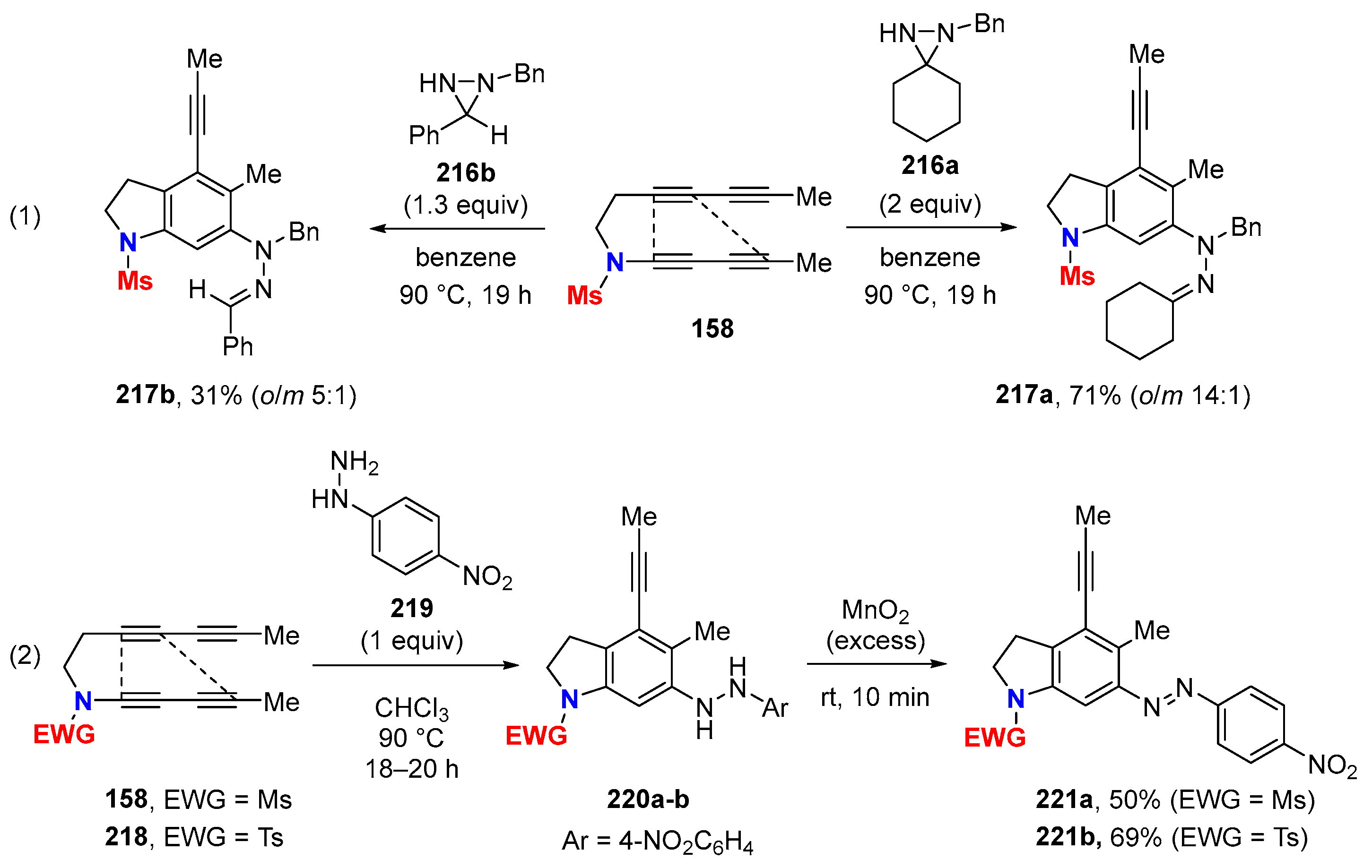
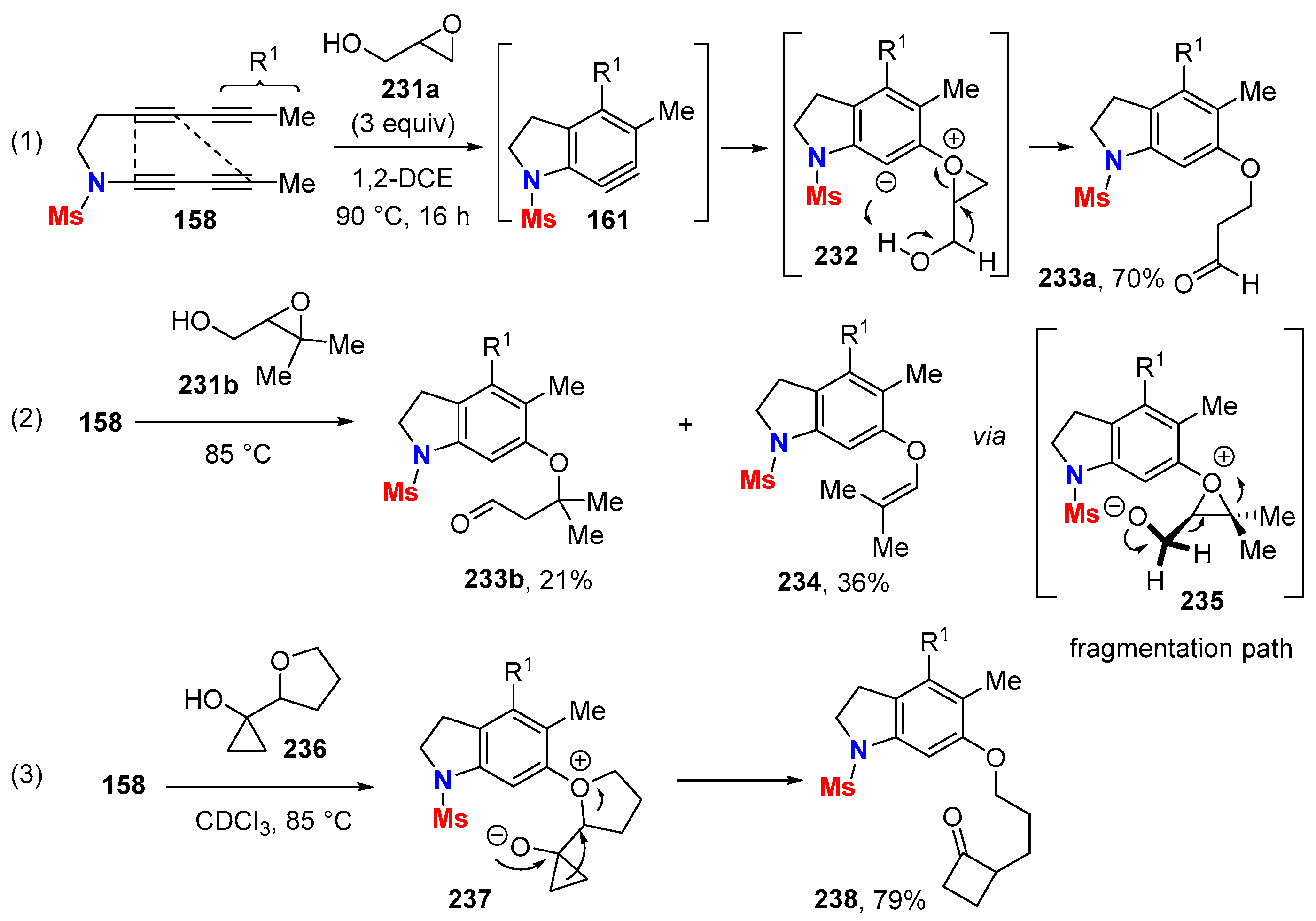
6.1. Sole Thermally Induced HDDA Cascade Reactions
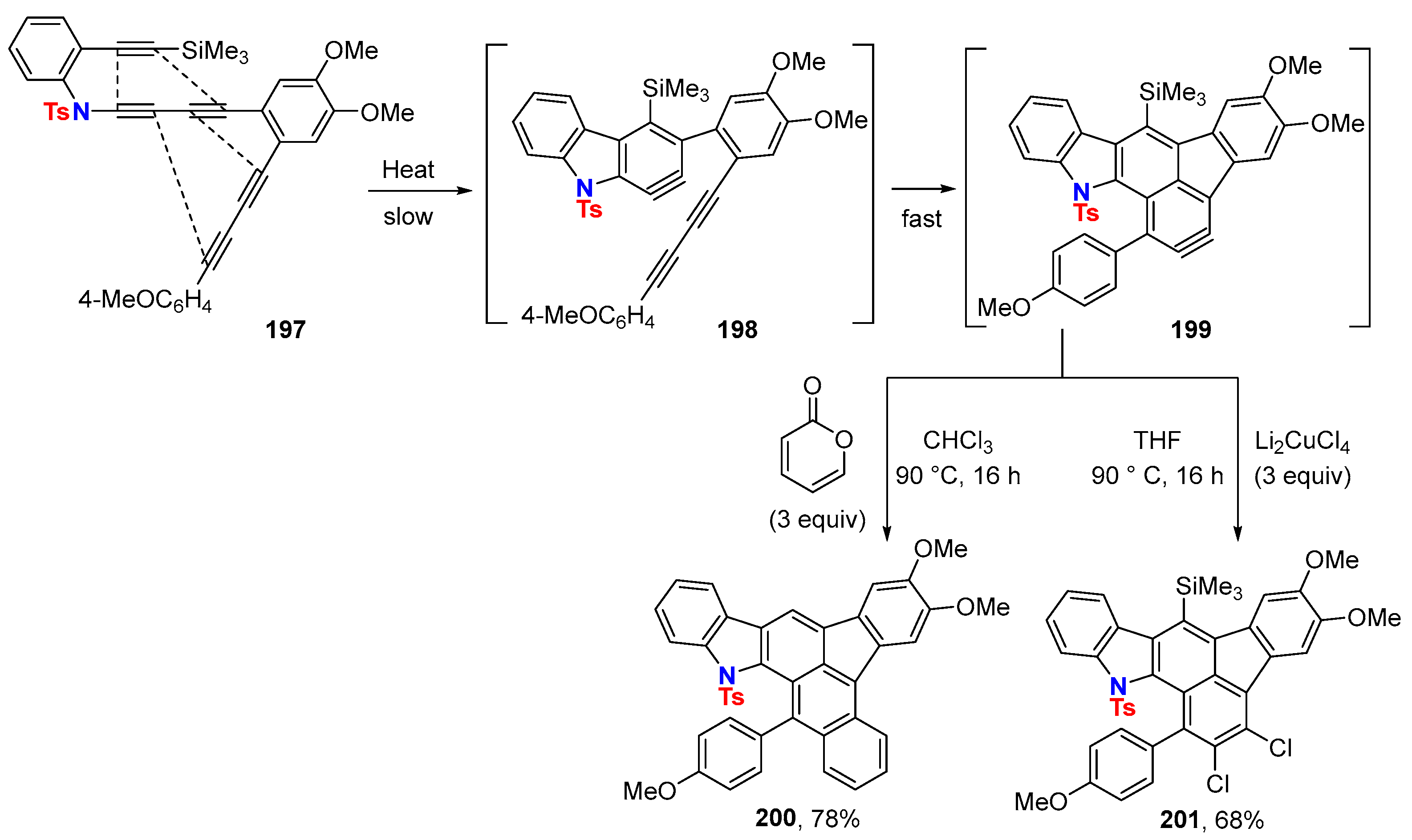
6.2. Silver-Catalyzed HDDA Reactions
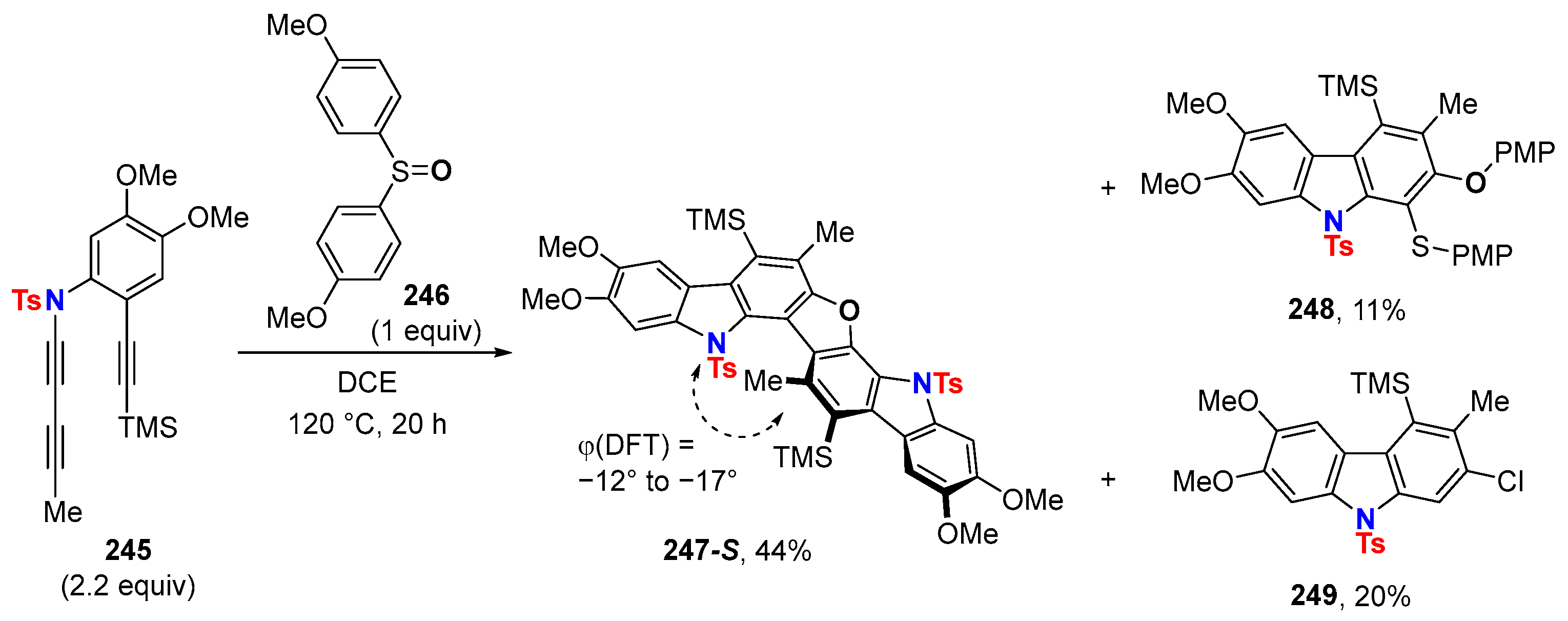
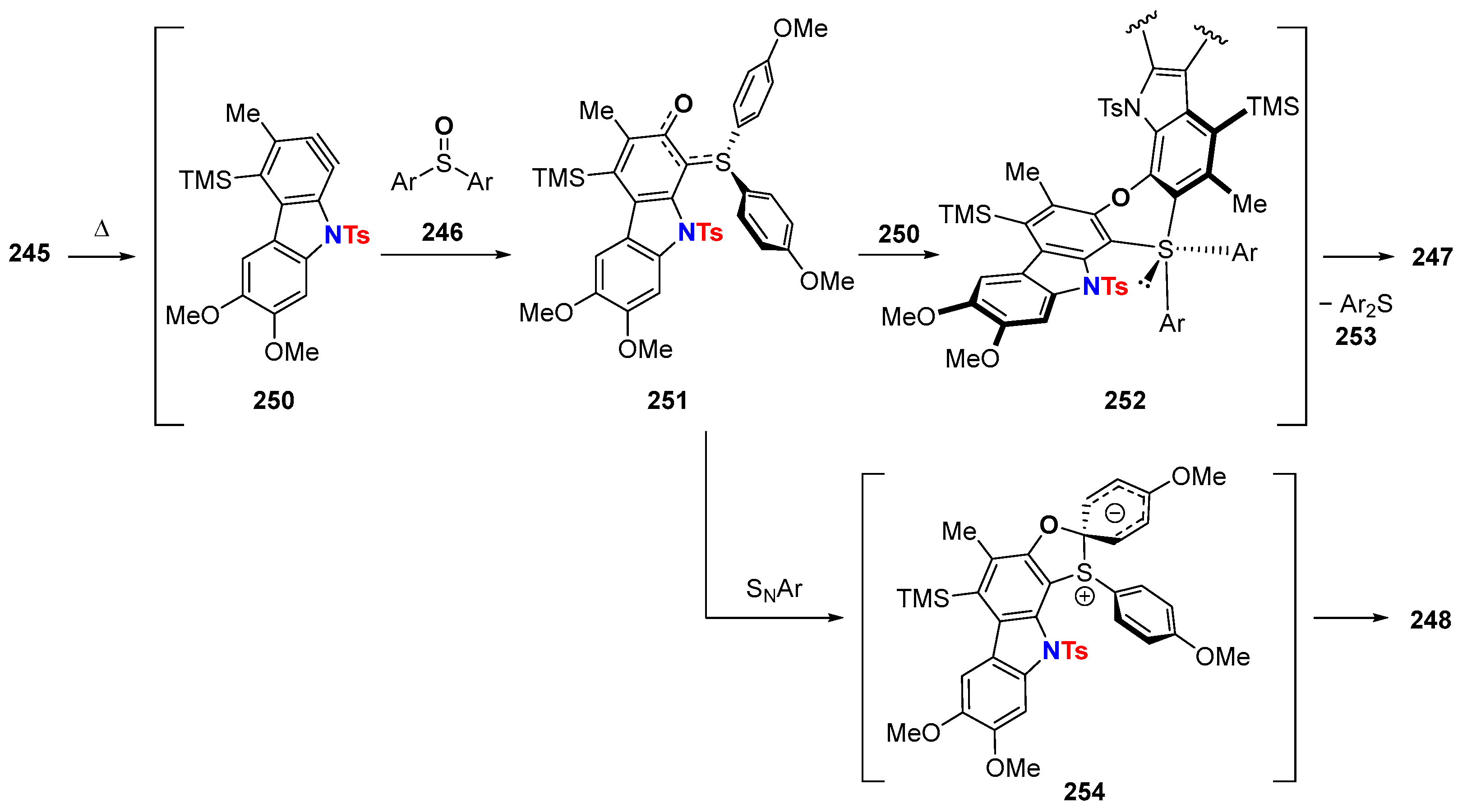
6.3. HDDA Reactions Catalyzed by Other Metals Than Silver
7. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Witulski, B.; Stengel, T. N-Functionalized 1-Alkynylamides: New Building Blocks for Transition Metal Mediated Inter- and Intramolecular [2+2+1] Cycloadditions. Angew. Chem. Int. Ed. 1998, 37, 489–492. [Google Scholar] [CrossRef]
- Iftikhar, R.; Mazhar, A.; Iqbal, M.S.; Khan, F.Z.; Askary, S.H.; Sibtain, H. Ring Forming Transformation of Ynamides via Cycloaddition. RSC. Adv. 2023, 13, 10715–10756. [Google Scholar] [CrossRef]
- Hu, Y.-C.; Zhao, Y.; Wan, B.; Chen, Q.-A. Reactivity of Ynamides in Catalytic Intermolecular Annulations. Chem. Soc. Rev. 2021, 50, 2582–2625. [Google Scholar] [CrossRef]
- Shandilya, S.; Gogoi, M.P.; Dutta, S.; Sahoo, A.K. Gold-Catalyzed Transformation of Ynamides. Chem. Rec. 2021, 21, 4123–4149. [Google Scholar] [CrossRef]
- Campeau, D.; Rayo, D.F.L.; Mansour, A.; Muratov, K.; Gagosz, F. Gold-Catalyzed Reactions of Specially Activated Alkynes, Allenes and Alkenes. Chem. Rev. 2021, 121, 8756–8867. [Google Scholar] [CrossRef]
- Chen, Y.-B.; Qian, P.-C.; Ye, L.-W. Brønsted Acid-Mediated Reactions of Ynamides. Chem. Soc. Rev. 2020, 49, 8897–8909. [Google Scholar] [CrossRef] [PubMed]
- Lynch, C.C.; Sripada, A.; Wolf, C. Asymmetric Synthesis with Ynamides: Unique Reaction Control, Chemical Diversity and Applications. Chem. Soc. Rev. 2020, 49, 8543–8583. [Google Scholar] [CrossRef] [PubMed]
- Tan, T.-D.; Wang, Z.-S.; Qian, P.-C.; Ye, L.-W. Radical Reactions of Ynamides. Small Methods 2021, 5, 2000673. [Google Scholar] [CrossRef] [PubMed]
- Mahe, C.; Cariou, K. Ynamides in Free Radical Reactions. Adv. Synth. Catal. 2020, 362, 4820–4832. [Google Scholar] [CrossRef]
- Hong, F.-L.; Ye, L.-W. Transition Metal-Catalyzed Tandem Reactions of Ynamides for Divergent N-Heterocycle Synthesis. Acc. Chem. Res. 2020, 53, 2003–2019. [Google Scholar] [CrossRef]
- Zhou, B.; Tan, T.-D.; Zhu, X.-Q.; Shang, M.; Ye, L.-W. Reversal of Regioselectivity in Ynamide Chemistry. ACS Catal. 2019, 9, 6393–6406. [Google Scholar] [CrossRef]
- Prabagar, B.; Ghosh, N.; Sahoo, A.K. Cyclization and Cycloisomerization of π-Tethered Ynamides: An Expedient Synthetic Method to Construct Carbo- and Heterocycles. Synlett 2017, 28, 2539–2555. [Google Scholar]
- Cook, A.M.; Wolf, C. Terminal Ynamides: Synthesis, Coupling Reactions, and Additions to Common Electrophiles. Tetrahedron Lett. 2015, 56, 2377–2392. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.-N.; Yeom, H.-S.; Fang, L.-C.; He, S.; Ma, Z.-X.; Kedrowski, B.L.; Hsung, R.P. Ynamides in Ring Forming Transformations. Acc. Chem. Res. 2014, 47, 560–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evano, G.; Coste, A.; Jouvin, K. Ynamides: Versatile Tools in Organic Synthesis. Angew. Chem. Int. Ed. 2010, 49, 2840–2859. [Google Scholar] [CrossRef] [PubMed]
