
Added Complexity!—Mechanistic Aspects of Heterobimetallic Complexes for Application in Homogeneous Catalysis

Abstract
:1. Introduction: Multimetallic Catalysis: From Enzymes to Complexes

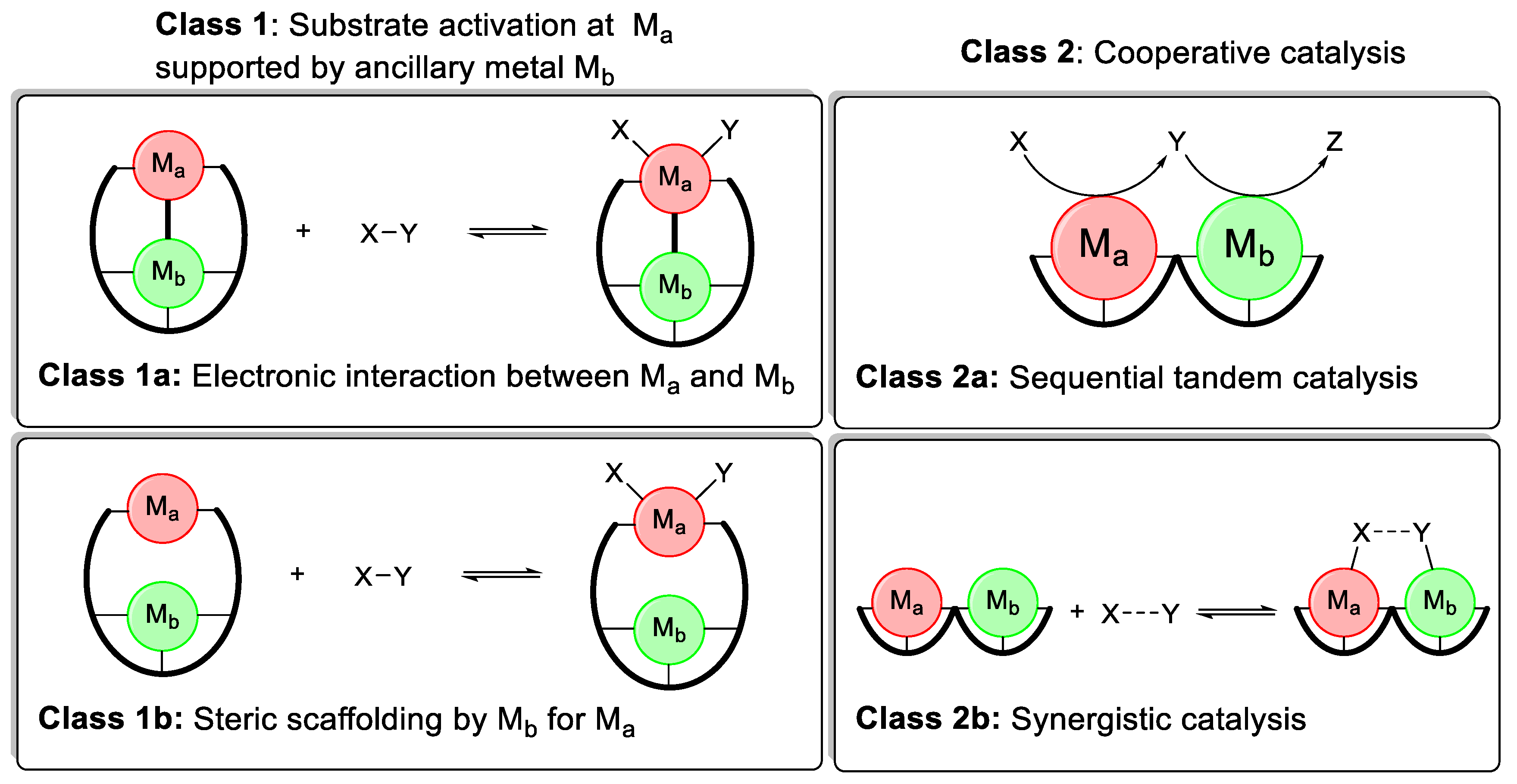
2. Heterobimetallic Complexes in Homogeneous Catalysis

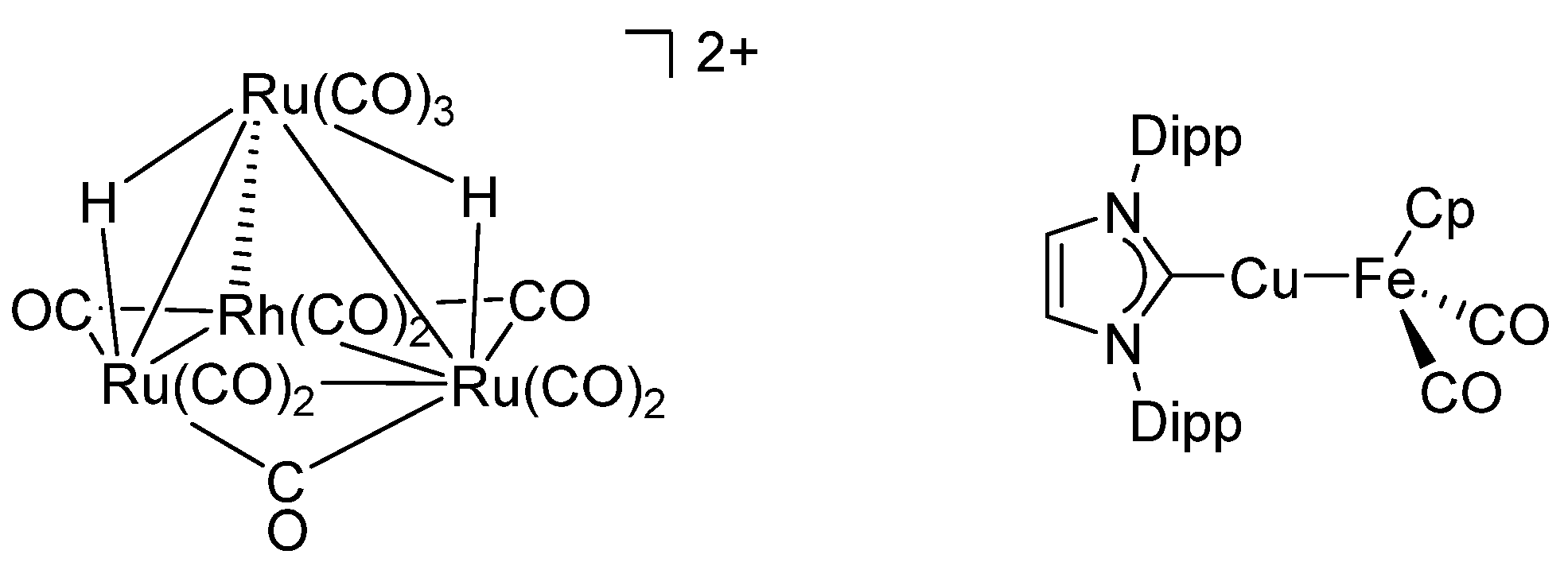
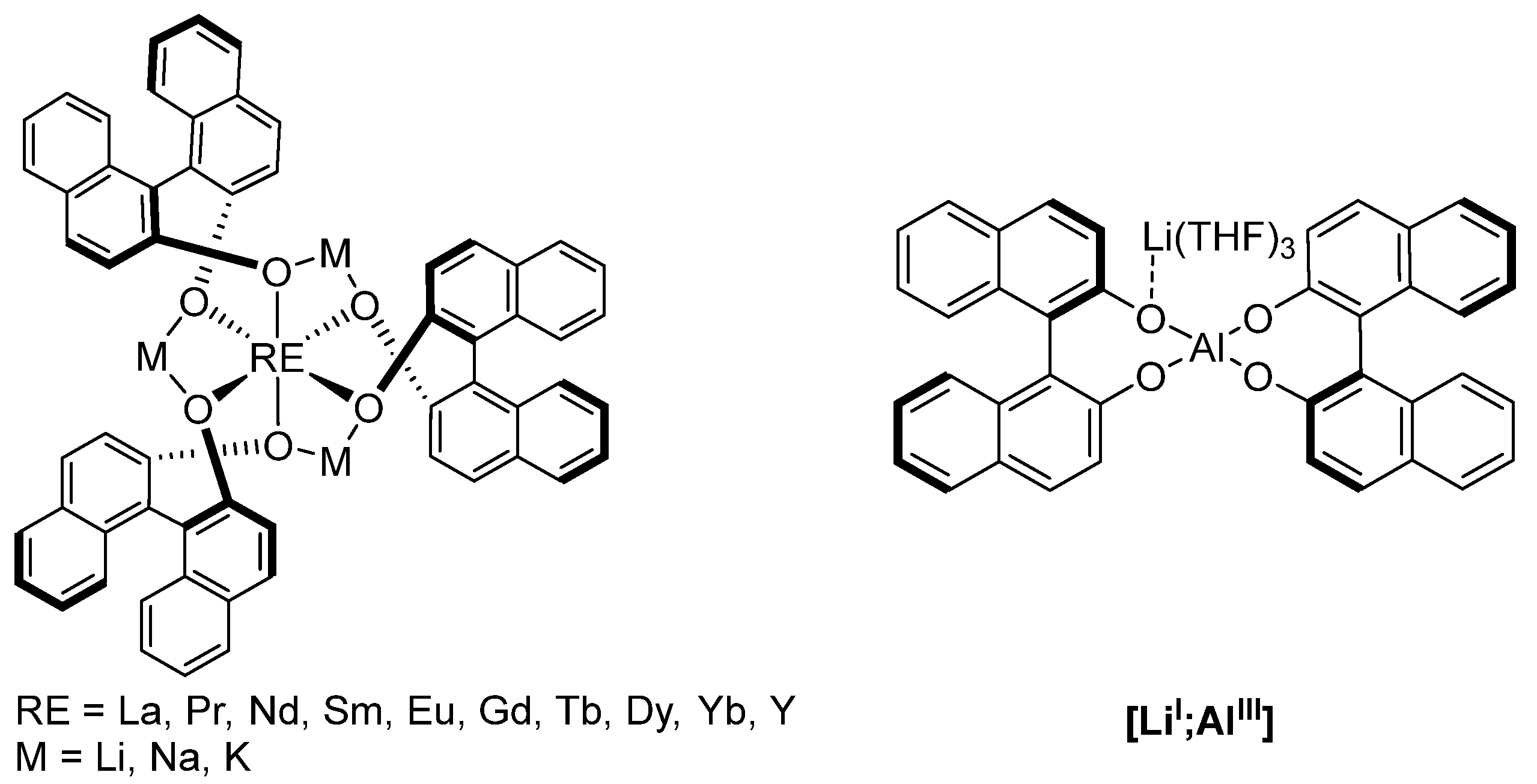
2.1. Electronic Interaction between Ma and Mb

2.2. Steric Scaffolding by Mb for Ma

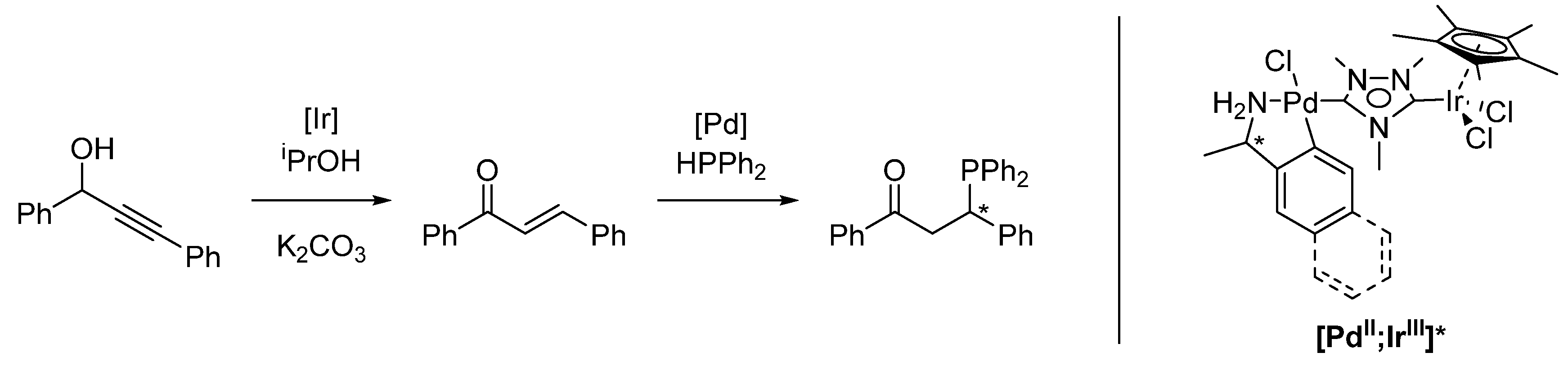
2.3. Sequential Tandem Catalysis
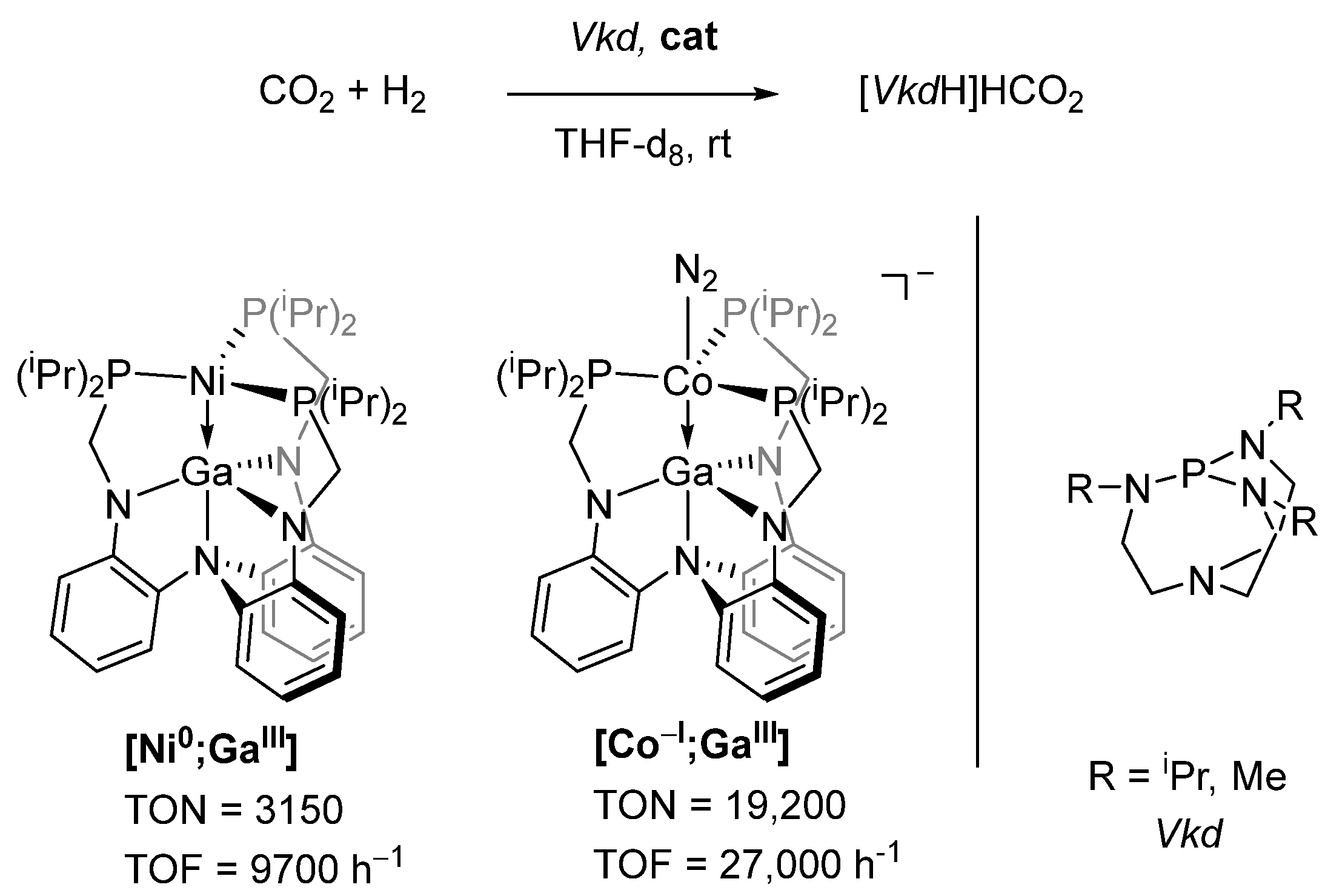
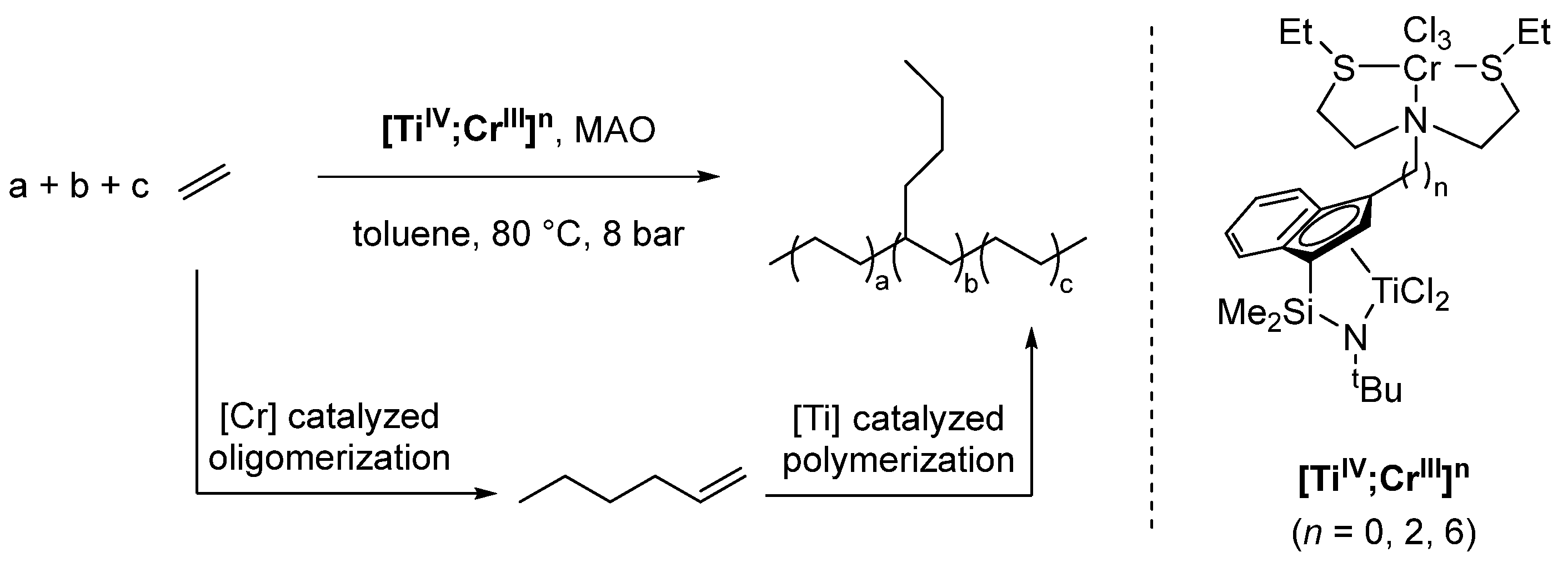
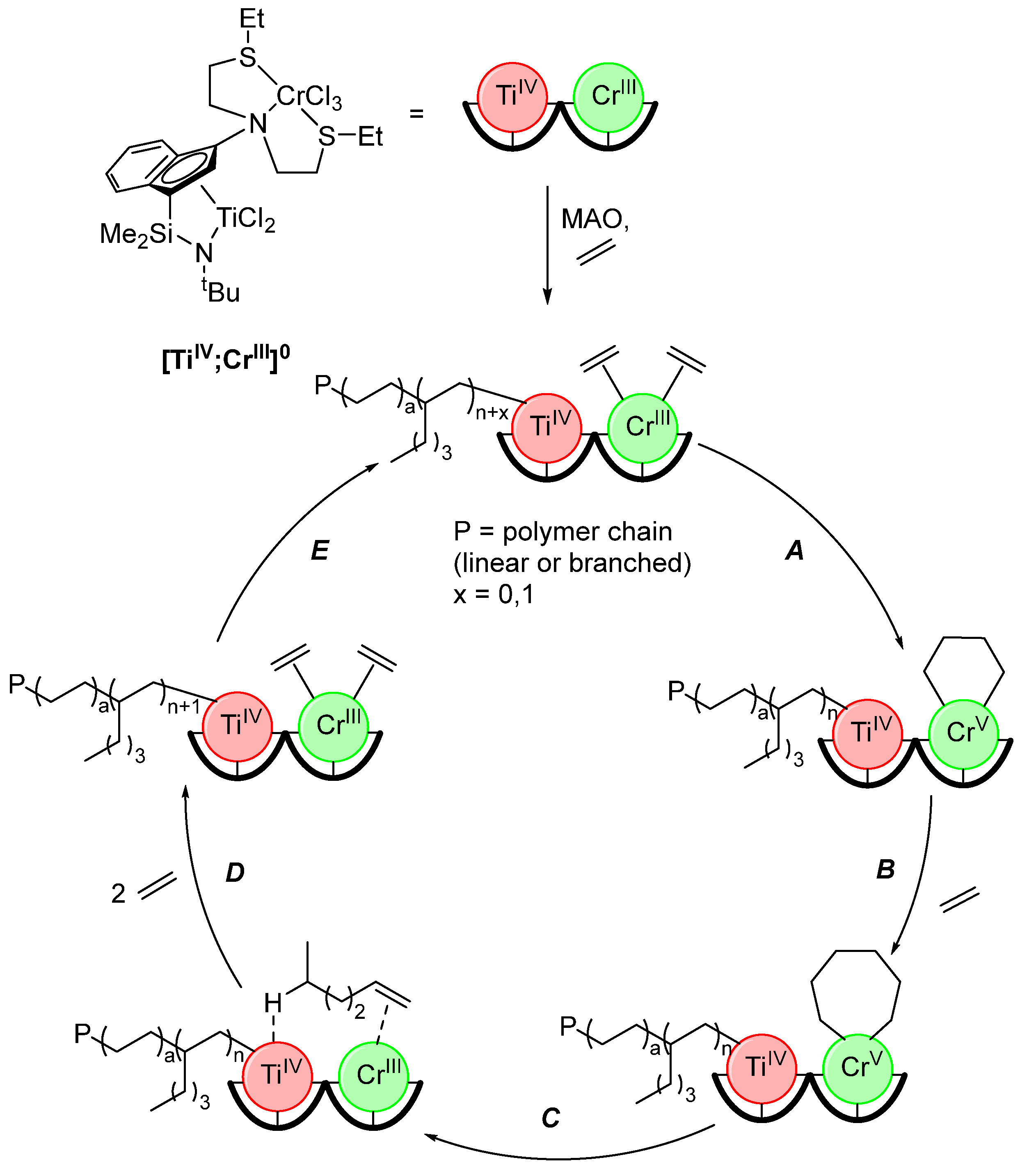
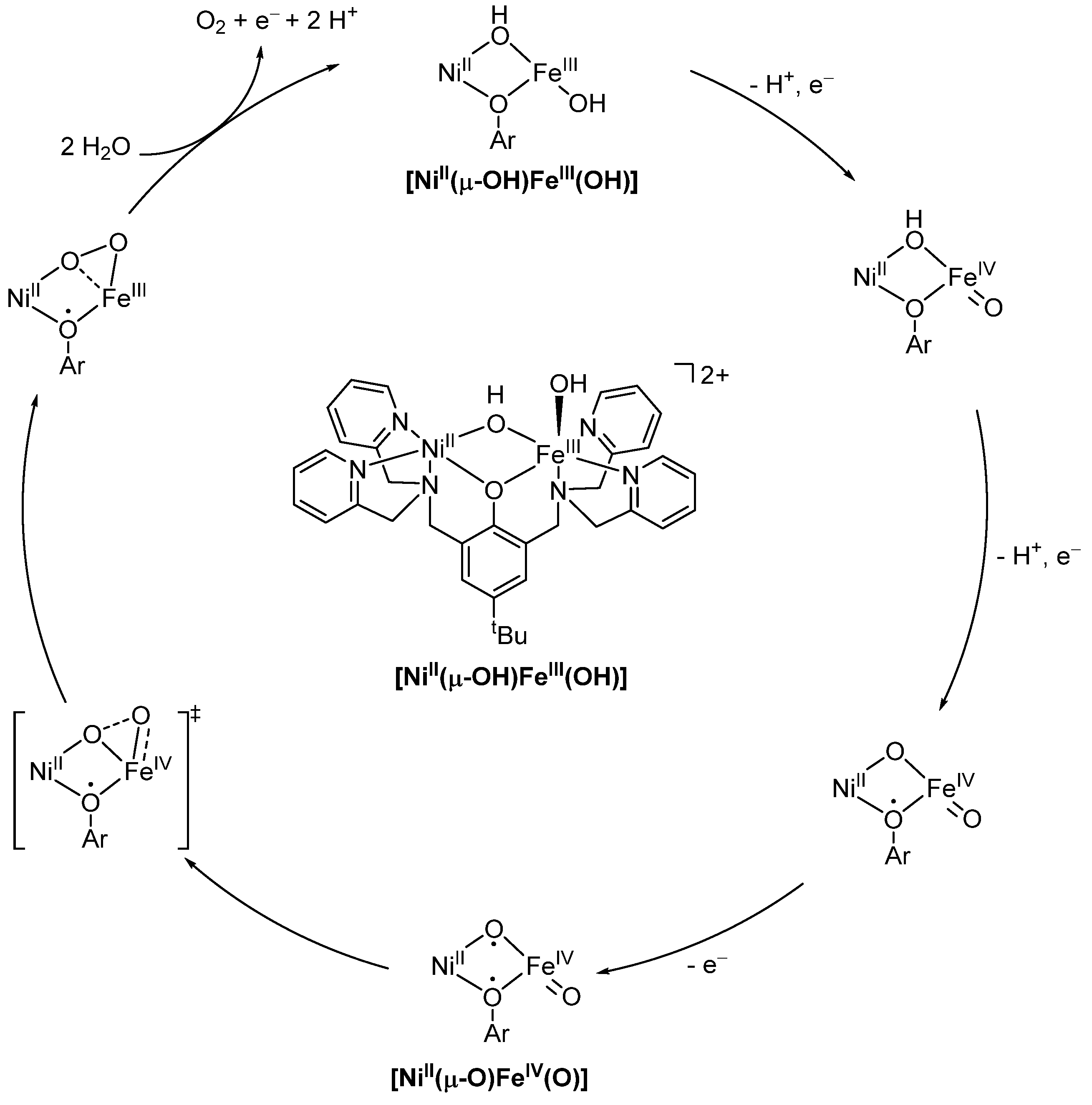
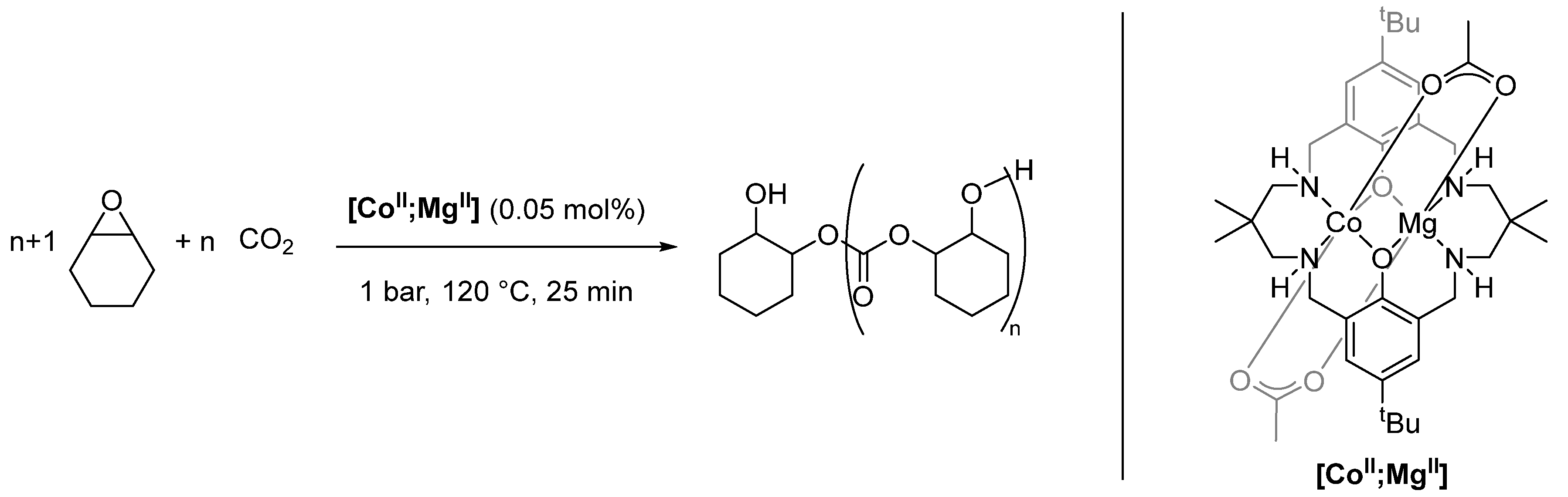
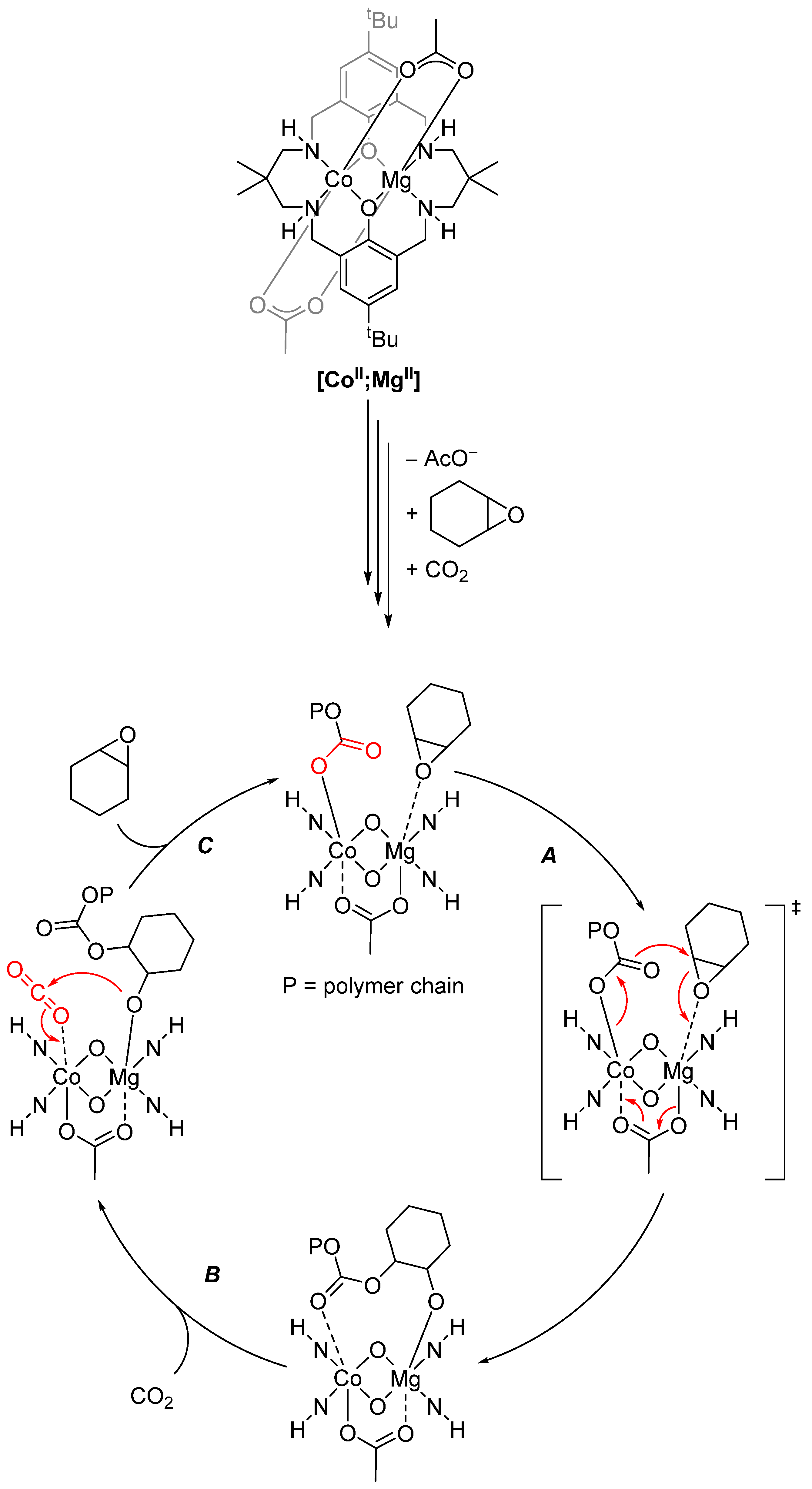
2.4. Synergistic Catalysis
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Cozzi, L.; Erdogan, M.; Arsalane, Y.; Bahar, H.; Barret, C.; Zavala, P.B.; Couse, J.; Criswell, T.; Dasgupta, A.; Gaugy, M.; et al. Global Energy Review 2021: Assessing the Effects of Economic Recoveries on Global Energy Demand and CO2 Emissions in 2021; International Energy Agency: Paris, France, 2021. [Google Scholar]
- Rockström, J.; Steffen, W.; Noone, K.; Persson, A.; Chapin, F.S.; Lambin, E.F.; Lenton, T.M.; Scheffer, M.; Folke, C.; Schellnhuber, H.J.; et al. A safe operating space for humanity. Nature 2009, 461, 472–475. [Google Scholar] [CrossRef] [PubMed]
- Plasseraud, L. Carbon Dioxide as Chemical Feedstock. Edited by Michele Aresta. ChemSusChem 2010, 3, 631–632. [Google Scholar] [CrossRef]
- Aresta, M.; Dibenedetto, A. Utilisation of CO2 as a chemical feedstock: Opportunities and challenges. Dalton Trans. 2007, 2975–2992. [Google Scholar] [CrossRef] [PubMed]
- Goeppert, A.; Czaun, M.; Jones, J.-P.; Surya Prakash, G.K.; Olah, G.A. Recycling of carbon dioxide to methanol and derived products—Closing the loop. Chem. Soc. Rev. 2014, 43, 7995–8048. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Huang, X.; Wang, X.; Wang, X. Progress in catalyst exploration for heterogeneous CO2 reduction and utilization: A critical review. J. Mater. Chem. A 2017, 5, 21625–21649. [Google Scholar] [CrossRef]
- Appel, A.M.; Bercaw, J.E.; Bocarsly, A.B.; Dobbek, H.; DuBois, D.L.; Dupuis, M.; Ferry, J.G.; Fujita, E.; Hille, R.; Kenis, P.J.A.; et al. Frontiers, opportunities, and challenges in biochemical and chemical catalysis of CO2 fixation. Chem. Rev. 2013, 113, 6621–6658. [Google Scholar] [CrossRef]
- Klankermayer, J.; Wesselbaum, S.; Beydoun, K.; Leitner, W. Selective Catalytic Synthesis Using the Combination of Carbon Dioxide and Hydrogen: Catalytic Chess at the Interface of Energy and Chemistry. Angew. Chem. Int. Ed. 2016, 55, 7296–7343. [Google Scholar] [CrossRef]
- Klankermayer, J.; Wesselbaum, S.; Beydoun, K.; Leitner, W. Selektive katalytische Synthesen mit Kohlendioxid und Wasserstoff: Katalyse-Schach an der Nahtstelle zwischen Energie und Chemie. Angew. Chem. 2016, 128, 7416–7467. [Google Scholar] [CrossRef]
- Gong, F.; Zhu, H.; Zhang, Y.; Li, Y. Biological carbon fixation: From natural to synthetic. J. CO2 Util. 2018, 28, 221–227. [Google Scholar] [CrossRef]
- Silverstein, A.; Silverstein, V.B.; Nunn, L.S. Photosynthesis; Twenty-First Century Books: Minneapolis, MN, USA, 2008; ISBN 9780822567981. [Google Scholar]
- Erb, T.J.; Zarzycki, J. A short history of RubisCO: The rise and fall (?) of Nature’s predominant CO2 fixing enzyme. Curr. Opin. Biotechnol. 2018, 49, 100–107. [Google Scholar] [CrossRef]
- Schulman, M.; Parker, D.; Ljungdahl, L.G.; Wood, H.G. Total synthesis of acetate from CO2 V. Determination by mass analysis of the different types of acetate formed from 13CO2 by heterotrophic bacteria. J. Bacteriol. 1972, 109, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Borrel, G.; Adam, P.S.; Gribaldo, S. Methanogenesis and the Wood-Ljungdahl Pathway: An Ancient, Versatile, and Fragile Association. Genome Biol. Evol. 2016, 8, 1706–1711. [Google Scholar] [CrossRef]
- Weiss, M.C.; Sousa, F.L.; Mrnjavac, N.; Neukirchen, S.; Roettger, M.; Nelson-Sathi, S.; Martin, W.F. The physiology and habitat of the last universal common ancestor. Nat. Microbiol. 2016, 1, 16116. [Google Scholar] [CrossRef] [PubMed]
- Ragsdale, S.W.; Pierce, E. Acetogenesis and the Wood-Ljungdahl pathway of CO2 fixation. Biochim. Biophys. Acta 2008, 1784, 1873–1898. [Google Scholar] [CrossRef] [PubMed]
- Jeoung, J.-H.; Dobbek, H. Carbon dioxide activation at the Ni,Fe-cluster of anaerobic carbon monoxide dehydrogenase. Science 2007, 318, 1461–1464. [Google Scholar] [CrossRef]
