3.1. Chemistry
Methods and Materials: All chemicals and anhydrous solvents were purchased from either Sigma-Aldrich (now Merck: Gillingham, UK) or Alfa Aesar (Heysham, UK). All organic solvents of AR grade were supplied by Fisher Scientific (Loughborough, UK). Melting points were determined using a Stanford Research Systems Optimelt MPA100 (Stanford Research Systems, Sunnyvale, CA, USA) and were uncorrected. Thin-layer chromatography (TLC) was performed on pre-coated aluminium plates (Merck, silica gel 60 F254). The products were visualised either by UV irradiation at 254 nm or by staining with 5% w/v phosphomolybdic acid in ethanol, followed by heating. Flash column chromatography was performed on pre-packed silica gel columns (RediSep Rf) and gradient elution (solvents indicated in text) on the Combiflash Rf system (Teledyne Isco). 1H NMR spectra were recorded with a Bruker 400 or 500 MHz spectrometer. The chemical shifts were reported in parts per million (ppm), either relative to the corresponding solvent residual peaks or tetramethylsilane (TMS) as an internal standard. High-resolution mass spectra (HRMS) were recorded on a Bruker MicroTOF with ESI. All compounds were ≥95% pure by 1H NMR spectroscopy.
General Procedure: Guanylation of substituted 3-(aminomethyl)phenols (11a–c): In a solution of the substituted 3-(aminomethyl)phenol hydrochloride 11a–c (6.0 mmol) in DMF (25 mL), S-methyl-N,N′-bis(tert-butoxycarbonyl) isothiourea (1.3 g, 4.5 mmol) was added, followed by Et3N (3.0 mL). The mixture was stirred at r.t. overnight and partitioned between EtOAc (100 mL) and citric acid (50 mL, 5% in water). The organic layer was washed with brine, dried (MgSO4), and concentrated in vacuo to give an off-white solid. Purification by flash column chromatography (petrol ether to petrol ether/EtOAc 11:9) afforded 12a–c.
tert-Butyl N-[{[(tert-butoxy)carbonyl]imino}({[(3-hydroxyphenyl)methyl]amino}) methyl]carbamate (12a): A white solid was obtained (75% yield), m.p. 159–161 °C. 1H NMR (400 MHz, CDCl3): δ 1.48 (9H, s, t-Bu), 1.50 (9H, s, t-Bu), 4.55 (2H, d, J = 5.2 Hz, CH2), 6.69–6.80 (3H, m, 3 × ArH), 7.15 (1H, t, J = 7.2 Hz, ArH), 8.69 (1H, t, J = 5.2 Hz, NH), and 11.6 (1H, s, NH). 13C NMR (100 MHz, CDCl3): δ 28.2, 28.4, 44.7, 79.7, 83.5, 114.6, 114.7, 119.6, 129.9, 139.1, 153.3, 156.3, 156.4, and 163.5. HRMS (ESI): Calcd. for C18H28N3O5 (M + H)+ 366.2029 and found 366.2008.
tert-Butyl N-{[(tert-butoxy)carbonyl]imino}({[(3-hydroxy-4-methoxyphenyl) methyl]amino})methyl]carbamate (12b): A white solid was obtained (69% yield), m.p. 127–129 °C. 1H NMR (400 MHz, CDCl3): δ 1.48 (9H, s, t-Bu), 1.50 (9H, s, t-Bu), 3.87 (3H, s, OCH3), 4.51 (2H, d, J = 5.2 Hz, CH2), 6.76–6.79 (2H, m, 2 × ArH), 6.88 (1H, d, J = 1.0 Hz, ArH), 8.49 (1H, br.s, NH), and 11.5 (1H, s, NH). HRMS (ESI): Calcd. for C19H30N3O6 (M + H)+ 396.2135 and found 396.2148.
tert-Butyl N-[{[(tert-butoxy)carbonyl]imino}({[(2,6-difluoro-3-hydroxyphenyl)methyl]amino})methyl]carbamate (12c): A white solid was obtained (59% yield), m.p. 132–134 °C. 1H NMR (400 MHz, CDCl3) δ 1.47 (9H, s, t-Bu), 1.52 (9H, s, t-Bu), 4.77 (2H, br.s, CH2), 6.79 (1H, m, ArH), 6.99 (1H, m, ArH), 8.70 (1H, br.s, NH), and 11.6 (1H, s, NH). HRMS (ESI): Calcd. for C18H26F2N3O5 (M + H)+ 402.1840 and found 402.1857.
