
cMolGPT: A Conditional Generative Pre-Trained Transformer for Target-Specific De Novo Molecular Generation

Abstract
:1. Introduction
2. Results and Discussion
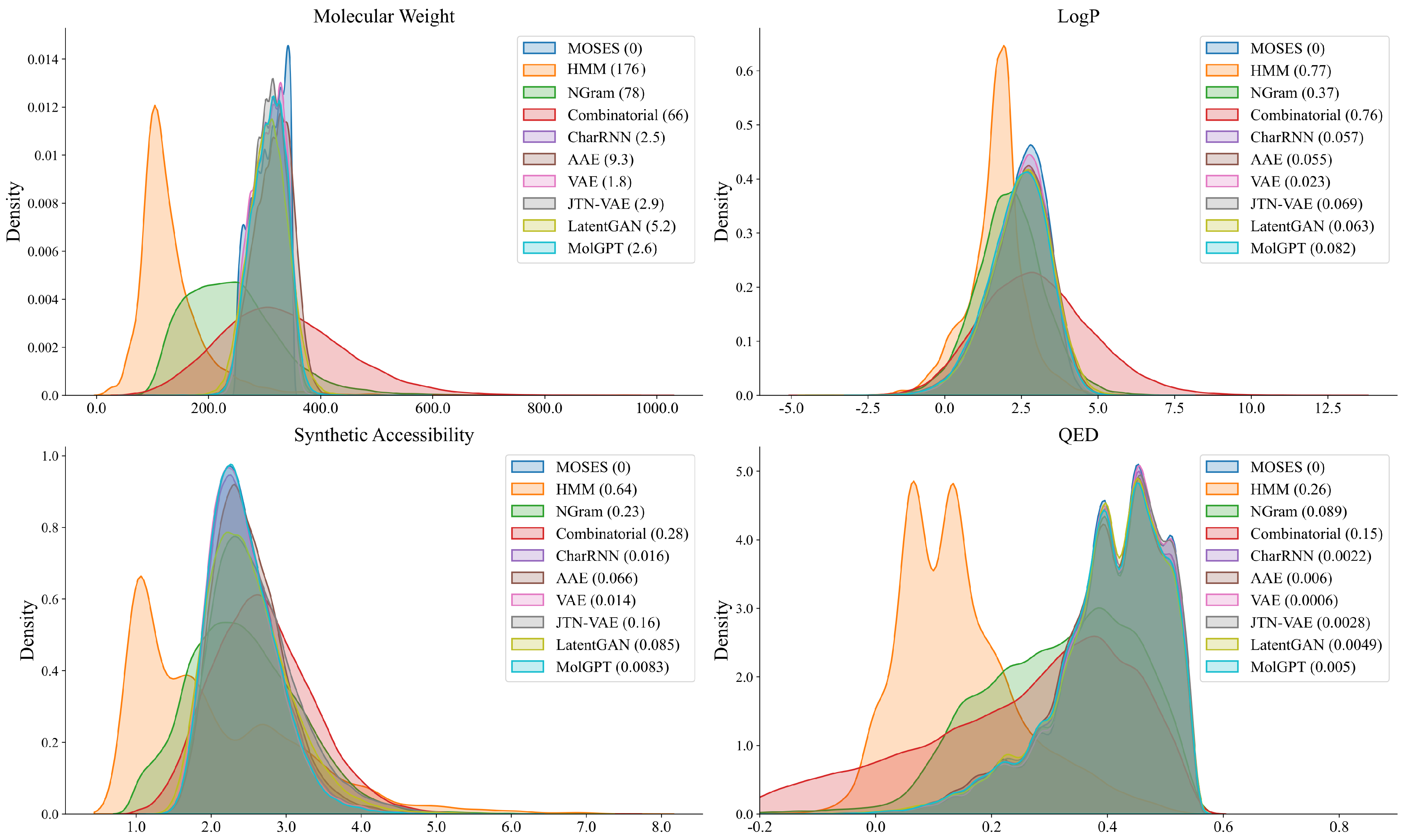
2.1. Generating Compound Libraries Using Pre-Trained cMolGPT
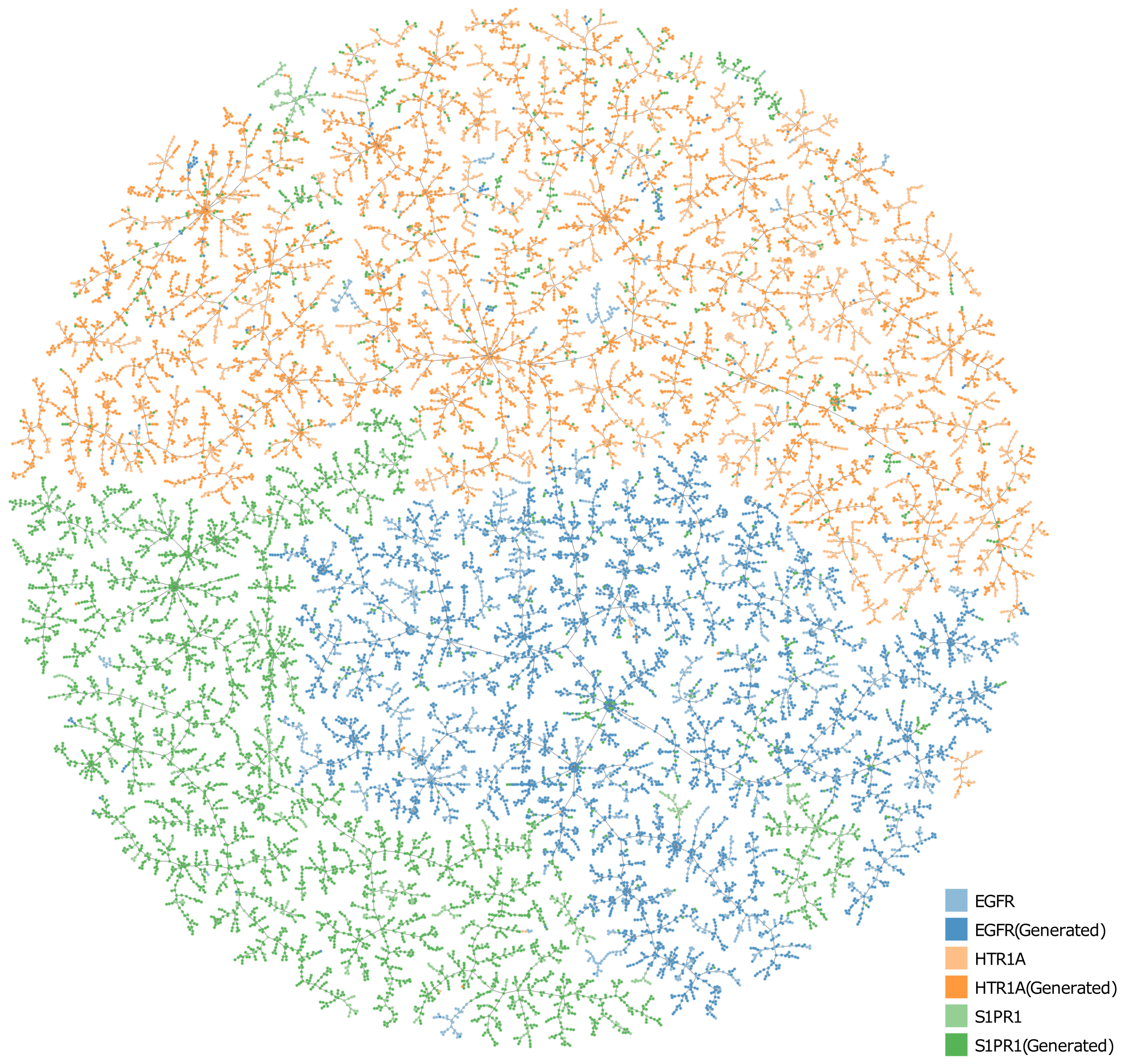
2.2. Generating Target-Specific Compound Libraries Using Conditional MolGPT
3. Methods and Materials
3.1. Problem Formulation
3.2. Transformer
3.3. Unsupervised Generative Pre-Training
3.4. Conditional Generative Pre-Trained Transformer
3.5. Workflow for Training and Sampling of the cMolGPT
- We first pre-trained the base model of cMolGPT by setting the target-specific embeddings as zero embeddings (without feeding target-specific information) on the MOSES database, as shown in Figure 5A. We did not place any target constraints on the sequential generation and solely focused on learning the drug-like structure from the data.