- Russell, W.K.; Lindahl, P.A. CO/CO2 potentiometric titrations of carbon monoxide dehydrogenase from Clostridium thermoaceticum and the effect of CO2. Biochemistry 1998, 37, 10016–10026. [Google Scholar] [CrossRef]
- Jeoung, J.-H.; Dobbek, H. Structural basis of cyanide inhibition of Ni, Fe-containing carbon monoxide dehydrogenase. J. Am. Chem. Soc. 2009, 131, 9922–9923. [Google Scholar] [CrossRef]
- Jeoung, J.-H.; Dobbek, H. n-Butyl isocyanide oxidation at the NiFe4S4OHx cluster of CO dehydrogenase. J. Biol. Inorg. Chem. 2012, 17, 167–173. [Google Scholar] [CrossRef]
- Seravalli, J.; Ragsdale, S.W. 13C NMR characterization of an exchange reaction between CO and CO2 catalyzed by carbon monoxide dehydrogenase. Biochemistry 2008, 47, 6770–6781. [Google Scholar] [CrossRef]
- Darnault, C.; Volbeda, A.; Kim, E.J.; Legrand, P.; Vernède, X.; Lindahl, P.A.; Fontecilla-Camps, J.C. NiZnFe4S4 and NiNiFe4S4 clusters in closed and open subunits of acetyl-CoA synthase/carbon monoxide dehydrogenase. Nat. Struct. Biol. 2003, 10, 271–279. [Google Scholar] [CrossRef]
- Möller, F.; Piontek, S.; Miller, R.G.; Apfel, U.-P. From Enzymes to Functional Materials-Towards Activation of Small Molecules. Chem. Eur. J. 2018, 24, 1471–1493. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.C.; Duboc, C.; Gennari, M. Synergy between metals for small molecule activation: Enzymes and bio-inspired complexes. Coord. Chem. Rev. 2021, 428, 213606. [Google Scholar] [CrossRef]
- Volbeda, A.; Charon, M.H.; Piras, C.; Hatchikian, E.C.; Frey, M.; Fontecilla-Camps, J.C. Crystal structure of the nickel-iron hydrogenase from Desulfovibrio gigas. Nature 1995, 373, 580–587. [Google Scholar] [CrossRef]
- Ogata, H.; Hirota, S.; Nakahara, A.; Komori, H.; Shibata, N.; Kato, T.; Kano, K.; Higuchi, Y. Activation process of NiFe hydrogenase elucidated by high-resolution X-ray analyses: Conversion of the ready to the unready state. Structure 2005, 13, 1635–1642. [Google Scholar] [CrossRef] [PubMed]
- Volbeda, A.; Martin, L.; Cavazza, C.; Matho, M.; Faber, B.W.; Roseboom, W.; Albracht, S.P.J.; Garcin, E.; Rousset, M.; Fontecilla-Camps, J.C. Structural differences between the ready and unready oxidized states of NiFe hydrogenases. J. Biol. Inorg. Chem. 2005, 10, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Volbeda, A.; Amara, P.; Darnault, C.; Mouesca, J.-M.; Parkin, A.; Roessler, M.M.; Armstrong, F.A.; Fontecilla-Camps, J.C. X-ray crystallographic and computational studies of the O2-tolerant NiFe-hydrogenase 1 from Escherichia coli. Proc. Natl. Acad. Sci. USA 2012, 109, 5305–5310. [Google Scholar] [CrossRef]
- Iwata, S.; Ostermeier, C.; Ludwig, B.; Michel, H. Structure at 2.8 A resolution of cytochrome c oxidase from Paracoccus denitrificans. Nature 1995, 376, 660–669. [Google Scholar] [CrossRef]
- Noodleman, L.; Han Du, W.-G.; Fee, J.A.; Götz, A.W.; Walker, R.C. Linking chemical electron-proton transfer to proton pumping in cytochrome c oxidase: Broken-symmetry DFT exploration of intermediates along the catalytic reaction pathway of the iron-copper dinuclear complex. Inorg. Chem. 2014, 53, 6458–6472. [Google Scholar] [CrossRef]
- Wikström, M.; Krab, K.; Sharma, V. Oxygen Activation and Energy Conservation by Cytochrome c Oxidase. Chem. Rev. 2018, 118, 2469–2490. [Google Scholar] [CrossRef]
- Yoshikawa, S.; Shimada, A. Reaction mechanism of cytochrome c oxidase. Chem. Rev. 2015, 115, 1936–1989. [Google Scholar] [CrossRef]
- Hough, M.A.; Hasnain, S.S. Crystallographic structures of bovine copper-zinc superoxide dismutase reveal asymmetry in two subunits: Functionally important three and five coordinate copper sites captured in the same crystal. J. Mol. Biol. 1999, 287, 579–592. [Google Scholar] [CrossRef] [PubMed]
- Ertl, G. (Ed.) Handbook of Heterogeneous Catalysis: 8 Volumes; Wiley-VCH: Weinheim, Germany, 1997; ISBN 978-3-527-31241-2. [Google Scholar]
- Gholami, Z.; Tišler, Z.; Rubáš, V. Recent advances in Fischer-Tropsch synthesis using cobalt-based catalysts: A review on supports, promoters, and reactors. Catal. Rev. 2021, 63, 512–595. [Google Scholar] [CrossRef]
- Kapteijn, F.; Rodriguez-Mirasol, J.; Moulijn, J.A. Heterogeneous catalytic decomposition of nitrous oxide. Appl. Catal. B 1996, 9, 25–64. [Google Scholar] [CrossRef]
- Wei, J.; Iglesia, E. Isotopic and kinetic assessment of the mechanism of reactions of CH4 with CO2 or H2O to form synthesis gas and carbon on nickel catalysts. J. Catal. 2004, 224, 370–383. [Google Scholar] [CrossRef]