General Procedure: Benzylation of substituted 3-(N,N′-di-Boc-guanydinomethyl)phenols (12a–c): In a solution of the substituted 3-(N,N′-di-Boc-guanydinomethyl)phenol (12a–c) (0.56 mmol) in acetone (8 mL), the substituted benzyl bromide (0.56 mmol) was added, followed by K2CO3 (96 mg). The mixture was stirred at r.t. overnight and partitioned between EtOAc (50 mL) and water (50 mL). The organic layer was washed with brine, dried (MgSO4), and concentrated in vacuo to give an off-white solid. Purification by flash column chromatography (petrol ether to petrol ether/EtOAc 3:1) afforded 13a–q.
tert-Butyl N-[({[3-(benzyloxy)phenyl]methyl}amino)({[(tert-butoxy) carbonyl] amino})methylidene]carbamate (13a): A white solid was obtained (85% yield), m.p. 90–91 °C. 1H NMR (400 MHz, CDCl3): δ 1.48 (9H, s, t-Bu), 1.51 (9H, s, t-Bu), 4.60 (2H, d, J = 5.2 Hz, CH2), 5.05 (2H, s, CH2), 6.88–6.92 (2H, m, 2 × ArH), 6.96 (1H, t, J = 2.0 Hz, ArH), 7.25 (1H, t, J = 8.0 Hz, ArH), 7.30–7.45 (5H, m, 5 × ArH), 8.59 (1H, br.s, NH), and 11.52 (1H, s, NH). HRMS (ESI): Calcd. for C25H33N3NaO5+ (M + Na)+ 478.2318 and found 478.2312.
tert-Butyl N-[{[(tert-butoxy)carbonyl]amino}[({3-[(4-chlorophenyl)methoxy]phenyl}methyl)amino]methylidene]carbamate (13b): A white solid was obtained (64% yield), m.p. 113–115 °C. 1H NMR (400 MHz, CDCl3): δ 1.48 (9H, s, t-Bu), 1.50 (9H, s, t-Bu), 4.59 (2H, d, J = 5.3 Hz, CH2), 5.01 (2H, s, CH2), 6.85–6.95 (3H, m, 3 × ArH), 7.22–7.35 (5H, m, 5 × ArH), 8.58 (1H, br.s, NH), and 11.53 (1H, s, NH). HRMS (ESI): Calcd. for C25H33ClN3O5 (M + H)+ 490.2109 and found 490.2122.
tert-Butyl N-[{[(tert-butoxy)carbonyl]amino}[({3-[(3-chlorophenyl)methoxy]phenyl}methyl)amino]methylidene]carbamate (13c): A clear oil was obtained (85 % yield). 1H NMR (400 MHz, CDCl3): δ 1.47 (9H, s, t-Bu), 1.50 (9H, s, t-Bu), 4.59 (2H, d, J = 5.5 Hz, CH2), 5.01 (2H, s, CH2), 6.85–6.95 (3H, m, 3 × ArH), 7.22–7.35 (4H, m, 4 × ArH), 7.43 (1H, s, ArH), 8.60 (1H, br.s, NH), and 11.52 (1H, s, NH). HRMS (ESI): Calcd. for C25H33ClN3O5 (M + H)+ 490.2109 and found 490.2135.