- To incorporate the target-specific information, we fine-tuned cMolGPT using <compound, target> pairs, which involved enforcing the conditions of the corresponding target by feeding target-specific embeddings to the attention layer as “memories”, as shown in Figure 5B. We used data from [33], where each SMILES sequence is manually tagged with a target (e.g., target proteins), indicating the specific physicochemical property of the small molecule.
- We generated a drug-like structure by auto-regressively sampling tokens from the trained decoder, as shown in Figure 5C. Optionally, we enforced the desired target by incorporating a target-specific embedding. The new generation will condition the target-specific information and likely has the desired property. The target-specific embeddings are denoted as .
3.6. Likelihood of Molecular Sequential Generation
3.7. Molecular Dataset and Target-Specific Dataset
3.8. ML-Based QSAR Model for Active Scoring
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Schneider, G.; Fechner, U. Computer-based de novo design of drug-like molecules. Nat. Rev. Drug Discov. 2005, 4, 649–663. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem substance and compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef] [PubMed]
- Reymond, J.L.; Van Deursen, R.; Blum, L.C.; Ruddigkeit, L. Chemical space as a source for new drugs. MedChemComm 2010, 1, 30–38. [Google Scholar] [CrossRef]
- Cheng, T.; Li, Q.; Zhou, Z.; Wang, Y.; Bryant, S.H. Structure-based virtual screening for drug discovery: A problem-centric review. AAPS J. 2012, 14, 133–141. [Google Scholar] [CrossRef]
- Scior, T.; Bender, A.; Tresadern, G.; Medina-Franco, J.L.; Martínez-Mayorga, K.; Langer, T.; Cuanalo-Contreras, K.; Agrafiotis, D.K. Recognizing pitfalls in virtual screening: A critical review. J. Chem. Inf. Model. 2012, 52, 867–881. [Google Scholar] [CrossRef]
- Shoichet, B.K. Virtual screening of chemical libraries. Nature 2004, 432, 862–865. [Google Scholar] [CrossRef]
- Gómez-Bombarelli, R.; Wei, J.N.; Duvenaud, D.; Hernández-Lobato, J.M.; Sánchez-Lengeling, B.; Sheberla, D.; Aguilera-Iparraguirre, J.; Hirzel, T.D.; Adams, R.P.; Aspuru-Guzik, A. Automatic chemical design using a data-driven continuous representation of molecules. ACS Cent. Sci. 2018, 4, 268–276. [Google Scholar] [CrossRef]
- Zhavoronkov, A.; Ivanenkov, Y.A.; Aliper, A.; Veselov, M.S.; Aladinskiy, V.A.; Aladinskaya, A.V.; Terentiev, V.A.; Polykovskiy, D.A.; Kuznetsov, M.D.; Asadulaev, A.; et al. Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat. Biotechnol. 2019, 37, 1038–1040. [Google Scholar] [CrossRef]
- Bilodeau, C.; Jin, W.; Jaakkola, T.; Barzilay, R.; Jensen, K.F. Generative models for molecular discovery: Recent advances and challenges. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2022, 12, e1608. [Google Scholar] [CrossRef]
- Cerchia, C.; Lavecchia, A. New avenues in artificial-intelligence-assisted drug discovery. Drug Discov. Today 2023, 28, 103516. [Google Scholar] [CrossRef]