- DeKorver, K.A.; Li, H.; Lohse, A.G.; Hayashi, R.; Lu, Z.; Zhang, Y.; Hsung, R.P. Ynamides: A Modern Functional Group for the New Millennium. Chem. Rev. 2010, 110, 5064–5106. [Google Scholar] [CrossRef] [Green Version]
- Fluegel, L.L.; Hoye, T.R. Hexadehydro-Diels–Alder Reaction: Benzyne Generation via Cycloisomerization of Tethered Triynes. Chem. Rev. 2021, 121, 2413–2444. [Google Scholar] [CrossRef]
- Hoye, T.R.; Baire, B.; Niu, D.; Willoughby, P.H.; Woods, B.P. The hexadehydro-Diels–Alder reaction. Nature 2012, 490, 208–212. [Google Scholar] [CrossRef] [Green Version]
- Holden (née Hall), C.; Greaney, M.F. The Hexadehydro-Diels–Alder Reaction: A New Chapter in Aryne Chemistry. Angew. Chem. Int. Ed. 2014, 53, 5746–5749. [Google Scholar] [CrossRef] [Green Version]
- Diamond, O.J.; Marder, T.B. Methodology and Applications of the Hexadehydro-Diels-Alder (HDDA) Reaction. Org. Chem. Front. 2017, 4, 891–910. [Google Scholar] [CrossRef]
- Yu, H.; Xu, F. Advances in the Synthesis of Nitrogen-Containing Heterocyclic Compounds by in situ Benzyne Cycloaddition. RSC Adv. 2023, 13, 8238–8253. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, D.; Castedo, L.; Saa, C. Homocoupling of 1-Alkynyl Tosylamides. Synlett 2004, 2004, 377–379. [Google Scholar] [CrossRef]
- Doan, T.-H.; Talbi, I.; Lohier, J.-F.; Touil, S.; Alayrac, C.; Witulski, B. Synthesis, Crystal Structure, Optical, Electrochemical and Thermal Properties of the Ynamide: Bis-(N-4-methylbenzenesulfonyl, N-n-butyl)-1,3-butadiyne-1,4-diamide. J. Mol. Struct. 2016, 1116, 127–134. [Google Scholar] [CrossRef]
- Takai, R.; Shimbo, D.; Tada, N.; Itoh, A. Ligand-Enabled Copper-Catalyzed N-Alkynylation of Sulfonamide with Alkynyl Benziodoxolone: Synthesis of Amino Acid-Derived Ynamide. J. Org. Chem. 2021, 86, 4699–4713. [Google Scholar] [CrossRef]
- Witulski, B.; Schweikert, T.; Schollmeyer, D.; Nemkovich, N.A. Synthesis and Molecular Properties of Donor-π-Spacer-Acceptor Ynamides with up to Four Conjugated Alkyne Units. Chem. Commun. 2010, 46, 2953–2955. [Google Scholar] [CrossRef]
- Talbi, I.; Alayrac, C.; Lohier, J.-F.; Touil, S.; Witulski, B. Application of Ynamides in the Synthesis of 2-(Tosylamido)- and 2,5-Bis(tosylamido)thiophenes. Org. Lett. 2016, 18, 2656–2659. [Google Scholar] [CrossRef]
- Wang, Y.-P.; Danheiser, R.L. Synthesis of 2-Iodoynamides and Regioselective [2+2] Cycloadditions with Ketene. Tetrahedron Lett. 2011, 52, 2111–2114. [Google Scholar] [CrossRef] [Green Version]
- Tracey, M.R.; Zhang, Y.; Frederick, M.O.; Mulder, J.A.; Hsung, R.P. Sonogashira Cross-Couplings of Ynamides. Syntheses of Urethane- and Sulfonamide-Terminated Conjugated Phenylacetylenic Systems. Org. Lett. 2004, 6, 2209–2212. [Google Scholar] [CrossRef]
- Dunetz, J.R.; Danheiser, R.L. Copper-Mediated N-Alkynylation of Carbamates, Ureas, and Sulfonamides. A General Method for the Synthesis of Ynamides. Org. Lett. 2003, 5, 4011–4014. [Google Scholar] [CrossRef] [Green Version]