- Gildner, P.G.; Colacot, T.J. Reactions of the 21st Century: Two Decades of Innovative Catalyst Design for Palladium-Catalyzed Cross-Couplings. Organometallics 2015, 34, 5497–5508. [Google Scholar] [CrossRef]
- Delmon, B. Catalyst Deactivation 1999: Proceedings of the 8th International Symposium, Brugge, Belgium, 10–13 October 1999, 1st ed.; Elsevier: Amsterdam, The Netherlands, 1999; ISBN 9780444502131. [Google Scholar]
- Pfaltz, A.; Blankenstein, J.; Hilgraf, R.; Hörmann, E.; McIntyre, S.; Menges, F.; Schönleber, M.; Smidt, S.P.; Wüstenberg, B.; Zimmermann, N. Iridium-Catalyzed Enantioselective Hydrogenation of Olefins. Adv. Synth. Catal. 2003, 345, 33–43. [Google Scholar] [CrossRef]
- Bradshaw, A.M. Structural studies of adsorbed molecules and molecular fragments: The surface-cluster analogy re-visited. Surf. Sci. 1995, 331–333, 978–988. [Google Scholar] [CrossRef]
- Muetterties, E.L. Molecular metal clusters. Science 1977, 196, 839–848. [Google Scholar] [CrossRef]
- Muetterties, E.L.; Rhodin, T.N.; Band, E.; Brucker, C.F.; Pretzer, W.R. Clusters and surfaces. Chem. Rev. 1979, 79, 91–137. [Google Scholar] [CrossRef]
- Dyson, P.J. Catalysis by low oxidation state transition metal (carbonyl) clusters. Coord. Chem. Rev. 2004, 248, 2443–2458. [Google Scholar] [CrossRef]
- Kakkonen, H.J.; Ahlgrèn, M.; Pakkanen, T.A.; Pursiainen, J. Synthesis and structural characterization of [N(PPh3)2][H2Ru3Rh(CO)12]. J. Organomet. Chem. 1994, 482, 279–283. [Google Scholar] [CrossRef]
- Mingos, D.M.P. High nuclearity clusters of the transition metals and a re-evaluation of the cluster surface analogy. J. Clust. Sci. 1992, 3, 397–409. [Google Scholar] [CrossRef]
- Zwart, J.; Snel, R. Metal carbonyl clusters in the catalytic hydrogenation of carbon monoxide. J. Mol. Cat. 1985, 30, 305–352. [Google Scholar] [CrossRef]
- Gade, L.H. Highly Polar Metal-Metal Bonds in “Early-Late” Heterodimetallic Complexes. Angew. Chem. Int. Ed. 2000, 39, 2658–2678. [Google Scholar] [CrossRef]
- Gade, L.H. Stark polare Metall-Metall-Bindungen in Heterodimetallkomplexen des „Early-Late”-Typs. Angew. Chem. 2000, 112, 2768–2789. [Google Scholar] [CrossRef]
- Buchwalter, P.; Rosé, J.; Braunstein, P. Multimetallic catalysis based on heterometallic complexes and clusters. Chem. Rev. 2015, 115, 28–126. [Google Scholar] [CrossRef]
- Knorr, M.; Jourdain, I. Activation of alkynes by diphosphine- and µ-phosphido-spanned heterobimetallic complexes. Coord. Chem. Rev. 2017, 350, 217–247. [Google Scholar] [CrossRef]
- Wheatley, N.; Kalck, P. Structure and Reactivity of Early-Late Heterobimetallic Complexes. Chem. Rev. 1999, 99, 3379–3420. [Google Scholar] [CrossRef]
- Cooper, B.G.; Napoline, J.W.; Thomas, C.M. Catalytic Applications of Early/Late Heterobimetallic Complexes. Catal. Rev. 2012, 54, 1–40. [Google Scholar] [CrossRef]
- Mazzacano, T.J.; Mankad, N.P. Base metal catalysts for photochemical C-H borylation that utilize metal-metal cooperativity. J. Am. Chem. Soc. 2013, 135, 17258–17261. [Google Scholar] [CrossRef]
- Wang, Q.; Brooks, S.H.; Liu, T.; Tomson, N.C. Tuning metal-metal interactions for cooperative small molecule activation. Chem. Commun. 2021, 57, 2839–2853. [Google Scholar] [CrossRef] [PubMed]
- Maity, R.; Birenheide, B.S.; Breher, F.; Sarkar, B. Cooperative Effects in Multimetallic Complexes Applied in Catalysis. ChemCatChem 2021, 13, 2337–2370. [Google Scholar] [CrossRef]
- Mata, J.A.; Hahn, F.E.; Peris, E. Heterometallic complexes, tandem catalysis and catalytic cooperativity. Chem. Sci. 2014, 5, 1723–1732. [Google Scholar] [CrossRef]
- Mankad, N.P. Selectivity Effects in Bimetallic Catalysis. Chem. Eur. J. 2016, 22, 5822–5829. [Google Scholar] [CrossRef]
- Mankad, N.P. Catalysis with Multinuclear Complexes. In Non-Noble Metal Catalysis: Molecular Approaches and Reactions; Moret, M.-E., Gebbink, R.J.M., Eds.; Wiley-VCH: Weinheim, Germany, 2019; pp. 49–68. ISBN 9783527699087. [Google Scholar]
- Park, J.; Hong, S. Cooperative bimetallic catalysis in asymmetric transformations. Chem. Soc. Rev. 2012, 41, 6931–6943. [Google Scholar] [CrossRef] [PubMed]
- Page, M.J.; Walker, D.B.; Messerle, B.A. Alkyne Activation Using Bimetallic Catalysts. In Homo- and Heterobimetallic Complexes in Catalysis; Kalck, P., Ed.; Springer International Publishing: Cham, Switzerland, 2016; pp. 103–137. ISBN 978-3-319-34182-8. [Google Scholar]
- Chaudhary, A.; Singh, A.; Kamboj, R.C. Heterobimetallic Complexes as Promising Catalysts. Chem. Sci. Rev. Lett. 2016, 5, 170–192. [Google Scholar]
