
Nitric Oxide Synthases in Rheumatoid Arthritis

Abstract
:1. Introduction
2. NO/NOS Pathway Overview
3. The Pathological Changes of RA
Histological Changes
4. The Pathogenesis of RA
4.1. RA and Autoimmunity
4.2. RA and Cytokines
5. NOS/NO Pathway in RA Pathogenesis
5.1. NOS/NO Pathway Involved in RA Autoimmunity
5.2. NOS/NO Pathway Involved in RA Oxidative Stress
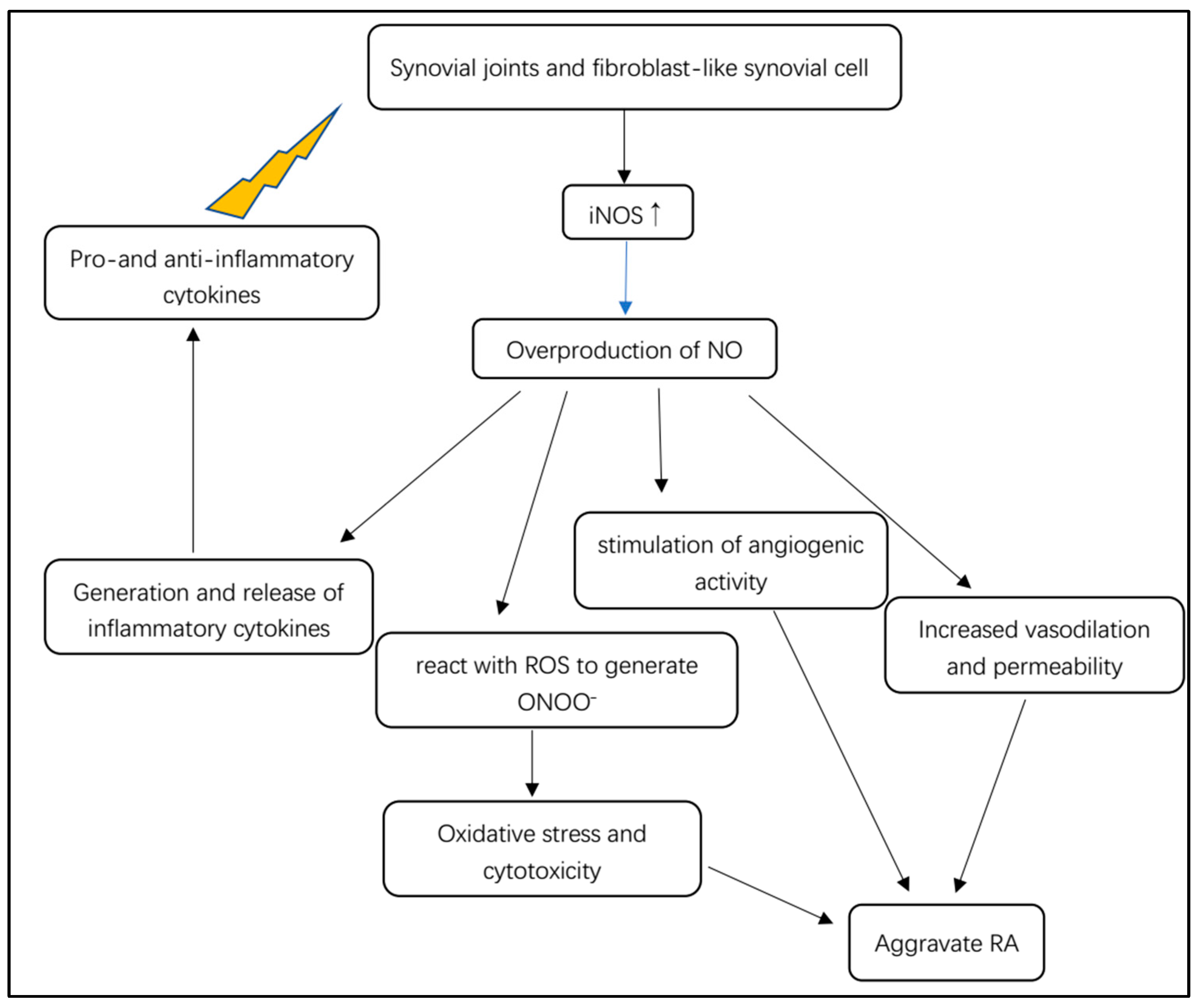
5.3. NOS/NO Pathway Involved in RA Inflammatory Cascade
5.4. NOS/NO Pathway Involved in RA Circadian Rhythm
5.5. NOS/NO Pathway Involved in RA Complications
5.5.1. The Effect of NOS/NO Pathway in Cardiovascular Manifestation of RA
5.5.2. The Effect of NOS/NO Pathway in Neurological Manifestations of RA
6. Disease-Modifying Antirheumatic Drugs Targeting NOS/NO Pathway
6.1. Conventional DMARDs
6.2. Novel DMARDs
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- England, B.R.; Roul, P.; Yang, Y.; Sayles, H.; Yu, F.; Michaud, K.; Xie, F.; Curtis, J.R.; Mikuls, T.R. Burden and trajectory of multimorbidity in rheumatoid arthritis: A matched cohort study from 2006 to 2015. Ann. Rheum. Dis. 2021, 80, 286–292. [Google Scholar] [CrossRef]
- Bullock, J.; Rizvi, S.A.A.; Saleh, A.M.; Ahmed, S.S.; Do, D.P.; Ansari, R.A.; Ahmed, J. Rheumatoid Arthritis: A Brief Overview of the Treatment. Med. Princ. Pract. 2018, 27, 501–507. [Google Scholar] [CrossRef]
- Serhal, L.; Lwin, M.N.; Holroyd, C.; Edwards, C.J. Rheumatoid arthritis in the elderly: Characteristics and treatment considerations. Autoimmun. Rev. 2020, 19, 102528. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, E.; Kondo, Y.; Kanazawa, N.; Akutsu, T.; Nishiyama, K.; Iizuka, T. Autoimmune Encephalitis as an Extra-articular Manifestation of Rheumatoid Arthritis. Intern. Med. 2019, 58, 1007–1009. [Google Scholar] [CrossRef]
- Napoli, C.; Paolisso, G.; Casamassimi, A.; Al-Omran, M.; Barbieri, M.; Sommese, L.; Infante, T.; Ignarro, L.J. Effects of nitric oxide on cell proliferation: Novel insights. J. Am. Coll. Cardiol. 2013, 62, 89–95. [Google Scholar] [CrossRef]
- Farrell, A.J.; Blake, D.R.; Palmer, R.M.; Moncada, S. Increased concentrations of nitrite in synovial fluid and serum samples suggest increased nitric oxide synthesis in rheumatic diseases. Ann. Rheum. Dis. 1992, 51, 1219–1222. [Google Scholar] [CrossRef] [PubMed]
- Nagy, G.; Koncz, A.; Telarico, T.; Fernandez, D.; Ersek, B.; Buzás, E.; Perl, A. Central role of nitric oxide in the pathogenesis of rheumatoid arthritis and systemic lupus erythematosus. Arthritis Res. Ther. 2010, 12, 210. [Google Scholar] [CrossRef] [PubMed]
- Onur, O.; Akinci, A.S.; Akbiyik, F.; Unsal, I. Elevated levels of nitrate in rheumatoid arthritis. Rheumatol. Int. 2001, 20, 154–158. [Google Scholar] [CrossRef]
- Pham, T.N.; Rahman, P.; Tobin, Y.M.; Khraishi, M.M.; Hamilton, S.F.; Alderdice, C.; Richardson, V.J. Elevated serum nitric oxide levels in patients with inflammatory arthritis associated with co-expression of inducible nitric oxide synthase and protein kinase C-eta in peripheral blood monocyte-derived macrophages. J. Rheumatol. 2003, 30, 2529–2534. [Google Scholar]
- Costa, D.; Benincasa, G.; Lucchese, R.; Infante, T.; Nicoletti, G.F.; Napoli, C. Effect of nitric oxide reduction on arterial thrombosis. Scand. Cardiovasc. J. 2019, 53, 1–8. [Google Scholar] [CrossRef]