- Kohnen, A.L.; Dunetz, J.R.; Danheiser, R.L. Synthesis of Ynamides by N-Alkynylation of Amine Derivatives. Preparation of N-Allyl-N-(Methoxycarbonyl)-1,3-Decadiynylamine. Org. Synth. 2007, 84, 88–101. [Google Scholar]
- Ross, S.P.; Hoye, T.R. Multiheterocyclic Motifs via Three-Component Reactions of Benzynes, Cyclic Amines, and Protic Nucleophiles. Org. Lett. 2018, 20, 100–103. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Hsung, R.P.; Tracey, M.R.; Kurtz, K.C.M.; Vera, E.L. Copper Sulfate-Pentahydrate-1,10-Phenanthroline Catalyzed Amidations of Alkynyl Bromides. Synthesis of Heteroaromatic Amine Substituted Ynamides. Org. Lett. 2004, 6, 1151–1154. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Niu, D.; Hoye, T.R. The Hexadehydro-Diels–Alder Cycloisomerization Reaction Proceeds by a Stepwise Mechanism. J. Am. Chem. Soc. 2016, 138, 7832–7835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mansfield, S.J.; Smith, R.C.; Yong, J.R.J.; Garry, O.L.; Anderson, E.A. A General Copper-Catalyzed Synthesis of Ynamides from 1,2-Dichloroenamides. Org. Lett. 2019, 21, 2918–2922. [Google Scholar] [CrossRef]
- Kawakarni, R.; Usui, S.; Tada, N.; Itoh, A. Late-Stage Diversification Strategy for Synthesizing Ynamides through Copper-Catalyzed Diynylation and Azide-Alkyne Cycloaddition. Chem. Commun. 2023, 59, 450–453. [Google Scholar]
- Gohrai, S.; Lee, D. Selectivity for Alkynyl or Allenyl Imidamides and Imidates in Copper-Catalyzed Reactions of Terminal 1,3-Diynes and Azides. Org. Lett. 2021, 23, 697–701. [Google Scholar] [CrossRef]
- Garner, M.H.; Hoffmann, R.; Rettrup, S.; Solomon, G.C. Coarctate and Möbius: The Helical Orbitals of Allene and Other Cumulenes. ACS Cent. Sci. 2018, 4, 688–700. [Google Scholar] [CrossRef]
- Garner, M.H.; Jensen, A.; Hyllested, L.O.H.; Solomon, G.C. Helical Orbitals and Circular Currents in Linear Carbon Wires. Chem. Sci. 2019, 10, 4598–4608. [Google Scholar] [CrossRef] [Green Version]
- Garner, M.H.; Corminboeuf, C. Helical Electronic Transitions of Spiroconjugated Molecules. Chem. Commun. 2021, 57, 6408–6411. [Google Scholar] [CrossRef]
- Ohashi, S.; Inagaki, S. Orbital Phase Control of Conformations of Alkyne Derivatives. Tetrahedron 2001, 57, 5361–5367. [Google Scholar] [CrossRef]
- Hendon, C.H.; Tiana, D.; Murray, A.T.; Carbery, D.R.; Walsh, A. Helical Frontier Orbitals of Conjugated Linear Molecules. Chem. Sci. 2013, 4, 4278–4284. [Google Scholar] [CrossRef] [Green Version]
- Balakrishnan, A.R.; Suresh, R.; Vijayakumar, S. Amine Terminated Polyynes as Candidates for Molecular Wire Applications: A DFT Study. Phys. E Low-Dimens. Syst. Nanostruct. 2022, 137, 115045. [Google Scholar] [CrossRef]
- Bro-Jorgensen, W.; Garner, M.H.; Solomon, G.C. Quantification of the Helicality of Helical Molecular Orbitals. J. Phys. Chem. A 2021, 125, 8107–8115. [Google Scholar] [CrossRef] [PubMed]
- Petrov, A.R.; Daniliuc, C.G.; Jones, P.G.; Tamm, M. A Novel Synthetic Approach to Diaminoacetylenes: Structural Characterization and Reactivity of Aromatic an Aliphatic Ynediamines. Chem. Eur. J. 2010, 16, 11804–11808. [Google Scholar] [CrossRef]