- Chatterjee, B.; Chang, W.-C.; Jena, S.; Werlé, C. Implementation of Cooperative Designs in Polarized Transition Metal Systems—Significance for Bond Activation and Catalysis. ACS Catal. 2020, 10, 14024–14055. [Google Scholar] [CrossRef]
- Platten, A.W.J.; Borys, A.M.; Hevia, E. Hydrophosphinylation of Styrenes Catalysed by Well-Defined s-Block Bimetallics. ChemCatChem 2022, 14, e202101853. [Google Scholar] [CrossRef]
- Takaya, J. Catalysis using transition metal complexes featuring main group metal and metalloid compounds as supporting ligands. Chem. Sci. 2021, 12, 1964–1981. [Google Scholar] [CrossRef]
- Nishad, R.C.; Kumar, S.; Rit, A. Hetero- and Homobimetallic Complexes Bridged by a Bis(NHC) Ligand: Synthesis via Selective Sequential Metalation and Catalytic Applications in Tandem Organic Transformations. Organometallics 2021, 40, 915–926. [Google Scholar] [CrossRef]
- Kalck, P. (Ed.) Homo- and Heterobimetallic Complexes in Catalysis; Springer International Publishing: Cham, Switzerland, 2016; ISBN 978-3-319-34182-8. [Google Scholar]
- Stevens, M.A.; Colebatch, A.L. Cooperative approaches in catalytic hydrogenation and dehydrogenation. Chem. Soc. Rev. 2022, 51, 1881–1898. [Google Scholar] [CrossRef] [PubMed]
- van den Beuken, E.K.; Feringa, B.L. Bimetallic catalysis by late transition metal complexes. Tetrahedron 1998, 54, 12985–13011. [Google Scholar] [CrossRef]
- Cotton, F.A.; Curtis, N.F.; Harris, C.B.; Johnson, B.F.; Lippard, S.J.; Mague, J.T.; Robinson, W.R.; Wood, J.S. Mononuclear and Polynuclear Chemistry of Rhenium(III): Its Pronounced Homophilicity. Science 1964, 145, 1305–1307. [Google Scholar] [CrossRef]
- Berry, J.F.; Lu, C.C. Metal-Metal Bonds: From Fundamentals to Applications. Inorg. Chem. 2017, 56, 7577–7581. [Google Scholar] [CrossRef] [PubMed]
- Berry, J.F.; Thomas, C.M. Multimetallic complexes: Synthesis and applications. Dalton Trans. 2017, 46, 5472–5473. [Google Scholar] [CrossRef]
- Greenwood, B.P.; Forman, S.I.; Rowe, G.T.; Chen, C.-H.; Foxman, B.M.; Thomas, C.M. Multielectron redox activity facilitated by metal-metal interactions in early/late heterobimetallics: Co/Zr complexes supported by phosphinoamide ligands. Inorg. Chem. 2009, 48, 6251–6260. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Napoline, J.W.; Thomas, C.M. A Catalytic Application of Co/Zr Heterobimetallic Complexes: Kumada Coupling of Unactivated Alkyl Halides with Alkyl Grignard Reagents. Eur. J. Inorg. Chem. 2011, 2011, 2029–2033. [Google Scholar] [CrossRef]
- Sue, T.; Sunada, Y.; Nagashima, H. Zirconium(IV) Tris(phosphinoamide) Complexes as a Tripodal-Type Metalloligand: A Route to Zr–M (M = Cu, Mo, Pt) Heterodimetallic Complexes. Eur. J. Inorg. Chem. 2007, 2007, 2897–2908. [Google Scholar] [CrossRef]
- Luh, T.Y.; Leung Mk, M.K.; Wong, K.T. Transition metal-catalyzed activation of aliphatic C-X bonds in carbon-carbon bond formation. Chem. Rev. 2000, 100, 3187–3204. [Google Scholar] [CrossRef]
- Tsuji, T.; Yorimitsu, H.; Oshima, K. Cobalt-Catalyzed Coupling Reaction of Alkyl Halides with Allylic Grignard Reagents. Angew. Chem. Int. Ed. 2002, 41, 4137–4139. [Google Scholar] [CrossRef]
- Tsuji, T.; Yorimitsu, H.; Oshima, K. Cobalt-Catalyzed Coupling Reaction of Alkyl Halides with Allylic Grignard Reagents. Angew. Chem. 2002, 114, 4311–4313. [Google Scholar] [CrossRef]
- Ohmiya, H.; Tsuji, T.; Yorimitsu, H.; Oshima, K. Cobalt-catalyzed cross-coupling reactions of alkyl halides with allylic and benzylic Grignard reagents and their application to tandem radical cyclization/cross-coupling reactions. Chem. Eur. J. 2004, 10, 5640–5648. [Google Scholar] [CrossRef] [PubMed]
- Greenwood, B.P.; Rowe, G.T.; Chen, C.-H.; Foxman, B.M.; Thomas, C.M. Metal-metal multiple bonds in early/late heterobimetallics support unusual trigonal monopyramidal geometries at both Zr and Co. J. Am. Chem. Soc. 2010, 132, 44–45. [Google Scholar] [CrossRef] [PubMed]
- Setty, V.N.; Zhou, W.; Foxman, B.M.; Thomas, C.M. Subtle differences between Zr and Hf in early/late heterobimetallic complexes with cobalt. Inorg. Chem. 2011, 50, 4647–4655. [Google Scholar] [CrossRef]
- Tsutsumi, H.; Sunada, Y.; Shiota, Y.; Yoshizawa, K.; Nagashima, H. Nickel(II), Palladium(II), and Platinum(II) η3-Allyl Complexes Bearing a Bidentate Titanium(IV) Phosphinoamide Ligand: A Ti←M2 Dative Bond Enhances the Electrophilicity of the π-Allyl Moiety. Organometallics 2009, 28, 1988–1991. [Google Scholar] [CrossRef]