- Polykovskiy, D.; Zhebrak, A.; Sanchez-Lengeling, B.; Golovanov, S.; Tatanov, O.; Belyaev, S.; Kurbanov, R.; Artamonov, A.; Aladinskiy, V.; Veselov, M.; et al. Molecular sets (MOSES): A benchmarking platform for molecular generation models. Front. Pharmacol. 2020, 11, 565644. [Google Scholar] [CrossRef]
- Brown, N.; Fiscato, M.; Segler, M.H.; Vaucher, A.C. GuacaMol: Benchmarking models for de novo molecular design. J. Chem. Inf. Model. 2019, 59, 1096–1108. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Ryu, S.; Kim, J.W.; Kim, W.Y. Molecular generative model based on conditional variational autoencoder for de novo molecular design. J. Cheminform. 2018, 10, 31. [Google Scholar] [CrossRef] [PubMed]
- Yuan, W.; Jiang, D.; Nambiar, D.K.; Liew, L.P.; Hay, M.P.; Bloomstein, J.; Lu, P.; Turner, B.; Le, Q.T.; Tibshirani, R.; et al. Chemical space mimicry for drug discovery. J. Chem. Inf. Model. 2017, 57, 875–882. [Google Scholar] [CrossRef] [PubMed]
- Bjerrum, E.J.; Threlfall, R. Molecular generation with recurrent neural networks (RNNs). arXiv 2017, arXiv:1705.04612. [Google Scholar]
- Gupta, A.; Müller, A.T.; Huisman, B.J.; Fuchs, J.A.; Schneider, P.; Schneider, G. Generative recurrent networks for de novo drug design. Mol. Inform. 2018, 37, 1700111. [Google Scholar] [CrossRef]
- Segler, M.H.; Kogej, T.; Tyrchan, C.; Waller, M.P. Generating focused molecule libraries for drug discovery with recurrent neural networks. ACS Cent. Sci. 2018, 4, 120–131. [Google Scholar] [CrossRef]
- Tong, X.; Liu, X.; Tan, X.; Li, X.; Jiang, J.; Xiong, Z.; Xu, T.; Jiang, H.; Qiao, N.; Zheng, M. Generative models for De Novo drug design. J. Med. Chem. 2021, 64, 14011–14027. [Google Scholar] [CrossRef]
- Arús-Pous, J.; Blaschke, T.; Ulander, S.; Reymond, J.L.; Chen, H.; Engkvist, O. Exploring the GDB-13 chemical space using deep generative models. J. Cheminform. 2019, 11, 20. [Google Scholar] [CrossRef]
- Kotsias, P.C.; Arús-Pous, J.; Chen, H.; Engkvist, O.; Tyrchan, C.; Bjerrum, E.J. Direct steering of de novo molecular generation with descriptor conditional recurrent neural networks. Nat. Mach. Intell. 2020, 2, 254–265. [Google Scholar] [CrossRef]
- He, J.; You, H.; Sandström, E.; Nittinger, E.; Bjerrum, E.J.; Tyrchan, C.; Czechtizky, W.; Engkvist, O. Molecular optimization by capturing chemist’s intuition using deep neural networks. J. Cheminform. 2021, 13, 26. [Google Scholar] [CrossRef] [PubMed]
- Vaswani, A.; Shazeer, N.; Parmar, N.; Uszkoreit, J.; Jones, L.; Gomez, A.N.; Kaiser, Ł.; Polosukhin, I. Attention is all you need. In Proceedings of the Advances in Neural Information Processing Systems, Long Beach, CA, USA, 4–9 December 2017; pp. 5998–6008. [Google Scholar]
- Li, Y.; Zhang, L.; Liu, Z. Multi-objective de novo drug design with conditional graph generative model. J. Cheminform. 2018, 10, 33. [Google Scholar] [CrossRef] [PubMed]
- Popova, M.; Isayev, O.; Tropsha, A. Deep reinforcement learning for de novo drug design. Sci. Adv. 2018, 4, eaap7885. [Google Scholar] [CrossRef] [PubMed]
- Olivecrona, M.; Blaschke, T.; Engkvist, O.; Chen, H. Molecular de-novo design through deep reinforcement learning. J. Cheminform. 2017, 9, 48. [Google Scholar] [CrossRef]