- Tokutome, Y.; Okuno, T. Preparations, Crystal Polymorphs and DFT Calculations of N1,N1,N4,N4-Tetraphenylbuta-1,3-diyne-1,4-diamine. J. Mol. Struct. 2013, 1047, 136–142. [Google Scholar] [CrossRef]
- Mayerle, J.J.; Flandera, M.A. Bis(1-carbazolyl)butadiyne. Acta Cryst. 1978, B34, 1374–1376. [Google Scholar] [CrossRef]
- Matsuda, H.; Nakanishi, H.; Hosomi, T.; Kato, M. Synthesis and Solid-State Polymerization of a New Diacetylene: 1-(N-Carbazolyl)penta-1,3-diyn-5-ol. Macromolecules 1988, 21, 1238–1240. [Google Scholar] [CrossRef]
- Tabata, H.; Kuwamoto, K.; Okuno, T. Conformational Polymorphs and Solid-State Polymerization of 9-(1,3-Butadiynyl)carbazole Derivatives. J. Mol. Struct. 2016, 1106, 452–459. [Google Scholar] [CrossRef] [Green Version]
- Ide, M.; Ohashi, K.; Mihara, S.; Iwasaha, T. Regio- and Stereoselective hydrohalogenation of Ynamide Components in 1,3-Butadiynes with in situ Generated HX. Tetrahedron Lett. 2014, 55, 2130–2133. [Google Scholar] [CrossRef]
- De la Vega-Hernandez, K.; Romain, E.; Coffinet, A.; Bijouard, K.; Gontard, G.; Chemla, F.; Ferreira, F.; Jackowski, O.; Perez-Luna, A. Radical Germylzincation of α-Heteroatom-Substituted Alkynes. J. Am. Chem. Soc. 2018, 140, 17632–17642. [Google Scholar] [CrossRef] [Green Version]
- Kramer, S.; Madsen, J.L.H.; Rottländer, M.; Skrydstrup, T. Access to 2,5-Diamidopyrroles and 2,5-Diamidofurans by Au(I)-Catalyzed Double Hydroamination or Hydration of 1,3-Diynes. Org. Lett. 2010, 12, 2758–2761. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, S.; Gayyur; Ghosh, N. Cu(II)-Catalyzed [4+1] and [4+3] Annulation Reactions: A Modular Approach to N-Aryl/Alkyl Substituted 2,5-Diamidopyrroles and Diazepines. Org. Biomol. Chem. 2022, 20, 7017–7021. [Google Scholar] [CrossRef] [PubMed]
- Dunetz, J.R.; Danheiser, R.L. Synthesis of Highly Substituted Indolines and Indoles via Intramolecular [4+2] Cycloaddition of Ynamides and Conjugated Enynes. J. Am. Chem. Soc. 2005, 127, 5776–5777. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Lu, Q.; Chen, G.; Yu, Y.; Li, C.; Huang, X. Rhodium-Catalyzed Azide-Alkyne Cycloaddition of Internal Ynamides: Regioselective Assembly of 5-Amino-Triazoles under Mild Conditions. ACS Catal. 2017, 7, 7529–7534. [Google Scholar] [CrossRef]
- Davies, P.W.; Cremonesi, A.; Dumitrescu, L. Intermolecular and Selective Synthesis of 2,4,5-Trisubstituted Oxazoles by a Gold-Catalyzed Formal [3+2] Cycloaddition. Angew. Chem. Int. Ed. 2011, 50, 8931–8935. [Google Scholar] [CrossRef]
- Skaria, M.; Hsu, Y.-C.; Jiang, Y.-T.; Lu, M.-Y.; Kuo, T.-C.; Cheng, M.-J.; Liu, R.-S. Gold-Catalyzed Oxidations of 1,3-Diynamides with C(1) versus C(3) Regioselectivity: Catalyst-Dependent Oxidative Cyclizations in the C(3) Oxidation. Org. Lett. 2020, 22, 4478–4482. [Google Scholar] [CrossRef]
- Lee, D.; Kim, M. Advances in the Metallotropic [1,3]-Shift of Alkynyl Carbenoids. Org. Biomol. Chem. 2007, 5, 3418–3427. [Google Scholar] [CrossRef]
- Yanai, H.; Kawazoe, T.; Ishii, N.; Witulski, B.; Matsumoto, T. Regioselective Synthesis of 4-Aryl-1,3-dihydroxy-2-naphthoates through 1,2-Aryl-Migrative Ring Rearrangement Reaction and their Photoluminescence Properties. Chem. Eur. J. 2021, 27, 11442–11449. [Google Scholar] [CrossRef]