- Król, M.; Kepinska, M. Human Nitric Oxide Synthase-Its Functions, Polymorphisms, and Inhibitors in the Context of Inflammation, Diabetes and Cardiovascular Diseases. Int. J. Mol. Sci. 2020, 22, 56. [Google Scholar] [CrossRef] [PubMed]
- Mondal, P.; Ishigami, I.; Yeh, S.R.; Wijeratne, G.B. The Role of Heme Peroxo Oxidants in the Rational Mechanistic Modeling of Nitric Oxide Synthase: Characterization of Key Intermediates and Elucidation of the Mechanism. Angew. Chem. (Int. Ed. Engl.) 2022, 61, e202211521. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.Y.; Hong, F.F.; Yang, S.L. The Roles of Nitric Oxide Synthase/Nitric Oxide Pathway in the Pathology of Vascular Dementia and Related Therapeutic Approaches. Int. J. Mol. Sci. 2021, 22, 4540. [Google Scholar] [CrossRef]
- Thai, T.; Zhong, F.; Dang, L.; Chan, E.; Ku, J.; Malle, E.; Geczy, C.L.; Keaney, J.F., Jr.; Thomas, S.R. Endothelial-transcytosed myeloperoxidase activates endothelial nitric oxide synthase via a phospholipase C-dependent calcium signaling pathway. Free Radic. Biol. Med. 2021, 166, 255–264. [Google Scholar] [CrossRef]
- Yeh, C.F.; Cheng, S.H.; Lin, Y.S.; Shentu, T.P.; Huang, R.T.; Zhu, J.; Chen, Y.T.; Kumar, S.; Lin, M.S.; Kao, H.L.; et al. Targeting mechanosensitive endothelial TXNDC5 to stabilize eNOS and reduce atherosclerosis in vivo. Sci. Adv. 2022, 8, eabl8096. [Google Scholar] [CrossRef]
- Saini, R.; Azam, Z.; Sapra, L.; Srivastava, R.K. Neuronal Nitric Oxide Synthase (nNOS) in Neutrophils: An Insight. Rev. Physiol. Biochem. Pharmacol. 2021, 180, 49–83. [Google Scholar] [CrossRef] [PubMed]
- Balke, J.E.; Zhang, L.; Percival, J.M. Neuronal nitric oxide synthase (nNOS) splice variant function: Insights into nitric oxide signaling from skeletal muscle. Nitric Oxide 2019, 82, 35–47. [Google Scholar] [CrossRef]
- Scallan, J.P.; Bouta, E.M.; Rahimi, H.; Kenney, H.M.; Ritchlin, C.T.; Davis, M.J.; Schwarz, E.M. Ex vivo Demonstration of Functional Deficiencies in Popliteal Lymphatic Vessels From TNF-Transgenic Mice with Inflammatory Arthritis. Front. Physiol. 2021, 12, 745096. [Google Scholar] [CrossRef]
- Hong, F.F.; Liang, X.Y.; Liu, W.; Lv, S.; He, S.J.; Kuang, H.B.; Yang, S.L. Roles of eNOS in atherosclerosis treatment. Inflamm. Res. 2019, 68, 429–441. [Google Scholar] [CrossRef]
- Radi, R. Oxygen radicals, nitric oxide, and peroxynitrite: Redox pathways in molecular medicine. Proc. Natl. Acad. Sci. USA 2018, 115, 5839–5848. [Google Scholar] [CrossRef]
- Shu, X.; Keller, T.C.T.; Begandt, D.; Butcher, J.T.; Biwer, L.; Keller, A.S.; Columbus, L.; Isakson, B.E. Endothelial nitric oxide synthase in the microcirculation. Cell. Mol. Life Sci. 2015, 72, 4561–4575. [Google Scholar] [CrossRef]
- Lind, M.; Hayes, A.; Caprnda, M.; Petrovic, D.; Rodrigo, L.; Kruzliak, P.; Zulli, A. Inducible nitric oxide synthase: Good or bad? Biomed. Pharmacother. 2017, 93, 370–375. [Google Scholar] [CrossRef]
- Cinelli, M.A.; Do, H.T.; Miley, G.P.; Silverman, R.B. Inducible nitric oxide synthase: Regulation, structure, and inhibition. Med. Res. Rev. 2020, 40, 158–189. [Google Scholar] [CrossRef]