- Walker, W.K.; Kay, B.M.; Michaelis, S.A.; Anderson, D.L.; Smith, S.J.; Ess, D.H.; Michaelis, D.J. Origin of fast catalysis in allylic amination reactions catalyzed by Pd-Ti heterobimetallic complexes. J. Am. Chem. Soc. 2015, 137, 7371–7378. [Google Scholar] [CrossRef] [PubMed]
- Walker, W.K.; Anderson, D.L.; Stokes, R.W.; Smith, S.J.; Michaelis, D.J. Allylic aminations with hindered secondary amine nucleophiles catalyzed by heterobimetallic Pd-Ti complexes. Org. Lett. 2015, 17, 752–755. [Google Scholar] [CrossRef]
- Carlsen, R.W.; Ess, D.H. Allylic amination reactivity of Ni, Pd, and Pt heterobimetallic and monometallic complexes. Dalton Trans. 2016, 45, 9835–9840. [Google Scholar] [CrossRef] [PubMed]
- Cammarota, R.C.; Vollmer, M.V.; Xie, J.; Ye, J.; Linehan, J.C.; Burgess, S.A.; Appel, A.M.; Gagliardi, L.; Lu, C.C. A Bimetallic Nickel-Gallium Complex Catalyzes CO2 Hydrogenation via the Intermediacy of an Anionic d10 Nickel Hydride. J. Am. Chem. Soc. 2017, 139, 14244–14250. [Google Scholar] [CrossRef]
- Vollmer, M.V.; Ye, J.; Linehan, J.C.; Graziano, B.J.; Preston, A.; Wiedner, E.S.; Lu, C.C. Cobalt-Group 13 Complexes Catalyze CO2 Hydrogenation via a Co(−I)/Co(I) Redox Cycle. ACS Catal. 2020, 10, 2459–2470. [Google Scholar] [CrossRef]
- Ye, J.; Cammarota, R.C.; Xie, J.; Vollmer, M.V.; Truhlar, D.G.; Cramer, C.J.; Lu, C.C.; Gagliardi, L. Rationalizing the Reactivity of Bimetallic Molecular Catalysts for CO2 Hydrogenation. ACS Catal. 2018, 8, 4955–4968. [Google Scholar] [CrossRef]
- Dübner, F.; Knochel, P. Highly enantioselective copper-catalyzed substitution of allylic chlorides with diorganozincs. Tetrahedron Lett. 2000, 41, 9233–9237. [Google Scholar] [CrossRef]
- Hu, X.; Dai, H.; Bai, C.; Chen, H.; Zheng, Z. Novel ferrocenylphosphine-imines containing a pyridine unit as a new family of chiral ligands: The important influence of the position of the pyridine N-atom on the reactivity and enantioselectivity in palladium-catalyzed asymmetric allylic alkylations. Tetrahedron Asymmetry 2004, 15, 1065–1068. [Google Scholar] [CrossRef]
- Ito, Y.; Sawamura, M.; Hayashi, T. Catalytic asymmetric aldol reaction: Reaction of aldehydes with isocyanoacetate catalyzed by a chiral ferrocenylphosphine-gold(I) complex. J. Am. Chem. Soc. 1986, 108, 6405–6406. [Google Scholar] [CrossRef]
- Lu, S.-M.; Han, X.-W.; Zhou, Y.-G. Asymmetric Hydrogenation of Quinolines Catalyzed by Iridium with Chiral Ferrocenyloxazoline Derived N,P Ligands. Adv. Synth. Catal. 2004, 346, 909–912. [Google Scholar] [CrossRef]
- Dai, L.-X.; Hou, X.-L. (Eds.) Chiral Ferrocenes in Asymmetric Catalysis: Synthesis and Applications; Wiley-VCH: Weinheim, Germany, 2010; ISBN 978-3527322800. [Google Scholar]
- Blaser, H.-U.; Brieden, W.; Pugin, B.; Spindler, F.; Studer, M.; Togni, A. Solvias Josiphos Ligands: From Discovery to Technical Applications. Top. Catal. 2002, 19, 3–16. [Google Scholar] [CrossRef]
- Maddox, A.F.; Rheingold, A.L.; Golen, J.A.; Scott Kassel, W.; Nataro, C. Taniaphos and Walphos ligands: Oxidative electrochemistry and complexation. Synthesis, characterization, oxidative electrochemistry and X-ray structures of [(Taniaphos/Walphos)MCl2] (M=Pd or Pt). Inorg. Chim. Acta 2008, 361, 3283–3293. [Google Scholar] [CrossRef]
- Peters, R. Chiral Ferrocenes in Asymmetric Catalysis. Synthesis and Applications. Edited by Li-Xin Dai and Xue-Long Hou. Book review. Angew. Chem. Int. Ed. 2010, 49, 4163–4164. [Google Scholar] [CrossRef]
- Hayashi, T.; Kanehira, K.; Tsuchiya, H.; Kumada, M. New chiral ligands designed for palladium-catalysed asymmetric allylic alkylation. J. Chem. Soc. Chem. Commun. 1982, 1162–1164. [Google Scholar] [CrossRef]
- Shibasaki, M.; Yoshikawa, N. Lanthanide complexes in multifunctional asymmetric catalysis. Chem. Rev. 2002, 102, 2187–2210. [Google Scholar] [CrossRef]
- Matsunaga, S.; Ohshima, T.; Shibasaki, M. Linked-BINOL: An Approach towards Practical Asymmetric Multifunctional Catalysis. Adv. Synth. Catal. 2002, 344, 3–15. [Google Scholar] [CrossRef]
- Shibasaki, M.; Matsunaga, S. Design and application of linked-BINOL chiral ligands in bifunctional asymmetric catalysis. Chem. Soc. Rev. 2006, 35, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Shibasaki, M.; Kanai, M.; Matsunaga, S.; Kumagai, N. Recent progress in asymmetric bifunctional catalysis using multimetallic systems. Acc. Chem. Res. 2009, 42, 1117–1127. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.M.A.; Yoshikawa, N.; Sasai, H.; Shibasaki, M. Direct Catalytic Asymmetric Aldol Reactions of Aldehydes with Unmodified Ketones. Angew. Chem. Int. Ed. Engl. 1997, 36, 1871–1873. [Google Scholar] [CrossRef]