- Kumar, M.; Kaliya, K.; Maurya, S.K. Recent Progress in the Homogeneous Gold-Catalysed Cycloisomerisation Reactions. Org. Biomol. Chem. 2023, 21, 3276–3295. [Google Scholar] [CrossRef]
- Liu, J.; Chakraborty, P.; Zhang, H.; Zhong, L.; Wang, Z.-X.; Huang, X. Gold-Catalyzed Atom-Economic Synthesis of Sulfone-Containing Pyrrolo [2,1-a]isoquinolines from Diynamides: Evidence for Consecutive Sulfonyl migration. ACS Catal. 2019, 9, 2610–2617. [Google Scholar] [CrossRef]
- Choudhary, S.; Gayyur; Ghosh, N. Gold(I)-Catalyzed and PTSA-Promoted Cycloisomerization of Ynamides to Access Pyrrole Substituted α,β-Unsaturated Ketones. Eur. J. Org. Chem. 2023, 26, e202201223. [Google Scholar] [CrossRef]
- Wang, H.F.; Guo, L.N.; Fan, Z.B.; Tang, T.H.; Zi, W. Gold-Catalyzed Formal Hexadehydro-Diels–Alder/Carboalkoxylation Reaction Cascades. Org. Lett. 2021, 23, 2676–2681. [Google Scholar] [CrossRef]
- Liu, N.; Sun, H.; Wang, J.; Zhang, Z.; Wang, T. Ag(I)-Catalyzed Synthesis of 2-Aminoquinolines from 1-Aminobutadiynes and Anilines. Adv. Synth. Catal. 2021, 363, 5443–5447. [Google Scholar] [CrossRef]
- Wang, J.; Du, J.; Wang, T. Synthesis of 2-Aminopyrrolo[1,2-b]pyridazines via Gold(I)-Catalyzed Chemoselective Hydroamination/Hydroarylation Cascade of 1,3-Diynamides with 1-Aminopyrroles. Adv. Synth. Catal. 2023, 365, 1088–1092. [Google Scholar] [CrossRef]
- Gayyur; Choudhary, S.; Kant, R.; Ghosh, N. Synergetic Copper/Zinc Catalysis: Synthesis of Aryl/Heteroaryl-Fused 1H-Pyrrolo[3,2-c]pyridines. Chem. Commun. 2022, 58, 1974–1977. [Google Scholar] [CrossRef]
- Pandit, Y.B.; Jiang, Y.-T.; Jian, J.-J.; Chen, T.-C.; Kuo, T.-C.; Cheng, M.-J.; Liu, R.-S. Gold-Catalyzed [5+2]-Annulations of 1,3-Diyn-1-amides with Anthranils Bearing no C(6)-Substituents. Org. Chem. Front. 2021, 8, 2563–2568. [Google Scholar] [CrossRef]
- Tian, X.; Song, L.; Farshadfar, K.; Rudolph, M.; Rominger, F.; Oeser, T.; Ariafard, A.; Hashmi, A.S.K. Acyl Migration versus Epoxidation in Gold Catalysis: Facile, Switchable, and Atom-Economic Synthesis of Acylindoles and Quinoline Derivatives. Angew. Chem. Int. Ed. 2020, 59, 471–478. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Zhu, L.; Wan, W.; Huang, X. Gold-Catalyzed Cascade Cyclization of 1,3-Diynamides: Polycyclic N-Heterocycle Synthesis via Construction of a Furopyridinyl Core. Org. Lett. 2020, 22, 3279–3285. [Google Scholar] [CrossRef]
- Xia, J.; Liu, J.; Yu, Y.; Zhang, J.; Huang, X. Divergent Access to Polycyclic N-Heterocyclic Compounds through Büchner-Type Dearomatisation Enabled Cycloisomerization of Diynamides under Gold Catalysis. Org. Lett. 2022, 24, 4298–4303. [Google Scholar] [CrossRef] [PubMed]
- Lenko, I.; Mamontov, A.; Alayrac, C.; Legay, R.; Witulski, B. Media-Driven Pd-Catalyzed Reaction Cascades with 1,3-Diynamides Leading Selectively to Either Indoles or Quinolines. Angew. Chem. Int. Ed. 2021, 60, 22729–22734. [Google Scholar] [CrossRef] [PubMed]
- Witulski, B.; Alayrac, C.; Tevzadze-Saeftel, L. Palladium-Catalyzed Synthesis of 2-Aminoindoles by a Heteroannulation Reaction. Angew. Chem. Int. Ed. 2003, 42, 4257–4260. [Google Scholar] [CrossRef] [PubMed]