- Kondo, T.; Otsuka, Y.; Aoki, H.; Goto, Y.; Kawaguchi, Y.; Waguri-Nagaya, Y.; Miyazawa, K.; Goto, S.; Aoyama, M. The Inducible Nitric Oxide Synthase Pathway Promotes Osteoclastogenesis under Hypoxic Culture Conditions. Am. J. Pathol. 2021, 191, 2072–2079. [Google Scholar] [CrossRef]
- Zaichko, K.; Zaichko, N.; Maievskyi, O.; Korotkyi, O.; Falalyeyeva, T.; Fagoonee, S.; Pellicano, R.; Abenavoli, L.; Stanislavchuk, M. Circadian Rhythms of Endothelial Nitric Oxide Synthase and Toll-like Receptors 2 Production in Females with Rheumatoid Arthritis Depending on NOS3 Gene Polymorphism. Rev. Recent Clin. Trials 2020, 15, 145–151. [Google Scholar] [CrossRef]
- Dariushnejad, H.; Chodari, L.; Sedighi, M.; Akbari, S.; Ghorbanzadeh, V. Rheumatoid arthritis: Current therapeutics compendium. Endocr. Regul. 2022, 56, 148–162. [Google Scholar] [CrossRef]
- Lin, Y.J.; Anzaghe, M.; Schülke, S. Update on the Pathomechanism, Diagnosis, and Treatment Options for Rheumatoid Arthritis. Cells 2020, 9, 880. [Google Scholar] [CrossRef] [PubMed]
- Cascão, R.; Moura, R.A.; Perpétuo, I.; Canhão, H.; Vieira-Sousa, E.; Mourão, A.F.; Rodrigues, A.M.; Polido-Pereira, J.; Queiroz, M.V.; Rosário, H.S.; et al. Identification of a cytokine network sustaining neutrophil and Th17 activation in untreated early rheumatoid arthritis. Arthritis Res. Ther. 2010, 12, R196. [Google Scholar] [CrossRef]
- Komatsu, N.; Takayanagi, H. Mechanisms of joint destruction in rheumatoid arthritis—Immune cell-fibroblast-bone interactions. Nat. Rev. Rheumatol. 2022, 18, 415–429. [Google Scholar] [CrossRef] [PubMed]
- Hugon, J. Rheumatoid arthritis and cognitive decline. Jt. Bone Spine 2022, 89, 105346. [Google Scholar] [CrossRef] [PubMed]
- Dey, P.; Panga, V.; Raghunathan, S. A Cytokine Signalling Network for the Regulation of Inducible Nitric Oxide Synthase Expression in Rheumatoid Arthritis. PLoS ONE 2016, 11, e0161306. [Google Scholar] [CrossRef]
- Li, R.L.; Duan, H.X.; Liang, Q.; Huang, Y.L.; Wang, L.Y.; Zhang, Q.; Wu, C.J.; Liu, S.Q.; Peng, W. Targeting matrix metalloproteases: A promising strategy for herbal medicines to treat rheumatoid arthritis. Front. Immunol. 2022, 13, 1046810. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Mao, M.; Nian, F. The Effect of TNF-α on CHD and the Relationship between TNF-α Antagonist and CHD in Rheumatoid Arthritis: A Systematic Review. Cardiol. Res. Pract. 2022, 2022, 6192053. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Takahashi, N.; Jimi, E.; Udagawa, N.; Takami, M.; Kotake, S.; Nakagawa, N.; Kinosaki, M.; Yamaguchi, K.; Shima, N.; et al. Tumor necrosis factor alpha stimulates osteoclast differentiation by a mechanism independent of the ODF/RANKL-RANK interaction. J. Exp. Med. 2000, 191, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Marahleh, A.; Kitaura, H.; Ohori, F.; Kishikawa, A.; Ogawa, S.; Shen, W.R.; Qi, J.; Noguchi, T.; Nara, Y.; Mizoguchi, I. TNF-α Directly Enhances Osteocyte RANKL Expression and Promotes Osteoclast Formation. Front. Immunol. 2019, 10, 2925. [Google Scholar] [CrossRef]
- Chabaud, M.; Lubberts, E.; Joosten, L.; van Den Berg, W.; Miossec, P. IL-17 derived from juxta-articular bone and synovium contributes to joint degradation in rheumatoid arthritis. Arthritis Res. 2001, 3, 168–177. [Google Scholar] [CrossRef]