tert-Butyl N-[{[(tert-butoxy)carbonyl]amino}[({3-[(2,4-dichlorophenyl)methoxy]-phenyl}methyl)amino]methylidene]carbamate (13d): A white solid was obtained (55% yield), m.p. 111–112 °C. 1H NMR (400 MHz, CDCl3): δ 1.48 (9H, s, t-Bu), 1.51 (9H, s, t-Bu), 4.60 (2H, d, J = 5.1 Hz, CH2), 5.11 (2H, s, CH2), 6.88–6.94 (3H, m, 3 × ArH), 7.24–7.28 (2H, m, 2 × ArH), 7.41 (1H, d, J = 1.9 Hz, ArH), 7.49 (1H, d, J = 8.2 Hz, ArH), 8.59 (1H, br.s, NH), and 11.54 (1H, s, NH). HRMS (ESI): Calcd. for C25H31Cl2N3NaO5 (M + Na)+ 546.1538 and found 546.1507.
tert-Butyl N-[{[(tert-butoxy)carbonyl]amino}[({3-[(3,4-dichlorophenyl) methoxy] phenyl}methyl)amino]methylidene]carbamate (13e): A white solid was obtained (52% yield), m.p. 131–132 °C. 1H NMR (400 MHz, CDCl3): δ 1.48 (9H, s, t-Bu), 1.51 (9H, s, t-Bu), 4.60 (2H, d, J = 5.2 Hz, CH2), 5.00 (2H, s, CH2), 6.75–6.90 (3H, m, 3 × ArH), 7.22–7.30 (2H, m, 2 × ArH), 7.45 (1H, d, J = 8.2 Hz, ArH), 7.53 (1H, d, J = 2.0 Hz, ArH), 8.59 (1H, br.s, NH), and 11.53 (1H, s, NH). HRMS (ESI): Calcd. for C25H31Cl2N3NaO5 (M + Na)+ 546.1538 and found 546.1525.
tert-Butyl N-[{[(tert-Butoxy)carbonyl]amino}[({3-[(2,5-dichlorophenyl) methoxy] phenyl}methyl)amino]methylidene]carbamate (13f): A white solid was obtained (69% yield), m.p. 127–129 °C. 1H NMR (400 MHz, CDCl3): δ 1.48 (9H, s, t-Bu), 1.51 (9H, s, t-Bu), 4.61 (2H, d, J = 5.1 Hz, CH2), 5.10 (2H, s, CH2), 6.89–6.95 (3H, m, 3 × ArH), 7.22–7.33 (3H, m, 3 × ArH), 7.58 (1H, d, J = 2.5 Hz, ArH), 8.60 (1H, br.s, NH), and 11.53 (1H, s, NH). HRMS (ESI): Calcd. for C25H31Cl2N3NaO5 (M + Na)+ 546.1538 and found 546.1506.
tert-Butyl N-[{[(tert-butoxy)carbonyl]amino}[({3-[(2,3-dichlorophenyl) methoxy] phenyl}methyl)amino]methylidene]carbamate (13g): A white solid was obtained (65% yield), m.p. 98–100 °C. 1H NMR (400 MHz, CDCl3): δ 1.48 (9H, s, t-Bu), 1.51 (9H, s, t-Bu), 4.59 (2H, d, J = 5.2 Hz, CH2), 5.16 (2H, s, CH2), 6.87–6.94 (3H, m, 3 × ArH), 7.26 (2H, m, 2 × ArH), 7.46 (2H, m, 2 × ArH), 8.59 (1H, br.s, NH), and 11.54 (1H, s, NH). 13C NMR (100 MHz, CDCl3): δ 28.2, 28.4, 45.0, 67.6, 79.6, 83.4, 114.0, 114.6, 120.9, 126.8, 127.6, 129.8, 130.1, 130.7, 133.2, 137.2, 139.2, 153.3, 156.3, 158.7, and 163.7. HRMS (ESI): Calcd. for C25H31Cl2N3NaO5 (M + Na)+ 546.1538 and found 546.1529.