- Radford, A.; Narasimhan, K.; Salimans, T.; Sutskever, I. Improving language understanding by generative pre-training. 2018; preprint. [Google Scholar]
- Radford, A.; Wu, J.; Child, R.; Luan, D.; Amodei, D.; Sutskever, I. Language models are unsupervised multitask learners. OpenAI Blog 2019, 1, 9. [Google Scholar]
- Brown, T.; Mann, B.; Ryder, N.; Subbiah, M.; Kaplan, J.D.; Dhariwal, P.; Neelakantan, A.; Shyam, P.; Sastry, G.; Askell, A.; et al. Language models are few-shot learners. Adv. Neural Inf. Process. Syst. 2020, 33, 1877–1901. [Google Scholar]
- Rabiner, L.; Juang, B. An introduction to hidden Markov models. IEEE ASSP Mag. 1986, 3, 4–16. [Google Scholar] [CrossRef]
- Kingma, D.P.; Welling, M. Auto-encoding variational bayes. arXiv 2013, arXiv:1312.6114. [Google Scholar]
- Makhzani, A.; Shlens, J.; Jaitly, N.; Goodfellow, I.; Frey, B. Adversarial autoencoders. arXiv 2015, arXiv:1511.05644. [Google Scholar]
- Jin, W.; Barzilay, R.; Jaakkola, T. Junction tree variational autoencoder for molecular graph generation. In Proceedings of the International Conference on Machine Learning, PMLR, Stockholm, Sweden, 10–15 July 2018; pp. 2323–2332. [Google Scholar]
- Prykhodko, O.; Johansson, S.V.; Kotsias, P.C.; Arús-Pous, J.; Bjerrum, E.J.; Engkvist, O.; Chen, H. A de novo molecular generation method using latent vector based generative adversarial network. J. Cheminform. 2019, 11, 74. [Google Scholar] [CrossRef]
- Probst, D.; Reymond, J.L. A probabilistic molecular fingerprint for big data settings. J. Cheminform. 2018, 10, 66. [Google Scholar] [CrossRef]
- Probst, D.; Reymond, J.L. Visualization of very large high-dimensional data sets as minimum spanning trees. J. Cheminform. 2020, 12, 12. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Jin, X.; Xuan, Y.; Zhou, X.; Chen, W.; Wang, Y.X.; Yan, X. Enhancing the locality and breaking the memory bottleneck of transformer on time series forecasting. Adv. Neural Inf. Process. Syst. 2019, 32, 5244–5254. [Google Scholar]
- Jiang, C.; Li, J.; Wang, W.; Ku, W.S. Modeling real estate dynamics using temporal encoding. In Proceedings of the 29th International Conference on Advances in Geographic Information Systems, Beijing, China, 2–5 November 2021; pp. 516–525. [Google Scholar]
- Liu, P.J.; Saleh, M.; Pot, E.; Goodrich, B.; Sepassi, R.; Kaiser, L.; Shazeer, N. Generating Wikipedia by Summarizing Long Sequences. In Proceedings of the 6th International Conference on Learning Representations, ICLR 2018, Vancouver, BC, Canada, 30 April–3 May 2018. [Google Scholar]
- Graves, A. Generating sequences with recurrent neural networks. arXiv 2013, arXiv:1308.0850. [Google Scholar]
- Sutskever, I.; Vinyals, O.; Le, Q.V. Sequence to sequence learning with neural networks. In Proceedings of the Advances in Neural Information Processing Systems, Montreal, QC, Canada, 8–12 December 2014; pp. 3104–3112. [Google Scholar]