- Chabaud, M.; Garnero, P.; Dayer, J.M.; Guerne, P.A.; Fossiez, F.; Miossec, P. Contribution of interleukin 17 to synovium matrix destruction in rheumatoid arthritis. Cytokine 2000, 12, 1092–1099. [Google Scholar] [CrossRef] [PubMed]
- Kokkonen, H.; Söderström, I.; Rocklöv, J.; Hallmans, G.; Lejon, K.; Rantapää Dahlqvist, S. Up-regulation of cytokines and chemokines predates the onset of rheumatoid arthritis. Arthritis Rheum. 2010, 62, 383–391. [Google Scholar] [CrossRef]
- Tang, M.; Tian, L.; Luo, G.; Yu, X. Interferon-Gamma-Mediated Osteoimmunology. Front. Immunol. 2018, 9, 1508. [Google Scholar] [CrossRef]
- Lasselin, J. Back to the future of psychoneuroimmunology: Studying inflammation-induced sickness behavior. Brain Behav. Immun.-Health 2021, 18, 100379. [Google Scholar] [CrossRef]
- Paulo, M.; Costa, D.; Bonaventura, D.; Lunardi, C.N.; Bendhack, L.M. Nitric Oxide Donors as Potential Drugs for the Treatment of Vascular Diseases Due to Endothelium Dysfunction. Curr. Pharm. Des. 2020, 26, 3748–3759. [Google Scholar] [CrossRef] [PubMed]
- Angiolilli, C.; Kabala, P.A.; Grabiec, A.M.; Van Baarsen, I.M.; Ferguson, B.S.; García, S.; Malvar Fernandez, B.; McKinsey, T.A.; Tak, P.P.; Fossati, G.; et al. Histone deacetylase 3 regulates the inflammatory gene expression programme of rheumatoid arthritis fibroblast-like synoviocytes. Ann. Rheum. Dis. 2017, 76, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Cao, Y.; Ma, L.; Pei, H.; Rausch, W.D.; Li, H. Dysfunction of Cerebrovascular Endothelial Cells: Prelude to Vascular Dementia. Front. Aging Neurosci. 2018, 10, 376. [Google Scholar] [CrossRef]
- Nagy, G.; Clark, J.M.; Buzas, E.I.; Gorman, C.L.; Cope, A.P. Nitric oxide, chronic inflammation and autoimmunity. Immunol. Lett. 2007, 111, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Ibiza, S.; Víctor, V.M.; Boscá, I.; Ortega, A.; Urzainqui, A.; O’Connor, J.E.; Sánchez-Madrid, F.; Esplugues, J.V.; Serrador, J.M. Endothelial nitric oxide synthase regulates T cell receptor signaling at the immunological synapse. Immunity 2006, 24, 753–765. [Google Scholar] [CrossRef] [PubMed]
- Talaat, R.M.; Mohamed, S.F.; Bassyouni, I.H.; Raouf, A.A. Th1/Th2/Th17/Treg cytokine imbalance in systemic lupus erythematosus (SLE) patients: Correlation with disease activity. Cytokine 2015, 72, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Okeke, E.B.; Uzonna, J.E. The Pivotal Role of Regulatory T Cells in the Regulation of Innate Immune Cells. Front. Immunol. 2019, 10, 680. [Google Scholar] [CrossRef]
- Beltran, B.; Quintero, M.; Garcia-Zaragoza, E.; O’Connor, E.; Esplugues, J.V.; Moncada, S. Inhibition of mitochondrial respiration by endogenous nitric oxide: A critical step in Fas signaling. Proc. Natl. Acad. Sci. USA 2002, 99, 8892–8897. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Xiao, X.H.; Hu, L.B.; Jie, H.Y.; Wang, Y.; Ye, W.C.; Li, M.M.; Liu, Z. Anhuienoside C Ameliorates Collagen-Induced Arthritis through Inhibition of MAPK and NF-κB Signaling Pathways. Front. Pharmacol. 2017, 8, 299. [Google Scholar] [CrossRef]
- Costa, N.T.; Scavuzzi, B.M.; Iriyoda, T.M.V.; Lozovoy, M.A.B.; Alfieri, D.F.; de Medeiros, F.A.; de Sa, M.C.; Micheletti, P.L.; Sekiguchi, B.A.; Reiche, E.M.V.; et al. Metabolic syndrome and the decreased levels of uric acid by leflunomide favor redox imbalance in patients with rheumatoid arthritis. Clin. Exp. Med. 2018, 18, 363–372. [Google Scholar] [CrossRef]