tert-Butyl N-[{[(tert-butoxy)carbonyl]amino}({[(3-{[4-(trifluoromethyl)phenyl] methoxy}phenyl)methyl]amino})methylidene]carbamate (13h): A white solid was obtained (79% yield), m.p. 108–110 °C. 1H NMR (400 MHz, CDCl3) δ 1.48 (9H, s, t-Bu), 1.51 (9H, s, t-Bu), 4.59 (2H, d, J = 5.2 Hz, CH2), 5.12 (2H, s, CH2), 6.88–6.95 (3H, m, 3 × ArH), 7.25 (1H, t, J = 8.0 Hz, ArH), 7.55 (2H, d, J = 8.2 Hz, 2 × ArH), 7.65 (2H, d, J = 8.2 Hz, 2 × ArH), 8.59 (1H, br.s, NH), and 11.53 (1H, s, NH). HRMS (ESI): Calcd. for C26H33F3N3O5 (M + H)+ 524.2372 and found 524.2354.
tert-Butyl N-[{[(tert-butoxy)carbonyl]amino}({[(3-{[3-(trifluoromethyl)phenyl] methoxy}phenyl)methyl]amino})methylidene]carbamate (13i): A clear oil was obtained (71% yield). 1H NMR (400 MHz, CDCl3): δ 1.48 (9H, s, t-Bu), 1.51 (9H, s, t-Bu), 4.60 (2H, d, J = 5.2 Hz, CH2), 5.12 (2H, s, CH2), 6.87–6.97 (3H, m, 3 × ArH), 7.27 (1H, t, J = 7.8 Hz, ArH), 7.50 (1H, t, J = 7.6 Hz, ArH), 7.58 (1H, d, J = 8.5 Hz, ArH), 7.62 (1H, d, J = 8.5 Hz, ArH), 7.70 (1H, s, ArH), 8.59 (1H, br.s, NH), and 11.54 (1H, s, NH). HRMS (ESI): Calcd. for C26H32F3N3NaO5 (M + Na)+ 546.2192 and found 546.2206.
tert-Butyl N-[{[(tert-butoxy)carbonyl]amino}[({3-[(4-bromophenyl)methoxy] phenyl}methyl)amino]methylidene]carbamate (13j): A white solid was obtained (59% yield), m.p. 121–122 °C. 1H NMR (400 MHz, CDCl3): δ 1.48 (9H, s, t-Bu), 1.50 (9H, s, t-Bu), 4.59 (2H, d, J = 5.3 Hz, CH2), 5.00 (2H, s, CH2), 6.84–6.94 (3H, m, 3 × ArH), 7.25 (1H, t, J = 7.9 Hz, ArH), 7.29–7.33 (2H, m, 2 × ArH), 7.50 (2H, d, J = 8.5 Hz, 2 × ArH), 8.58 (1H, br.s, NH), and 11.52 (1H, s, NH). HRMS (ESI): Calcd. for C25H32BrN3NaO5 (M + Na)+ 556.1423 and found 556.1405.
tert-Butyl N-[{[(tert-butoxy)carbonyl]amino}[({3-[(4-fluorophenyl)methoxy] phenyl}methyl)amino]methylidene]carbamate (13k): A white solid was obtained (57% yield), m.p. 98–99 °C. 1H NMR (400 MHz, CDCl3): δ 1.49 (9H, s, t-Bu), 1.51 (9H, s, t-Bu), 4.59 (2H, d, J = 5.3 Hz, CH2), 5.01 (2H, s, CH2), 6.86–6.94 (3H, m, 3 × ArH), 7.04–7.08 (2H, m, 2 × ArH), 7.25 (1H, t, J = 8.2 Hz, ArH), 7.38–7.42 (2H, m, 2 × ArH), 8.58 (1H, br.s, NH), and 11.53 (1H, s, NH). HRMS (ESI): Calcd. for C25H33FN3O5 (M + H)+ 474.2404 and found 474.2425.