- Bradley, A.Z.; Johnson, R.P. Thermolysis of 1,3,8-Nonatriyne: Evidence for Intramolecular [2+4] Cycloaromatization to a Benzyne Intermediate. J. Am. Chem. Soc. 1997, 119, 9917–9918. [Google Scholar] [CrossRef]
- Miyawaki, K.; Suzuki, R.; Kawano, T.; Ueda, I. Cycloaromatization of a Non-Conjugated Polyenyne System: Synthesis of 5H-Benzo[d]fluoreno[3,2-b]pyrans via Diradicals Generated from 1-[2-{4-(2-Alkoxymethylphenyl)butan-1,3-diynyl}]phenylpentan-2,4-diyn-1-ols and Trapping Evidence for the 1,2-Didehydrobenzene Diradical. Tetrahedron Lett. 1997, 38, 3943–3946. [Google Scholar]
- Liang, Y.; Hong, X.; Yu, P.; Houk, K.N. Why Alkynyl Substituents Dramatically Accelerate Hexadehydro-Diels–Alder (HDDA) Reactions: Stepwise Mechanisms of HDDA Cycloadditions. Org. Lett. 2014, 16, 5702–5705. [Google Scholar] [CrossRef] [PubMed]
- Karmakar, R.; Yun, S.Y.; Wang, K.-P.; Lee, D. Regioselectivity in the Nucleophile Trapping of Arynes: The Electronic and Steric Effects of Nucleophiles and Substituents. Org. Lett. 2014, 16, 6–9. [Google Scholar] [CrossRef]
- Wang, T.; Hoye, T.R. Hexadehydro-Diels–Alder (HDDA)-Enabled Carbazolyne Chemistry: Single Step, de Novo Construction of the Pyranocarbazole Core of Alkaloids of the Murraya koenigii (Curry Tree) Family. J. Am. Chem. Soc. 2016, 138, 13870–13873. [Google Scholar] [CrossRef]
- Ross, S.P.; Hoye, T.R. Reactions of Hexadehydro-Diels–Alder Benzynes with Structurally Complex Multifunctional Natural Products. Nat. Chem. 2017, 9, 523–530. [Google Scholar] [CrossRef]
- Chen, M.; He, C.Q.; Houk, K.N. Mechanism and Regioselectivity of an Unsymmetrical Hexadehydro-Diels–Alder (HDDA) Reaction. J. Org. Chem. 2019, 84, 1959–1963. [Google Scholar] [CrossRef]
- Cheong, P.H.-Y.; Paton, R.S.; Bronner, S.M.; Im, G.-Y.J.; Garg, N.K.; Houk, K.N. Indolyne and Aryne Distortions and Nucleophilic Regioselectivities. J. Am. Chem. Soc. 2010, 132, 1267–1269. [Google Scholar] [CrossRef] [Green Version]
- Karmakar, R.; Lee, D. Reactions of Arynes Promoted by Silver Ions. Chem. Soc. Rev. 2016, 45, 4459–4470. [Google Scholar] [CrossRef]
- Zhang, J.; Niu, D.; Brinker, V.A.; Hoye, T.R. The Phenol–Ene Reaction: Biaryl Synthesis via Trapping Reactions between HDDA-Generated Benzynes and Phenolics. Org. Lett. 2016, 18, 5596–5599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, S.; Xie, P.; Xia, Y.; Lee, D. Reactivity and Selectivity in the Intermolecular Alder–Ene Reactions of Arynes with Functionalized Alkenes. Org. Lett. 2017, 19, 5162–5165. [Google Scholar] [CrossRef]
- Karmakar, R.; Mamidipalli, P.; Yun, S.Y.; Lee, D. Alder-Ene Reactions of Arynes. Org. Lett. 2013, 15, 1938–1941. [Google Scholar] [CrossRef] [PubMed]
- Karmakar, R.; Le, A.; Xie, P.; Xia, Y.; Lee, D. Reactivity of Arynes for Arene Dearomatization. Org. Lett. 2018, 20, 4168–4172. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Lin, Y.; Xia, Y.; Wink, D.J.; Lee, D. Alder-Ene Reactions Driven by High Steric Strain and Bond Angle Distortion to Form Benzocyclobutenes. Chem. Sci. 2019, 10, 2212–2217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, A.; Lee, D. Selectivity between an Alder-ene Reaction and a [2+2] Cycloaddition in the Intramolecular Reactions of Allene-Tethered Arynes. Org. Chem. Front. 2021, 8, 3390–3397. [Google Scholar] [CrossRef]