- Grandvuillemin, I.; Buffat, C.; Boubred, F.; Lamy, E.; Fromonot, J.; Charpiot, P.; Simoncini, S.; Sabatier, F.; Dignat-George, F.; Peyter, A.C.; et al. Arginase upregulation and eNOS uncoupling contribute to impaired endothelium-dependent vasodilation in a rat model of intrauterine growth restriction. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 315, R509–R520. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.; Sang, X.; Cheng, H. Total glucosides of peony induce fibroblast-like synovial apoptosis, and ameliorate cartilage injury via blocking the NF-κB/STAT3 pathway. Ann. Transl. Med. 2022, 10, 51. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Ji, S. Cellular senescence: Molecular mechanisms and pathogenicity. J. Cell. Physiol. 2018, 233, 9121–9135. [Google Scholar] [CrossRef] [PubMed]
- Behera, J.; Nagarajan, S.; Saran, U.; Kumar, R.; Keshri, G.K.; Suryakumar, G.; Chatterjee, S. Nitric oxide restores peripheral blood mononuclear cell adhesion against hypoxia via NO-cGMP signalling. Cell Biochem. Funct. 2020, 38, 319–329. [Google Scholar] [CrossRef]
- Liu, X.Z.; Gao, Y.; Qi, K.; Zhao, D.B. Expression of dishevelled-2 in cartilage of rheumatoid arthritis and its effect on cartilage destruction. Zhonghua Nei Ke Za Zhi 2018, 57, 674–678. [Google Scholar] [CrossRef]
- Zaichko, K.; Stanislavchuk, M.; Zaichko, N. Circadian fluctuations of endothelial nitric oxide synthase activity in females with rheumatoid arthritis: A pilot study. Rheumatol. Int. 2020, 40, 549–554. [Google Scholar] [CrossRef]
- Zaichko, K.; Stanislavchuk, M.; Zaichko, N.; Khomenko, V. Associations between efficacy of the therapy and circadian fluctuations of endothelial nitric oxide synthase with toll-like receptors 2 expression, and nos3 polymorphism in females with rheumatoid arthritis. Georgian Med. News 2020, 299, 93–100. [Google Scholar]
- Luczak, A.; Madej, M.; Kasprzyk, A.; Doroszko, A. Role of the eNOS Uncoupling and the Nitric Oxide Metabolic Pathway in the Pathogenesis of Autoimmune Rheumatic Diseases. Oxid. Med. Cell. Longev. 2020, 2020, 1417981. [Google Scholar] [CrossRef]
- Zafari, P.; Zarifian, A.; Alizadeh-Navaei, R.; Taghadosi, M.; Rafiei, A.; Samimi, Z.; Niksolat, F. Asymmetric and symmetric dimethylarginine concentration as an indicator of cardiovascular diseases in rheumatoid arthritis patients: A systematic review and meta-analysis of case-control studies. Clin. Rheumatol. 2020, 39, 127–134. [Google Scholar] [CrossRef]
- Mangoni, A.A.; Tommasi, S.; Sotgia, S.; Zinellu, A.; Paliogiannis, P.; Piga, M.; Cauli, A.; Pintus, G.; Carru, C.; Erre, G.L. Asymmetric Dimethylarginine: A Key Player in the Pathophysiology of Endothelial Dysfunction, Vascular Inflammation and Atherosclerosis in Rheumatoid Arthritis? Curr. Pharm. Des. 2021, 27, 2131–2140. [Google Scholar] [CrossRef]
- Dimitroulas, T.; Kitas, G.D. Genetic regulation of dimethylarginines and endothelial dysfunction in rheumatoid arthritis. Amino Acids 2019, 51, 983–990. [Google Scholar] [CrossRef]
- Pankowski, D.; Wytrychiewicz-Pankowska, K.; Janowski, K.; Pisula, E. Cognitive impairment in patients with rheumatoid arthritis: A systematic review and meta-analysis. Jt. Bone Spine 2022, 89, 105298. [Google Scholar] [CrossRef] [PubMed]