tert-Butyl N-[{[(tert-butoxy)carbonyl]amino}({[(3-{[2-chloro-3-(trifluoromethyl) phenyl]methoxy}phenyl)methyl]amino})methylidene]carbamate (13m): A white solid was obtained (76% yield), m.p. 100–102 °C. 1H NMR (400 MHz, CDCl3): δ 1.49 (9H, s, t-Bu), 1.52 (9H, s, t-Bu), 4.73 (2H, d, J = 5.2 Hz, CH2), 5.21 (2H, s, CH2), 6.91 (1H, d, J = 7.9 Hz, ArH), 6.97 (1H, d, J = 7.9 Hz, ArH), 6.99 (1H, s, ArH), 7.29 (1H, t, J = 7.8 Hz, ArH), 7.40 (1H, t, J = 7.8 Hz, ArH), 7.68 (1H, d, J = 7.8 Hz, ArH), 7.79 (1H, t, J = 7.8 Hz, ArH), 8.65 (1H, br.s, NH), and 11.55 (1H, s, NH). HRMS (ESI): Calcd. for C26H32ClF3N3O5 (M + H)+ 558.1983 and found 558.1971.
tert-Butyl N-[{[(tert-butoxy)carbonyl]amino}[({3-[(3-chlorophenyl)methoxy]-4-methoxyphenyl}methyl)amino]methylidene]carbamate (13n): A white solid was obtained (82% yield), m.p. 79–81 °C. 1H NMR (400 MHz, CDCl3): δ 1.47 (9H, s, t-Bu), 1.51 (9H, s, t-Bu), 3.87 (3H, s, OCH3), 4.49 (2H, d, J = 5.1 Hz, CH2), 5.09 (2H, s, CH2), 6.75–6.80 (3H, m, 3 × ArH), 7.25–7.35 (3H, m, 3 × ArH), 7.45 (1H, s, ArH), 8.49 (1H, br.s, NH), and 11.52 (1H, s, NH). HRMS (ESI): Calcd. for C26H34ClN3NaO6 (M + Na)+ 542.2034 and found 542.2014.
tert-Butyl N-[{[(tert-butoxy)carbonyl]amino}[({3-[(4-chlorophenyl)methoxy]-4-methoxyphenyl}methyl)amino]methylidene]carbamate (13o): A white solid was obtained (79% yield), m.p. 126–128 °C. 1H NMR (400 MHz, CDCl3): δ 1.47 (9H, s, t-Bu), 1.51 (9H, s, t-Bu), 3.80 (3H, s, OCH3), 4.49 (2H, d, J = 5.1 Hz, CH2), 5.09 (2H, s, CH2), 6.85–6.90 (3H, m, 3 × ArH), 7.30–7.40 (4H, m, 4 × ArH), 8.49 (1H, br.s, NH), and 11.53 (1H, s, NH). HRMS (ESI): Calcd. for C26H34ClN3NaO6 (M + Na)+ 542.2034 and found 542.2050.
tert-Butyl N-[{[(tert-butoxy)carbonyl]amino}[({3-[(2,3-dichlorophenyl)methoxy]-2,6-difluorophenyl}methyl)amino]methylidene]carbamate (13p): A white solid was obtained (65% yield), m.p. 140–142 °C. 1H NMR (400 MHz, CDCl3): δ 1.47 (9H, s, t-Bu), 1.52 (9H, s, t-Bu), 4.74 (2H, d, J = 5.1 Hz, CH2), 5.19 (2H, s, CH2), 6.82–6.89 (2H, m, 2 × ArH), 7.25 (1H, t, J = 8.2 Hz, ArH), 7.42 (1H, d, J = 8.2 Hz, ArH), 7.50 (1H, d, J = 8.1 Hz, ArH), 8.52 (1H, br.s, NH), and 11.54 (1H, s, NH). HRMS (ESI): Calcd. for C25H30Cl2F2N3O5 (M + H)+ 560.1530 and found 560.1511.