- Yamada, M.A.Y.; Yoshikawa, N.; Sasai, H.; Shibasaki, M. Direkte katalytische asymmetrische Aldolreaktionen von Aldehyden mit nicht modifizierten Ketonen. Angew. Chem. 1997, 109, 1942–1944. [Google Scholar] [CrossRef]
- Sasai, H.; Suzuki, T.; Arai, S.; Arai, T.; Shibasaki, M. Basic character of rare earth metal alkoxides. Utilization in catalytic carbon-carbon bond-forming reactions and catalytic asymmetric nitroaldol reactions. J. Am. Chem. Soc. 1992, 114, 4418–4420. [Google Scholar] [CrossRef]
- Sasai, H.; Arai, T.; Satow, Y.; Houk, K.N.; Shibasaki, M. The First Heterobimetallic Multifunctional Asymmetric Catalyst. J. Am. Chem. Soc. 1995, 117, 6194–6198. [Google Scholar] [CrossRef]
- Yamada, K.; Harwood, S.J.; Gröger, H.; Shibasaki, M. Erste katalytische asymmetrische Nitro-Mannich-Reaktion mit einem neuen Heterodimetallkomplex als Katalysator. Angew. Chem. 1999, 111, 3713–3715. [Google Scholar] [CrossRef]
- Yamada, K.; Harwood, S.J.; Gröger, H.; Shibasaki, M. The First Catalytic Asymmetric Nitro-Mannich-Type Reaction Promoted by a New Heterobimetallic Complex. Angew. Chem. Int. Ed. 1999, 38, 3504–3506. [Google Scholar] [CrossRef]
- Arai, T.; Sasai, H.; Yamaguchi, K.; Shibasaki, M. Regioselective Catalytic Asymmetric Reaction of Horner−Wadsworth−Emmons Reagents with Enones: The Odyssey of Chiral Aluminum Catalysts. J. Am. Chem. Soc. 1998, 120, 441–442. [Google Scholar] [CrossRef]
- Arai, T.; Sasai, H.; Aoe, K.; Okamura, K.; Date, T.; Shibasaki, M. A New Multifunctional Heterobimetallic Asymmetric Catalyst for Michael Additions and Tandem Michael–Aldol Reactions. Angew. Chem. Int. Ed. Engl. 1996, 35, 104–106. [Google Scholar] [CrossRef]
- Arai, T.; Sasai, H.; Aoe, K.; Okamura, K.; Date, T.; Shibasaki, M. Ein neuartiger multifunktioneller asymmetrischer Hetero-Dimetall-Katalysator für Michael-Additionen und Tandem-Michael-Aldol-Reaktionen. Angew. Chem. 1996, 108, 103–105. [Google Scholar] [CrossRef]
- Shibasaki, M.; Sasai, H.; Arai, T. Asymmetric Catalysis with Heterobimetallic Compounds. Angew. Chem. Int. Ed. Engl. 1997, 36, 1236–1256. [Google Scholar] [CrossRef]
- Shibasaki, M.; Sasai, H.; Arai, T. Asymmetrische Katalyse mit Hetero-Dimetall-Verbindungen. Angew. Chem. 1997, 109, 1290–1311. [Google Scholar] [CrossRef]
- Ajamian, A.; Gleason, J.L. Two birds with one metallic stone: Single-pot catalysis of fundamentally different transformations. Angew. Chem. Int. Ed. 2004, 43, 3754–3760. [Google Scholar] [CrossRef] [PubMed]
- Ajamian, A.; Gleason, J.L. Zwei Fliegen mit einer Klappe: Katalyse völlig unterschiedlicher Transformationen im Eintopfverfahren. Angew. Chem. 2004, 116, 3842–3848. [Google Scholar] [CrossRef]
- Fogg, D.E.; dos Santos, E.N. Tandem catalysis: A taxonomy and illustrative review. Coord. Chem. Rev. 2004, 248, 2365–2379. [Google Scholar] [CrossRef]
- Wasilke, J.-C.; Obrey, S.J.; Baker, R.T.; Bazan, G.C. Concurrent tandem catalysis. Chem. Rev. 2005, 105, 1001–1020. [Google Scholar] [CrossRef]
- Zanardi, A.; Mata, J.A.; Peris, E. Well-defined Ir/Pd complexes with a triazolyl-diylidene bridge as catalysts for multiple tandem reactions. J. Am. Chem. Soc. 2009, 131, 14531–14537. [Google Scholar] [CrossRef]
- Sabater, S.; Mata, J.A.; Peris, E. Heterobimetallic Iridium–Ruthenium Assemblies through an Ambidentate Triazole-Diylidene Ligand: Electrochemical Properties and Catalytic Behavior in a Cascade Reaction. Organometallics 2012, 31, 6450–6456. [Google Scholar] [CrossRef]
- Robin, M.B.; Day, P. Mixed Valence Chemistry-A Survey and Classification; Elsevier: Amsterdam, The Netherlands, 1968; pp. 247–422. ISBN 9780120236107. [Google Scholar]
- Gusev, D.G. Donor Properties of a Series of Two-Electron Ligands. Organometallics 2009, 28, 763–770. [Google Scholar] [CrossRef]
- Gusev, D.G. Electronic and Steric Parameters of 76 N-Heterocyclic Carbenes in Ni(CO)3(NHC). Organometallics 2009, 28, 6458–6461. [Google Scholar] [CrossRef]
- Gusev, D.G.; Peris, E. The Tolman electronic parameter (TEP) and the metal-metal electronic communication in ditopic NHC complexes. Dalton Trans. 2013, 42, 7359–7364. [Google Scholar] [CrossRef]
- Sabater, S.; Mata, J.A.; Peris, E. Synthesis of Heterodimetallic Iridium–Palladium Complexes Containing Two Axes of Chirality: Study of Sequential Catalytic Properties. Eur. J. Inorg. Chem. 2013, 2013, 4764–4769. [Google Scholar] [CrossRef]
- Liu, S.; Motta, A.; Mouat, A.R.; Delferro, M.; Marks, T.J. Very large cooperative effects in heterobimetallic titanium-chromium catalysts for ethylene polymerization/copolymerization. J. Am. Chem. Soc. 2014, 136, 10460–10469. [Google Scholar] [CrossRef]