- DeQuattro, K.; Imboden, J.B. Neurologic Manifestations of Rheumatoid Arthritis. Rheum. Dis. Clin. N. Am. 2017, 43, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Joaquim, A.F.; Appenzeller, S. Neuropsychiatric manifestations in rheumatoid arthritis. Autoimmun. Rev. 2015, 14, 1116–1122. [Google Scholar] [CrossRef] [PubMed]
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef]
- Ally, A.; Powell, I.; Ally, M.M.; Chaitoff, K.; Nauli, S.M. Role of neuronal nitric oxide synthase on cardiovascular functions in physiological and pathophysiological states. Nitric Oxide 2020, 102, 52–73. [Google Scholar] [CrossRef]
- Tan, X.L.; Xue, Y.Q.; Ma, T.; Wang, X.; Li, J.J.; Lan, L.; Malik, K.U.; McDonald, M.P.; Dopico, A.M.; Liao, F.F. Partial eNOS deficiency causes spontaneous thrombotic cerebral infarction, amyloid angiopathy and cognitive impairment. Mol. Neurodegener. 2015, 10, 24. [Google Scholar] [CrossRef]
- Minhas, R.; Bansal, Y.; Bansal, G. Inducible nitric oxide synthase inhibitors: A comprehensive update. Med. Res. Rev. 2020, 40, 823–855. [Google Scholar] [CrossRef]
- Totoson, P.; Maguin-Gaté, K.; Prigent-Tessier, A.; Monnier, A.; Verhoeven, F.; Marie, C.; Wendling, D.; Demougeot, C. Etanercept improves endothelial function via pleiotropic effects in rat adjuvant-induced arthritis. Rheumatology 2016, 55, 1308–1317. [Google Scholar] [CrossRef]
- Yamashita, M. Auranofin: Past to Present, and repurposing. Int. Immunopharmacol. 2021, 101 Pt B, 108272. [Google Scholar] [CrossRef]
- Zhang, Z.; Ma, X.; Zha, Z.; Zhao, Z.; Li, J. The protective effects of allopurinol against IL-17A-induced inflammatory response in mast cells. Mol. Immunol. 2022, 141, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Gaafar, A.G.A.; Messiha, B.A.S.; Abdelkafy, A.M.L. Nicorandil and theophylline can protect experimental rats against complete Freund’s adjuvant-induced rheumatoid arthritis through modulation of JAK/STAT/RANKL signaling pathway. Eur. J. Pharmacol. 2018, 822, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, Z.; Meng, Q.; Zhang, P.; Yan, P.; Zhang, Z.; Zhang, H.; Pan, J.; Zhai, Y.; Liu, Y.; et al. Chronic Calcium Channel Inhibitor Verapamil Antagonizes TNF-α-Mediated Inflammatory Reaction and Protects Against Inflammatory Arthritis in Mice. Inflammation 2016, 39, 1624–1634. [Google Scholar] [CrossRef]
- Azouz, A.A.; Saleh, E.; Abo-Saif, A.A. Aliskiren, tadalafil, and cinnamaldehyde alleviate joint destruction biomarkers; MMP-3 and RANKL; in complete Freund’s adjuvant arthritis model: Downregulation of IL-6/JAK2/STAT3 signaling pathway. Saudi Pharm. J. 2020, 28, 1101–1111. [Google Scholar] [CrossRef] [PubMed]
- Moustafa Ahmed, Y.; Shehata Messiha, B.A.; El-Sayed El-Daly, M.; Abo-Saif, A.A. Effects of ticagrelor, empagliflozin and tamoxifen against experimentally-induced vascular reactivity defects in rats in vivo and in vitro. Pharmacol. Rep. 2019, 71, 1034–1043. [Google Scholar] [CrossRef]
- Pfänder, P.; Fidan, M.; Burret, U.; Lipinski, L.; Vettorazzi, S. Cdk5 Deletion Enhances the Anti-inflammatory Potential of GC-Mediated GR Activation During Inflammation. Front. Immunol. 2019, 10, 1554. [Google Scholar] [CrossRef]