- Xiao, X.; Hoye, T.R. The Domino Hexadehydro-Diels–Alder Reaction Transforms Polyynes to Benzynes to Naphthynes to Anthracynes to Tetracynes (and Beyond?). Nat. Chem. 2018, 10, 838–844. [Google Scholar] [CrossRef]
- Ross, S.P.; Baire, B.; Hoye, T.R. Mechanistic Duality in Tertiary Amine Additions to Thermally Generated Hexadehydro-Diels–Alder Benzynes. Org. Lett. 2017, 19, 5705–5708. [Google Scholar] [CrossRef]
- Arora, S.; Zhang, J.; Pogula, V.; Hoye, T.R. Reactions of Thermally Generated Benzynes with Six-Membered N-Heteroaromatics: Pathway and Product Diversity. Chem. Sci. 2019, 10, 9069–9076. [Google Scholar] [CrossRef] [Green Version]
- Arora, S.; Palani, V.; Hoye, T.R. Reactions of Diaziridines with Benzynes Give N-Arylhydrazones. Org. Lett. 2018, 20, 8082–8085. [Google Scholar] [CrossRef]
- Sneddon, D.S.; Hoye, T.R. Arylhydrazine Trapping of Benzynes: Mechanistic Insights and a Route to Azoarenes. Org. Lett. 2021, 23, 3432–3436. [Google Scholar] [CrossRef] [PubMed]
- Arora, S.; Sneddon, D.S.; Hoye, T.R. Reactions of HDDA Benzynes with C,N-Diarylimines (ArCH = NAr’). Eur. J. Org. Chem. 2020, 2020, 2379–2383. [Google Scholar] [CrossRef]
- Zhang, J.; Hoye, T.R. Divergent Reactivity during the Trapping of Benzynes by Glycidol Analogs: Ring Cleavage via Pinacol-Like Rearrangements vs. Oxirane Fragmentations. Org. Lett. 2019, 21, 2615–2619. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Oswood, C.J.; Hoye, T.R. Trapping of Hexadehydro-Diels–Alder Benzynes with Exocyclic, Conjugated Enals as a Route to Fused Spirocyclic Benzopyran Motifs. Synlett 2017, 28, 2933–2935. [Google Scholar] [CrossRef] [Green Version]
- Ritts, C.B.; Hoye, T.R. Sulfurane [S(IV)]-Mediated Fusion of Benzynes Leads to Helical Dibenzofurans. J. Am. Chem. Soc. 2021, 143, 13501–13506. [Google Scholar] [CrossRef]
- Chen, J.; Palani, V.; Hoye, T.R. Reactions of HDDA-Derived Benzynes with Sulfides: Mechanism, Modes, and Three-Component Reactions. J. Am. Chem. Soc. 2016, 138, 4318–4321. [Google Scholar] [CrossRef] [Green Version]
- Palani, V.; Chen, J.; Hoye, T.R. Reactions of Hexadehydro-Diels–Alder (HDDA)-Derived Benzynes with Thioamides: Synthesis of Dihydrobenzothiazino-Heterocyclics. Org. Lett. 2016, 18, 6312–6315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, T.; Curran, D.P.; Taniguchi, T. Hydroboration of Arynes Formed by Hexadehydro Diels–Alder Cyclizations with N-Heterocyclic Carbene Boranes. Org. Lett. 2015, 17, 3450–3453. [Google Scholar] [CrossRef]
- Yun, S.Y.; Wang, K.P.; Lee, N.-K.; Mamidipalli, P.; Lee, D. Alkane C–H Insertion by Aryne Intermediates with a Silver Catalyst. J. Am. Chem. Soc. 2013, 135, 4668–4671. [Google Scholar] [CrossRef]
- Mamidipalli, P.; Yun, S.Y.; Wang, K.-P.; Zhou, T.; Xia, Y.; Lee, D. Formal Hydrogenation of Arynes with Silyl Cβ–H Bonds as an Active Hydride Source. Chem. Sci. 2014, 5, 2362–2367. [Google Scholar] [CrossRef]