- Pfeffer, M.G.; Kowacs, T.; Wächtler, M.; Guthmuller, J.; Dietzek, B.; Vos, J.G.; Rau, S. Optimization of hydrogen-evolving photochemical molecular devices. Angew. Chem. Int. Ed. Engl. 2015, 54, 6627–6631. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, M.G.; Kowacs, T.; Wächtler, M.; Guthmuller, J.; Dietzek, B.; Vos, J.G.; Rau, S. Gezielte Optimierung von molekularen Photokatalysatoren zur Wasserstoffproduktion mit sichtbarem Licht. Angew. Chem. 2015, 127, 6727–6731. [Google Scholar] [CrossRef]
- Pfeffer, M.G.; Schäfer, B.; Smolentsev, G.; Uhlig, J.; Nazarenko, E.; Guthmuller, J.; Kuhnt, C.; Wächtler, M.; Dietzek, B.; Sundström, V.; et al. Palladium versus platinum: The metal in the catalytic center of a molecular photocatalyst determines the mechanism of the hydrogen production with visible light. Angew. Chem. Int. Ed. Engl. 2015, 54, 5044–5048. [Google Scholar] [CrossRef]
- Pfeffer, M.G.; Schäfer, B.; Smolentsev, G.; Uhlig, J.; Nazarenko, E.; Guthmuller, J.; Kuhnt, C.; Wächtler, M.; Dietzek, B.; Sundström, V.; et al. Palladium versus Platin—Das Metall im Katalysezentrum eines molekularen Photokatalysators bestimmt den Mechanismus der Wasserstoffproduktion mit sichtbarem Licht. Angew. Chem. 2015, 127, 5132–5136. [Google Scholar] [CrossRef]
- Rau, S.; Schäfer, B.; Gleich, D.; Anders, E.; Rudolph, M.; Friedrich, M.; Görls, H.; Henry, W.; Vos, J.G. Ein supramolekularer Photokatalysator zur Erzeugung von Wasserstoff und zur selektiven Hydrierung von Tolan. Angew. Chem. 2006, 118, 6361–6364. [Google Scholar] [CrossRef]
- Tschierlei, S.; Karnahl, M.; Presselt, M.; Dietzek, B.; Guthmuller, J.; González, L.; Schmitt, M.; Rau, S.; Popp, J. Photochemical fate: The first step determines efficiency of H2 formation with a supramolecular photocatalyst. Angew. Chem. Int. Ed. Engl. 2010, 49, 3981–3984. [Google Scholar] [CrossRef] [PubMed]
- Tschierlei, S.; Karnahl, M.; Presselt, M.; Dietzek, B.; Guthmuller, J.; González, L.; Schmitt, M.; Rau, S.; Popp, J. Photochemisches Schicksal: Der erste Schritt bestimmt die Effizienz der H2-Bildung mit einem supramolekularen Photokatalysator. Angew. Chem. 2010, 122, 4073–4076. [Google Scholar] [CrossRef]
- Zedler, L.; Wintergerst, P.; Mengele, A.K.; Müller, C.; Li, C.; Dietzek-Ivanšić, B.; Rau, S. Outpacing conventional nicotinamide hydrogenation catalysis by a strongly communicating heterodinuclear photocatalyst. Nat. Commun. 2022, 13, 2538. [Google Scholar] [CrossRef]
- Rau, S.; Schäfer, B.; Gleich, D.; Anders, E.; Rudolph, M.; Friedrich, M.; Görls, H.; Henry, W.; Vos, J.G. A supramolecular photocatalyst for the production of hydrogen and the selective hydrogenation of tolane. Angew. Chem. Int. Ed. 2006, 45, 6215–6218. [Google Scholar] [CrossRef] [PubMed]
- Zedler, L.; Mengele, A.K.; Ziems, K.M.; Zhang, Y.; Wächtler, M.; Gräfe, S.; Pascher, T.; Rau, S.; Kupfer, S.; Dietzek, B. Unraveling the Light-Activated Reaction Mechanism in a Catalytically Competent Key Intermediate of a Multifunctional Molecular Catalyst for Artificial Photosynthesis. Angew. Chem. Int. Ed. Engl. 2019, 58, 13140–13148. [Google Scholar] [CrossRef] [PubMed]
- Zedler, L.; Mengele, A.K.; Ziems, K.M.; Zhang, Y.; Wächtler, M.; Gräfe, S.; Pascher, T.; Rau, S.; Kupfer, S.; Dietzek, B. Unraveling the Light-Activated Reaction Mechanism in a Catalytically Competent Key Intermediate of a Multifunctional Molecular Catalyst for Artificial Photosynthesis. Angew. Chem. 2019, 131, 13274–13282. [Google Scholar] [CrossRef]
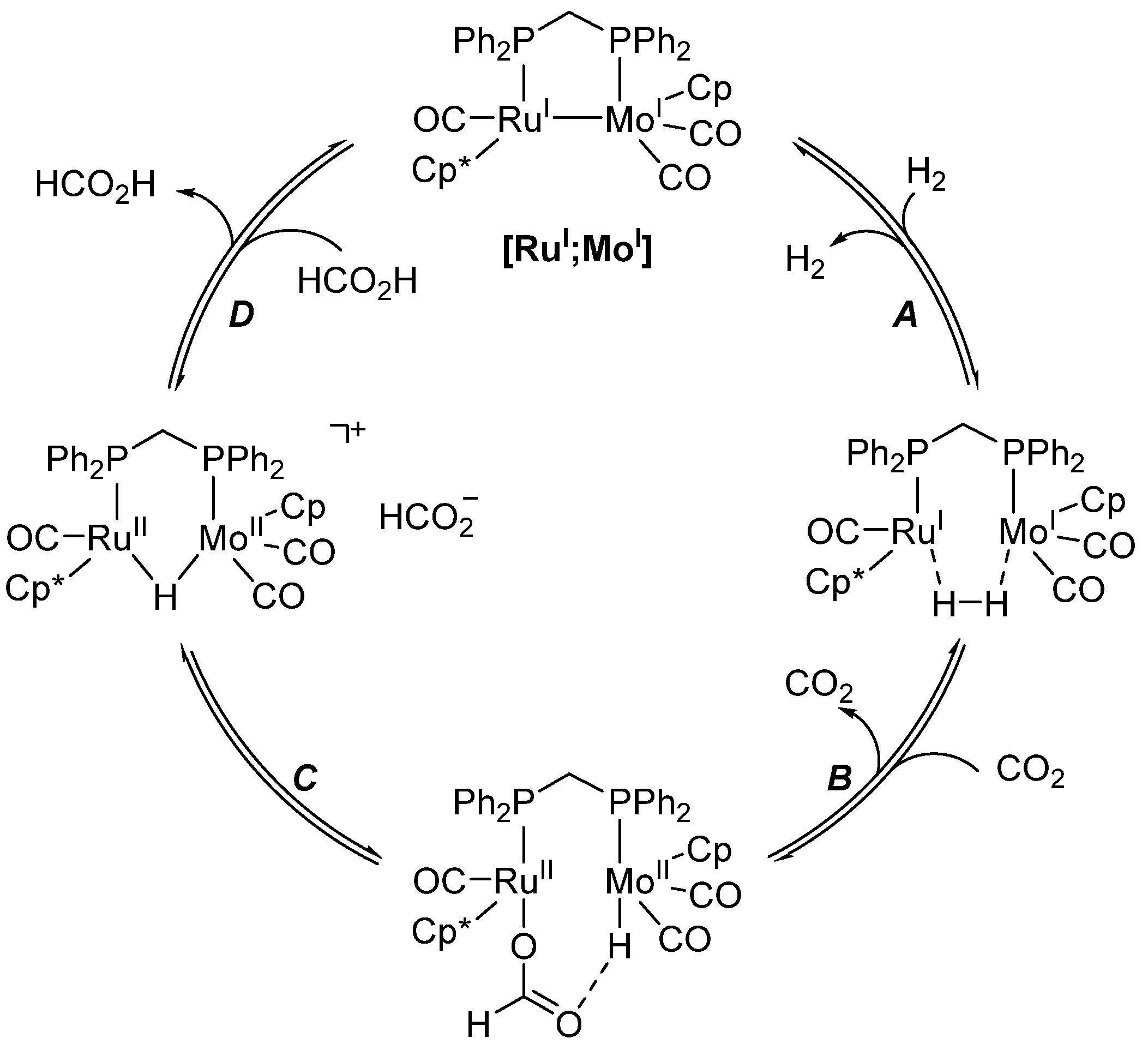
- Man, M.L.; Zhou, Z.; Ng, S.M.; Lau, C.P. Synthesis, characterization and reactivity of heterobimetallic complexes (η5-C5R5)Ru(CO)(μ-dppm)M(CO)2(η5-C5H5) (R = H, CH3; M = Mo, W). Interconversion of hydrogen/carbon dioxide and formic acid by these complexes. Dalton Trans. 2003, 20, 3727–3735. [Google Scholar] [CrossRef]
- Uyeda, C.; Peters, J.C. Selective nitrite reduction at heterobimetallic CoMg complexes. J. Am. Chem. Soc. 2013, 135, 12023–12031. [Google Scholar] [CrossRef]
- Fujii, I.; Semba, K.; Li, Q.-Z.; Sakaki, S.; Nakao, Y. Magnesiation of Aryl Fluorides Catalyzed by a Rhodium-Aluminum Complex. J. Am. Chem. Soc. 2020, 142, 11647–11652. [Google Scholar] [CrossRef]
- Seki, R.; Hara, N.; Saito, T.; Nakao, Y. Selective C-O Bond Reduction and Borylation of Aryl Ethers Catalyzed by a Rhodium-Aluminum Heterobimetallic Complex. J. Am. Chem. Soc. 2021, 143, 6388–6394. [Google Scholar] [CrossRef]