- Zhang, X.; Ye, G.; Wu, Z.; Zou, K.; He, X.; Xu, X.; Yao, J.; Wei, Q. The therapeutic effects of edaravone on collagen-induced arthritis in rats. J. Cell. Biochem. 2020, 121, 1463–1474. [Google Scholar] [CrossRef]
- Wahba, M.G.F.; Messiha, B.A.S.; El-Daly, M.E.; Abo-Saif, A.A. Vardenafil and cilostazol can improve vascular reactivity in rats with diabetes mellitus and rheumatoid arthritis co-morbidity. Life Sci. 2019, 229, 67–79. [Google Scholar] [CrossRef]
- Arroul-Lammali, A.; Rahal, F.; Chetouane, R.; Djeraba, Z.; Medjeber, O.; Ladjouze-Rezig, A.; Touil-Boukoffa, C. Ex vivo all-trans retinoic acid modulates NO production and regulates IL-6 effect during rheumatoid arthritis: A study in Algerian patients. Immunopharmacol. Immunotoxicol. 2017, 39, 87–96. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, R.; Li, L.; Zhu, L.; Gao, S.; Lu, Q.; Gu, Y.; Zhang, Y.; Yang, H.; Hou, T.; et al. Macrophage migration inhibitory factor (MIF) inhibitor, Z-590 suppresses cartilage destruction in adjuvant-induced arthritis via inhibition of macrophage inflammatory activation. Immunopharmacol. Immunotoxicol. 2018, 40, 149–157. [Google Scholar] [CrossRef]
- Kusuda, R.; Carreira, E.U.; Ulloa, L.; Cunha, F.Q.; Kanashiro, A.; Cunha, T.M. Choline attenuates inflammatory hyperalgesia activating nitric oxide/cGMP/ATP-sensitive potassium channels pathway. Brain Res. 2020, 1727, 146567. [Google Scholar] [CrossRef]
- Bai, J.; Ge, G.; Wang, Y.; Zhang, W.; Wang, Q.; Wang, W.; Guo, X.; Yu, B.; Xu, Y.; Yang, H.; et al. A selective CB(2) agonist protects against the inflammatory response and joint destruction in collagen-induced arthritis mice. Biomed. Pharmacother. 2019, 116, 109025. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, J.M.; Abdalla, H.B.; Basting, R.T.; Hammock, B.D.; Napimoga, M.H.; Clemente-Napimoga, J.T. Peripheral soluble epoxide hydrolase inhibition reduces hypernociception and inflammation in albumin-induced arthritis in temporomandibular joint of rats. Int. Immunopharmacol. 2020, 87, 106841. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Cheng, J.; Zong, S.; Yu, Y.; Wang, Y.; Song, Y.; He, R.; Yuan, S.; Chen, T.; Hu, M.; et al. The glycolysis inhibitor 2-deoxyglucose ameliorates adjuvant-induced arthritis by regulating macrophage polarization in an AMPK-dependent manner. Mol. Immunol. 2021, 140, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Minhas, R.; Bansal, G.; Bansal, Y. Novel Coupled Molecules from Active Structural Motifs of Synthetic and Natural Origin as Immunosuppressants. Med. Chem. 2020, 16, 544–554. [Google Scholar] [CrossRef] [PubMed]
- Emam, S.H.; Sonousi, A.; Osman, E.O.; Hwang, D.; Kim, G.D.; Hassan, R.A. Design and synthesis of methoxyphenyl- and coumarin-based chalcone derivatives as anti-inflammatory agents by inhibition of NO production and down-regulation of NF-κB in LPS-induced RAW264.7 macrophage cells. Bioorg. Chem. 2021, 107, 104630. [Google Scholar] [CrossRef]
- Shi, J.B.; Chen, L.Z.; Wang, B.S.; Huang, X.; Jiao, M.M.; Liu, M.M.; Tang, W.J.; Liu, X.H. Novel Pyrazolo[4,3-d]pyrimidine as Potent and Orally Active Inducible Nitric Oxide Synthase (iNOS) Dimerization Inhibitor with Efficacy in Rheumatoid Arthritis Mouse Model. J. Med. Chem. 2019, 62, 4013–4031. [Google Scholar] [CrossRef]