- Sterling, T.; Irwin, J.J. ZINC 15–ligand discovery for everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Jeliazkova, N.; Chupakhin, V.; Golib-Dzib, J.F.; Engkvist, O.; Carlsson, L.; Wegner, J.; Ceulemans, H.; Georgiev, I.; Jeliazkov, V.; et al. ExCAPE-DB: An integrated large scale dataset facilitating Big Data analysis in chemogenomics. J. Cheminform. 2017, 9, 17. [Google Scholar] [CrossRef] [PubMed]
- Ke, G.; Meng, Q.; Finley, T.; Wang, T.; Chen, W.; Ma, W.; Ye, Q.; Liu, T.Y. Lightgbm: A highly efficient gradient boosting decision tree. Adv. Neural Inf. Process. Syst. 2017, 30, 3146–3154. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Frag | SNN | |||||
|---|---|---|---|---|---|---|---|
| Valid | Unique@1k | Unique@10k | Test | TestSF | Test | TestSF | |
| HMM | 0.076 | 0.623 | 0.567 | 0.575 | 0.568 | 0.388 | 0.38 |
| NGram | 0.238 | 0.974 | 0.922 | 0.985 | 0.982 | 0.521 | 0.5 |
| Combinatorial | 1.0 | 0.998 | 0.991 | 0.991 | 0.99 | 0.451 | 0.439 |
| CharRNN | 0.975 | 1.0 | 0.999 | 1.0 | 0.998 | 0.601 | 0.565 |
| AAE | 0.937 | 1.0 | 0.997 | 0.991 | 0.99 | 0.608 | 0.568 |
| VAE | 0.977 | 1.0 | 0.998 | 0.999 | 0.998 | 0.626 | 0.578 |
| JTN-VAE | 1.0 | 1.0 | 1.0 | 0.997 | 0.995 | 0.548 | 0.519 |
| LatentGAN | 0.897 | 1.0 | 0.997 | 0.999 | 0.998 | 0.538 | 0.514 |
| cMolGPT * | 0.988 | 1.0 | 0.999 | 1.0 | 0.998 | 0.619 | 0.578 |
| Target | Model | Valid | Unique@10k | Novel |
|---|---|---|---|---|
| EGFR | cRNN | 0.921 | 0.861 | 0.662 |
| cMolGPT | 0.885 | 0.940 | 0.898 | |
| HTR1A | cRNN | 0.922 | 0.844 | 0.498 |
| cMolGPT | 0.905 | 0.896 | 0.787 | |
| S1PR1 | cRNN | 0.926 | 0.861 | 0.514 |
| cMolGPT | 0.926 | 0.838 | 0.684 |
| EGFR | 215.07 | 781.24 | 21.06 | 168.92 | −0.82 | 12.15 | 0.00 | 8.00 | 3.00 | 14.00 |
| HTR1A | 176.09 | 664.35 | 3.24 | 157.04 | −0.85 | 8.92 | 0.00 | 6.00 | 1.00 | 12.00 |
| S1PR1 | 263.22 | 716.17 | 6.48 | 227.06 | −2.56 | 13.25 | 0.00 | 8.00 | 1.00 | 14.00 |
| MW ([200, 500]) | TPSA ([20, 130]) | LogP ([−1, 6]) | HBD ([ , 5]) | HBA ([ , 10]) | QED ([0.4, ]) | SA ([ , 5]) | |
|---|---|---|---|---|---|---|---|
| EGFR | 57.94% | 96.68% | 62.38% | 99.38% | 98.08% | 39.02% | 99.92% |
| HTR1A | 93.80% | 96.54% | 91.76% | 99.96% | 99.72% | 86.52% | 99.84% |
| S1PR1 | 72.12% | 81.62% | 80.26% | 97.66% | 96.46% | 34.06% | 99.96% |
| Target | QSAR | ||||
|---|---|---|---|---|---|
| # of Active Mols | # of Mols | pXC50 | R | RMSE | |
| EGFR | 1381 | 5181 | |||
| HTR1A | 3485 | 6332 | |||
| S1PR1 | 795 | 1400 | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Zhao, H.; Sciabola, S.; Wang, W. cMolGPT: A Conditional Generative Pre-Trained Transformer for Target-Specific De Novo Molecular Generation. Molecules 2023, 28, 4430. https://doi.org/10.3390/molecules28114430
Wang Y, Zhao H, Sciabola S, Wang W. cMolGPT: A Conditional Generative Pre-Trained Transformer for Target-Specific De Novo Molecular Generation. Molecules. 2023; 28(11):4430. https://doi.org/10.3390/molecules28114430
Chicago/Turabian StyleWang, Ye, Honggang Zhao, Simone Sciabola, and Wenlu Wang. 2023. "cMolGPT: A Conditional Generative Pre-Trained Transformer for Target-Specific De Novo Molecular Generation" Molecules 28, no. 11: 4430. https://doi.org/10.3390/molecules28114430