- Zhang, H.-T.; Guo, Y.-H.; Xiao, Y.; Du, H.-Y.; Zhang, M.-T. Heterobimetallic NiFe Cooperative Molecular Water Oxidation Catalyst. Angew. Chem. 2023, 135, e202218859. [Google Scholar] [CrossRef]
- Zhang, H.-T.; Guo, Y.-H.; Xiao, Y.; Du, H.-Y.; Zhang, M.-T. Heterobimetallic NiFe Cooperative Molecular Water Oxidation Catalyst. Angew. Chem. Int. Ed. Engl. 2023, 62, e202218859. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.; Shimoyama, Y.; Ohgomori, Y.; Kanega, R.; Kotani, H.; Ishizuka, T.; Kon, Y.; Himeda, Y.; Kojima, T. Cooperative Effects of Heterodinuclear IrIII-MII Complexes on Catalytic H2 Evolution from Formic Acid Dehydrogenation in Water. Inorg. Chem. 2020, 59, 11976–11985. [Google Scholar] [CrossRef] [PubMed]
- Shimoyama, Y.; Ohgomori, Y.; Kon, Y.; Hong, D. Hydrogen peroxide production from oxygen and formic acid by homogeneous Ir-Ni catalyst. Dalton Trans. 2021, 50, 9410–9416. [Google Scholar] [CrossRef] [PubMed]
- Shimoyama, Y.; Kitagawa, Y.; Ohgomori, Y.; Kon, Y.; Hong, D. Formate-driven catalysis and mechanism of an iridium-copper complex for selective aerobic oxidation of aromatic olefins in water. Chem. Sci. 2021, 12, 5796–5803. [Google Scholar] [CrossRef] [PubMed]
- Haak, R.M.; Wezenberg, S.J.; Kleij, A.W. Cooperative multimetallic catalysis using metallosalens. Chem. Commun. 2010, 46, 2713–2723. [Google Scholar] [CrossRef]
- Apilardmongkol, P.; Ratanasak, M.; Hasegawa, J.; Parasuk, V. Exploring the Reaction Mechanism of Heterobimetallic Nickel-Alkali Catalysts for Ethylene Polymerization: Secondary-Metal-Ligand Cooperative Catalysis. ChemCatChem 2022, 14, e202200028. [Google Scholar] [CrossRef]
- Garden, J.A.; Saini, P.K.; Williams, C.K. Greater than the Sum of Its Parts: A Heterodinuclear Polymerization Catalyst. J. Am. Chem. Soc. 2015, 137, 15078–15081. [Google Scholar] [CrossRef]
- Rosetto, G.; Deacy, A.C.; Williams, C.K. Mg(II) heterodinuclear catalysts delivering carbon dioxide derived multi-block polymers. Chem. Sci. 2021, 12, 12315–12325. [Google Scholar] [CrossRef]
- Gruszka, W.; Garden, J.A. Advances in heterometallic ring-opening (co)polymerisation catalysis. Nat. Commun. 2021, 12, 3252. [Google Scholar] [CrossRef]
- Deacy, A.C.; Durr, C.B.; Garden, J.A.; White, A.J.P.; Williams, C.K. Groups 1, 2 and Zn(II) Heterodinuclear Catalysts for Epoxide/CO2 Ring-Opening Copolymerization. Inorg. Chem. 2018, 57, 15575–15583. [Google Scholar] [CrossRef] [PubMed]
- Deacy, A.C.; Durr, C.B.; Williams, C.K. Heterodinuclear complexes featuring Zn(II) and M = Al(III), Ga(III) or In(III) for cyclohexene oxide and CO2 copolymerisation. Dalton Trans. 2020, 49, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Deacy, A.C.; Moreby, E.; Phanopoulos, A.; Williams, C.K. Co(III)/Alkali-Metal(I) Heterodinuclear Catalysts for the Ring-Opening Copolymerization of CO2 and Propylene Oxide. J. Am. Chem. Soc. 2020, 142, 19150–19160. [Google Scholar] [CrossRef] [PubMed]
- Deacy, A.C.; Kilpatrick, A.F.R.; Regoutz, A.; Williams, C.K. Understanding metal synergy in heterodinuclear catalysts for the copolymerization of CO2 and epoxides. Nat. Chem. 2020, 12, 372–380. [Google Scholar] [CrossRef]
- Fickenscher, Z.B.G.; Lönnecke, P.; Müller, A.K.; Hollóczki, O.; Kirchner, B.; Hey-Hawkins, E. Synergistic Catalysis in Heterobimetallic Complexes for Homogeneous Carbon Dioxide Hydrogenation. Molecules 2023, 28, 2574. [Google Scholar] [CrossRef]
- Fickenscher, Z.B.G.; Lönnecke, P.; Müller, A.K.; Baumann, W.; Kirchner, B.; Hey-Hawkins, E. Stronger Together! Mechanistic investigation into synergistic effects during homogeneous carbon dioxide hydrogenation using a heterobimetallic catalyst. Inorg. Chem. 2023; submitted. [Google Scholar]
- Fickenscher, Z.B.G.; Torres-Texeira, L.; Lönnecke, P.; Hey-Hawkins, E. Synthesis and Reactivity of a Heterobimetallic Mo,Co Complex. Inorg. Chim. Acta, 2023; submitted. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fickenscher, Z.; Hey-Hawkins, E. Added Complexity!—Mechanistic Aspects of Heterobimetallic Complexes for Application in Homogeneous Catalysis. Molecules 2023, 28, 4233. https://doi.org/10.3390/molecules28104233
Fickenscher Z, Hey-Hawkins E. Added Complexity!—Mechanistic Aspects of Heterobimetallic Complexes for Application in Homogeneous Catalysis. Molecules. 2023; 28(10):4233. https://doi.org/10.3390/molecules28104233
Chicago/Turabian StyleFickenscher, Zeno, and Evamarie Hey-Hawkins. 2023. "Added Complexity!—Mechanistic Aspects of Heterobimetallic Complexes for Application in Homogeneous Catalysis" Molecules 28, no. 10: 4233. https://doi.org/10.3390/molecules28104233