- Lee, N.-K.; Yun, S.Y.; Mamidipalli, P.; Salzman, R.M.; Lee, D.; Zhou, T.; Xia, Y. Hydroarylation of Arynes Catalyzed by Silver for Biaryl Synthesis. J. Am. Chem. Soc. 2014, 136, 4363–4368. [Google Scholar] [CrossRef] [PubMed]
- Ghorai, S.; Lee, D. Aryne Formation via the Hexadehydro Diels–Alder Reaction and their Ritter-Type Transformations Catalyzed by a Cationic Silver Complex. Tetrahedron 2017, 73, 4062–4069. [Google Scholar] [CrossRef]
- Ghorai, S.; Lin, Y.; Xia, Y.; Wink, D.J.; Lee, D. Silver-Catalyzed Annulation of Arynes with Nitriles for Synthesis of Structurally Diverse Quinazolines. Org. Lett. 2020, 22, 626–630. [Google Scholar] [CrossRef]
- Karmakar, R.; Ghorai, S.; Xia, Y.; Lee, D. Synthesis of Phenolic Compounds by Trapping Arynes with a Hydroxy Surrogate. Molecules 2015, 20, 15862–15880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.-P.; Yun, S.Y.; Mamidipalli, P.; Lee, D. Silver-Mediated Fluorination, Trifluoromethylation, and Trifluoromethylthiolation of Arynes. Chem. Sci. 2013, 4, 3205–3211. [Google Scholar] [CrossRef]
- Karmakar, R.; Wang, K.P.; Yun, S.Y.; Mamidipalli, P.; Lee, D. Hydrohalogenative Aromatization of Multiynes Promoted by Ruthenium Alkylidene Complexes. Org. Biomol. Chem. 2016, 14, 4782–4788. [Google Scholar] [CrossRef]
- Xiao, X.; Wang, T.; Xu, F.; Hoye, T.R. CuI-Mediated Bromoalkynylation and Hydroalkynylation Reactions of Unsymmetrical Benzynes: Complementary Modes of Addition. Angew. Chem. Int. Ed. 2018, 57, 16564–16568. [Google Scholar] [CrossRef]


















































































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
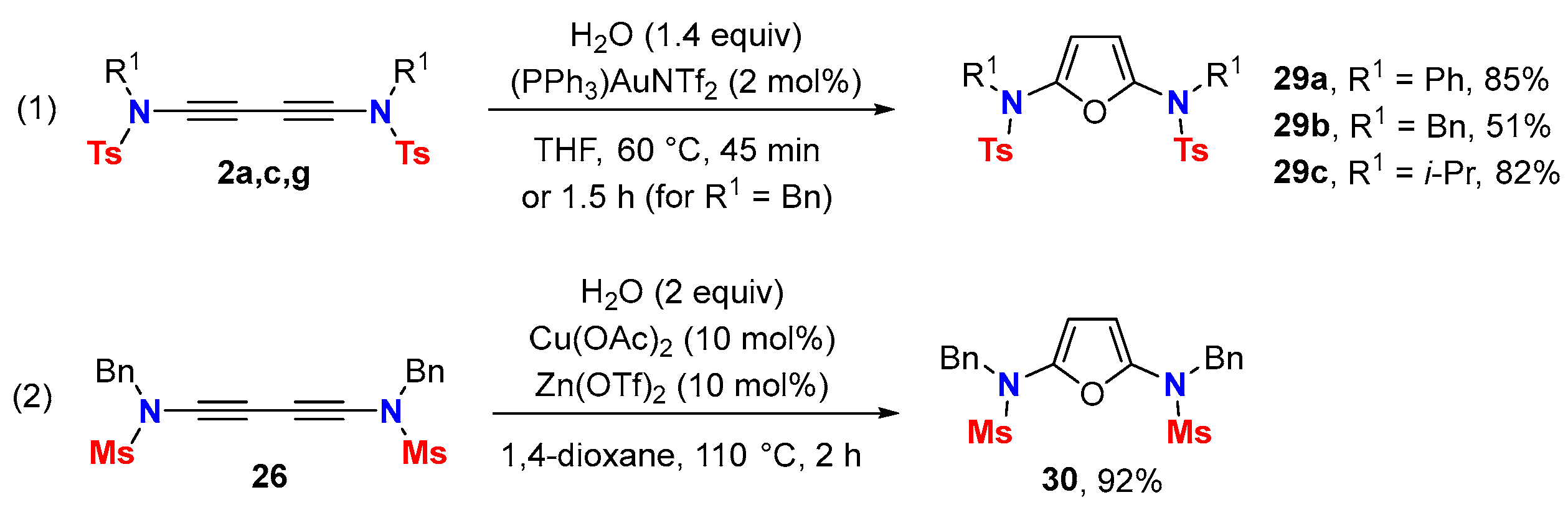{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
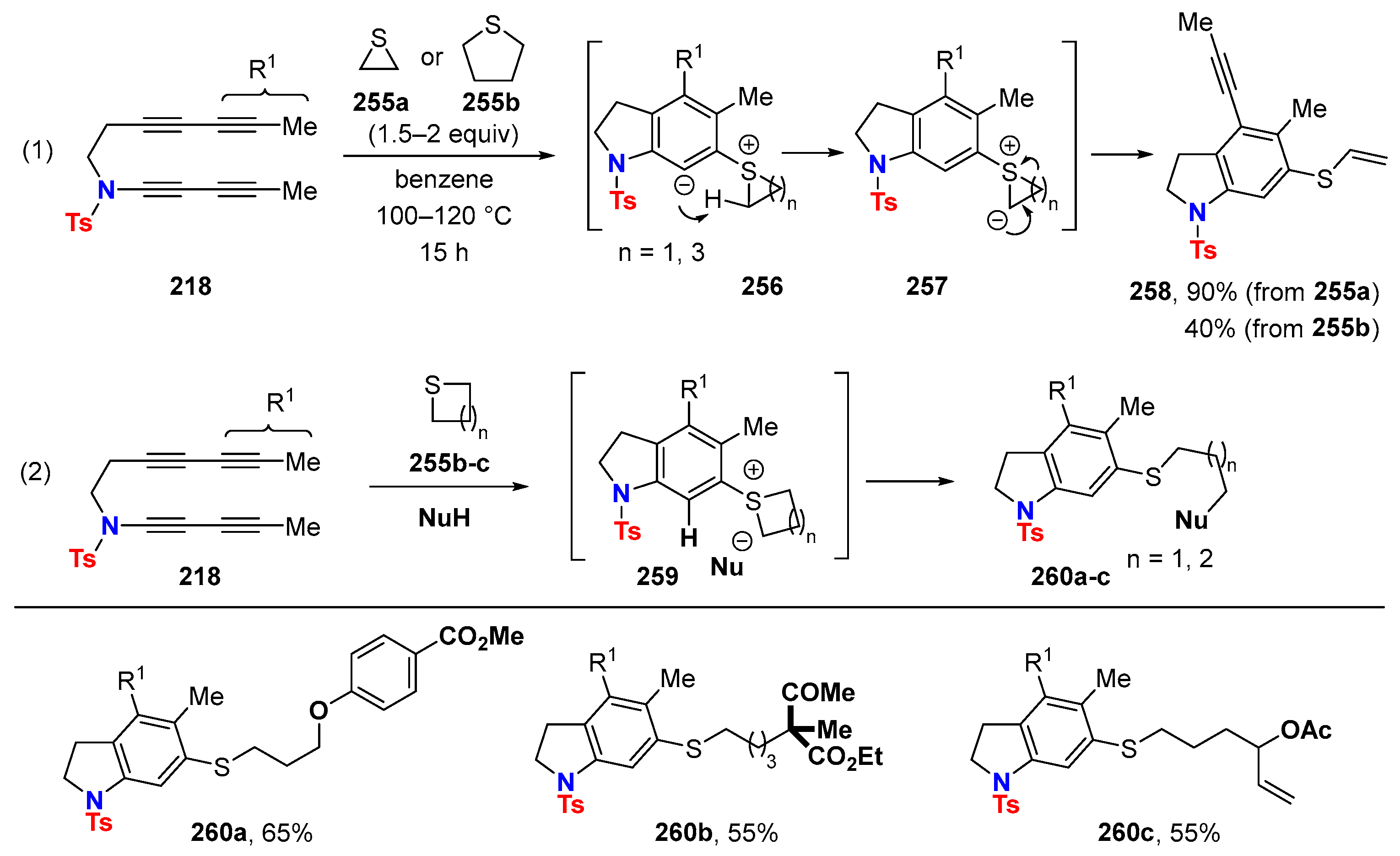{kind=link}
{kind=link}
{kind=link}
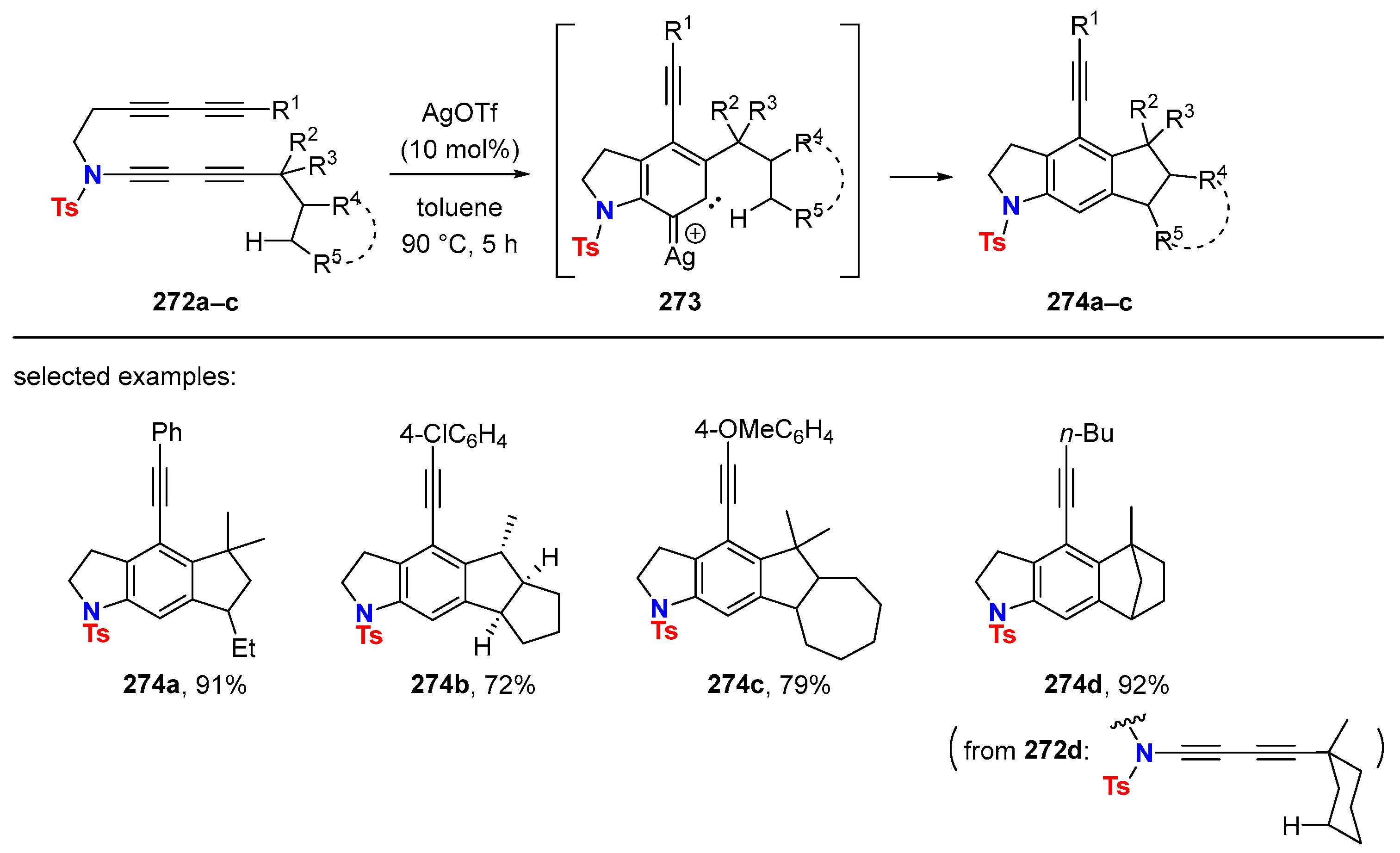{kind=link}
{kind=link}
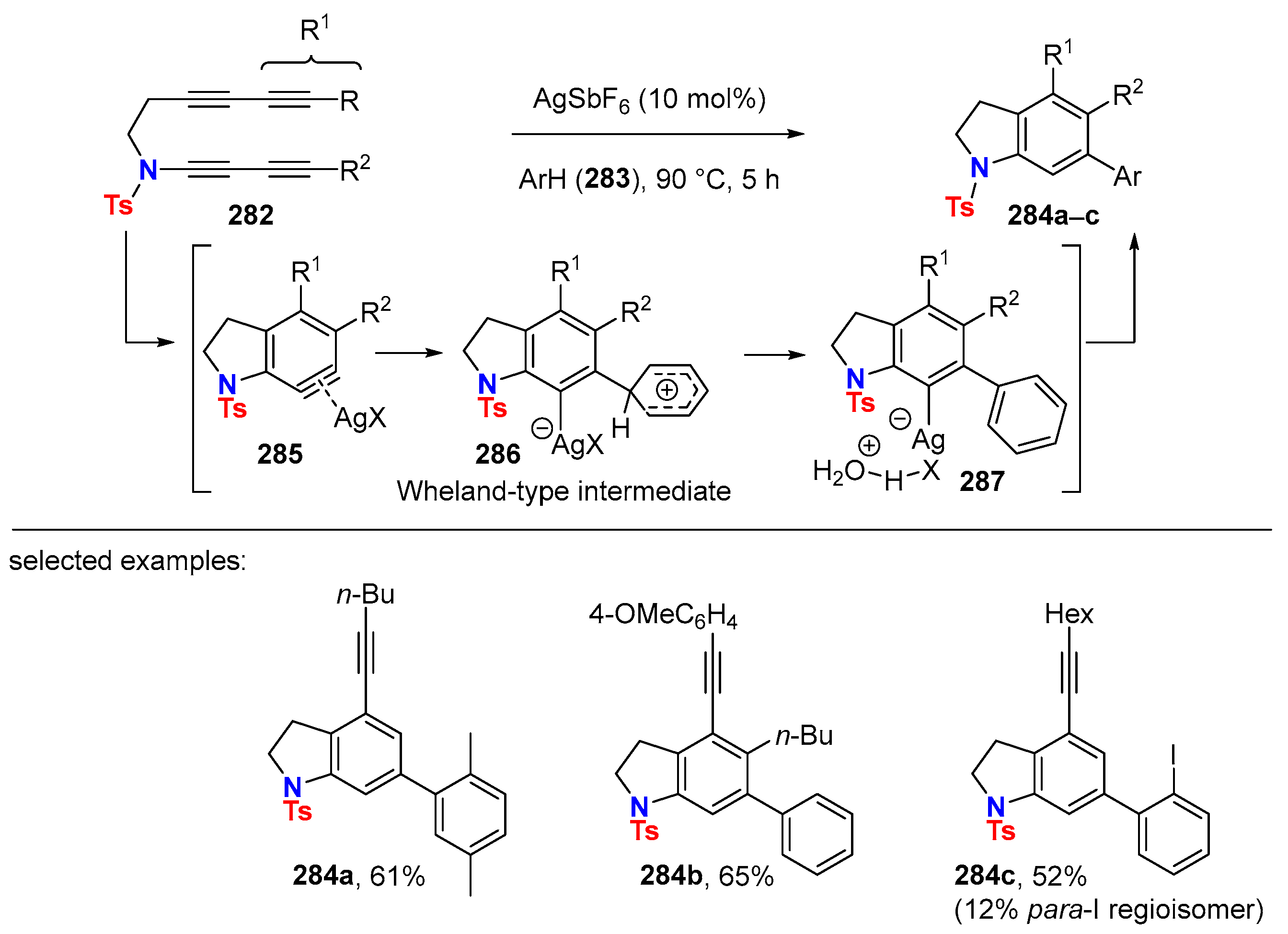{kind=link}
{kind=link}
{kind=link}
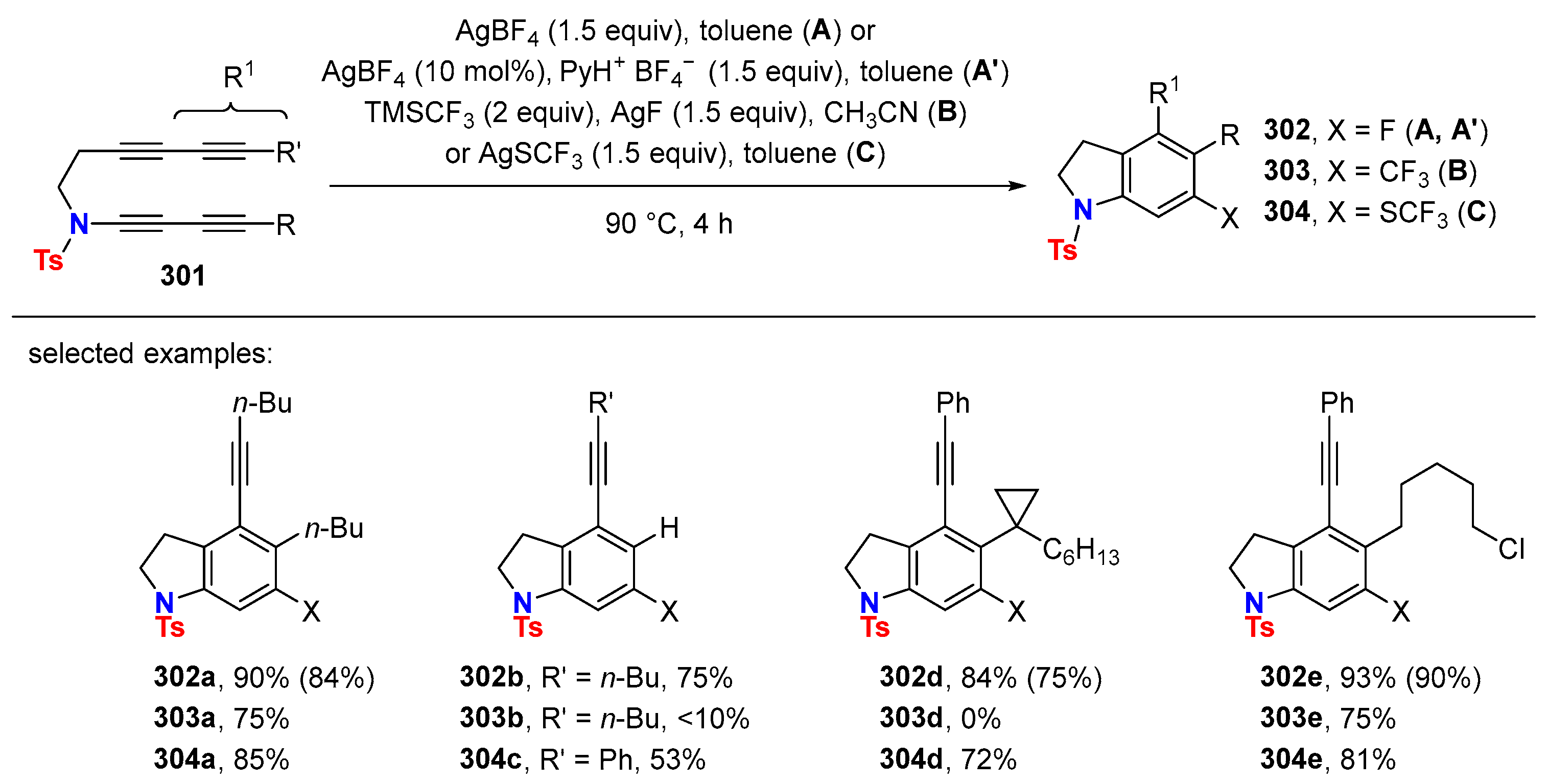{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ynamide | μg | Δaμ | μaFC |
|---|---|---|---|
| [10−30 Cm] in 1,4-dioxane | |||
 | 9.7 | 74.6 | 84.3 |
 | 9.5 | 92.3 | 101.8 |
 | 10.9 | 30.8 | 41.7 |
 | 11.8 | 69.7 | 81.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lenko, I.; Alayrac, C.; Bożek, I.; Witulski, B. 1,3-Butadiynamides the Ethynylogous Ynamides: Synthesis, Properties and Applications in Heterocyclic Chemistry. Molecules 2023, 28, 4564. https://doi.org/10.3390/molecules28114564
Lenko I, Alayrac C, Bożek I, Witulski B. 1,3-Butadiynamides the Ethynylogous Ynamides: Synthesis, Properties and Applications in Heterocyclic Chemistry. Molecules. 2023; 28(11):4564. https://doi.org/10.3390/molecules28114564
Chicago/Turabian StyleLenko, Illia, Carole Alayrac, Igor Bożek, and Bernhard Witulski. 2023. "1,3-Butadiynamides the Ethynylogous Ynamides: Synthesis, Properties and Applications in Heterocyclic Chemistry" Molecules 28, no. 11: 4564. https://doi.org/10.3390/molecules28114564