- Lee, Y.Z.; Guo, H.C.; Zhao, G.H.; Yang, C.W.; Chang, H.Y.; Yang, R.B.; Chen, L.; Lee, S.J. Tylophorine-based compounds are therapeutic in rheumatoid arthritis by targeting the caprin-1 ribonucleoprotein complex and inhibiting expression of associated c-Myc and HIF-1α. Pharmacol. Res. 2020, 152, 104581. [Google Scholar] [CrossRef]
- Cui, Z.; Lin, Y.; Liu, Y.; Cao, L.; Cui, L. Retinoic Acid-Platinum (II) Complex [RT-Pt(II)] Protects Against Rheumatoid Arthritis in Mice via MEK/Nuclear Factor kappa B (NF-κB) Pathway Downregulation. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2020, 26, e924787. [Google Scholar] [CrossRef]
- Khan, M.A.; Khan, M.J. Nano-gold displayed anti-inflammatory property via NF-kB pathways by suppressing COX-2 activity. Artif. Cells Nanomed. Biotechnol. 2018, 46 (Suppl. 1), 1149–1158. [Google Scholar] [CrossRef]
- Ansari, M.M.; Ahmad, A.; Kumar, A.; Alam, P.; Khan, T.H.; Jayamurugan, G.; Raza, S.S.; Khan, R. Aminocellulose-grafted-polycaprolactone coated gelatin nanoparticles alleviate inflammation in rheumatoid arthritis: A combinational therapeutic approach. Carbohydr. Polym. 2021, 258, 117600. [Google Scholar] [CrossRef] [PubMed]
- Tao, S.; Cheng, J.; Su, G.; Li, D.; Shen, Z.; Tao, F.; You, T.; Hu, J. Breathing Micelles for Combinatorial Treatment of Rheumatoid Arthritis. Angew. Chem. (Int. Ed. Engl.) 2020, 59, 21864–21869. [Google Scholar] [CrossRef] [PubMed]
- Yeo, J.; Lee, Y.M.; Lee, J.; Park, D.; Kim, K.; Kim, J.; Park, J.; Kim, W.J. Nitric Oxide-Scavenging Nanogel for Treating Rheumatoid Arthritis. Nano Lett. 2019, 19, 6716–6724. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Conventional | Novel | ||
|---|---|---|---|
| Name | Target | Name | Target |
| Etanercept | arginase-2 | MIF inhibitor compound (Z-590) | iNOS |
| Auranofin | Toll-like receptors | α7nachRs agonist (choline) | NO/cGMP/ATP-sensitive potassium channels |
| Allopurinol | NF-κB and iNOS | CB2 agonist (4Q3C) | iNOS |
| Indometacin | PPARγ-Akt-eNOS pathway | sEH enzyme inhibitor (TPPU) | iNOS |
| Nicocdil | iNOS and eNOS | glycolysis inhibitor (2-deoxyglucose) | NF-κB and iNOS |
| Aliskiren, tadalafil | NF-κB | coumarin moieties coupled to phenols (YR2e) | iNOS |
| Ticagrelor, Empagliflozin, Tamoxifen | eNOS | chalcone derivatives | NF-κB and phosphorylated Iκb |
| Roscovitine | iNOS | pyrimidine-based compounds tylophorine-based compounds | iNOS |
| Edaravone | NF-κB p65 | RT-Pt (II) | NF-κB and iNOS |
| Verapamil | NF-κB signaling pathway | AuNGs R-AuNPs | NF-κB and iNOS |
| All-trans retinoic acid | NF-κB and iNOS | PCL-AC | iNOS |
| Vardenafil and cilostazol | eNOS | BM NO-SCV gel | iNOS HO-1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, J.-B.; Chen, Z.-R.; Yang, S.-L.; Hong, F.-F. Nitric Oxide Synthases in Rheumatoid Arthritis. Molecules 2023, 28, 4414. https://doi.org/10.3390/molecules28114414
Huang J-B, Chen Z-R, Yang S-L, Hong F-F. Nitric Oxide Synthases in Rheumatoid Arthritis. Molecules. 2023; 28(11):4414. https://doi.org/10.3390/molecules28114414
Chicago/Turabian StyleHuang, Jia-Bao, Zhi-Ru Chen, Shu-Long Yang, and Fen-Fang Hong. 2023. "Nitric Oxide Synthases in Rheumatoid Arthritis" Molecules 28, no. 11: 4414. https://doi.org/10.3390/molecules28114414