tert-Butyl N-[{[(tert-butoxy)carbonyl]amino}({[(2,6-difluoro-3-{[3-(trifluoromethyl) phenyl]methoxy}phenyl)methyl]amino})methylidene]carbamate (13q): A white solid was obtained (62% yield), m.p. 85–86 °C. 1H NMR (400 MHz, CDCl3): δ 1.46 (9H, s, t-Bu), 1.51 (9H, s, t-Bu), 4.72 (2H, d, J = 5.1 Hz, CH2), 5.13 (2H, s, CH2), 6.82–6.90 (2H, m, 2 × ArH), 7.51 (1H, t, J = 8.1 Hz, ArH), 7.59–7.63 (2H, m, 2 × ArH), 7.69 (1H, s, ArH), 8.47 (1H, br.s, NH), and 11.53 (1H, s, NH). HRMS (ESI): Calcd. for C26H31F5N3O5 (M + H)+ 560.2184 and found 560.2172.
General Procedure: Synthesis of benzyl guanidine derivatives (9a–q): In a solution of the substituted N,N′-di-Boc-(guanydinomethyl)benzene (13a–q) (0.3 mmol) in CH2Cl2 (2 mL), TFA (1 mL) was added. The mixture was shaken at r.t. overnight and then evaporated to dryness. Et2O (1 mL) was added, and the precipitate was collected, washed with Et2O, and dried in vacuo to give 9a–q as a white or off-white solid.
1-(3-(Benzyloxy)benzyl)guanidinium 2,2,2-trifluoroacetate (9a): A white solid was obtained (95% yield). 1H NMR (400 MHz, CD3OD): δ 4.26 (2H, s, CH2), 4.98 (2H, s, CH2), 6.80–6.95 (2H, m, 2 × ArH), 7.18–7.29 (2H, m, 2 × ArH), 7.24 (2H, m, 2 × ArH), and 7.33 (2H, m, 2 × ArH). HRMS (ESI): Calcd. for C15H18N3O (M + H)+ 256.1450 and found 256.1454.
1-(3-(4-Chlorobenzyloxy)benzyl)guanidinium 2,2,2-trifluoroacetate (9b): An off-white solid was obtained (96% yield). 1H NMR (400 MHz, CD3OD): δ 4.39 (2H, s, CH2), 5.16 (2H, s, CH2), 6.98–7.05 (3H, m, 3 × ArH), 7.37 (1H, t, J = 8.3 Hz, ArH), and 7.43–7.50 (4H, m, 4 × ArH). HRMS (ESI): Calcd. for C15H17ClN3O (M + H)+ 290.1060 and found 290.1066.
1-(3-(3-Chlorobenzyloxy)benzyl)guanidinium 2,2,2-trifluoroacetate (9c): An off-white solid was obtained (85% yield). 1H NMR (400 MHz, CD3OD): δ 4.38 (2H, s, CH2), 5.16 (2H, s, CH2), 6.98–7.05 (3H, m, 3 × ArH), 7.32–7.40 (4H, m, 4 × ArH), and 7.51 (1H, s, ArH). HRMS (ESI): Calcd. for C15H17ClN3O (M + H)+ 290.1060 and found 290.1067.
1-(3-(2,4-Dichlorobenzyloxy)benzyl)guanidinium 2,2,2-trifluoroacetate (9d): A white solid was obtained (98% yield). 1H NMR (400 MHz, DMSO-d6): δ 4.40 (2H, d, J = 5.0 Hz, CH2), 5.20 (2H, s, CH2), 6.95–7.05 (3H, m, 3 × ArH), 7.38 (1H, t, J = 8.0 Hz, ArH), 7.55 (1H, dd, J = 7.9, 1.9 Hz, ArH), 7.67 (1H, d, J = 7.9 Hz, ArH), 7.78 (1H, t, J = 2.1 Hz, ArH), and 8.05 (1H, br.s, NH). HRMS (ESI): Calcd. for C15H16Cl2N3O (M + H)+ 324.0670 and found 324.0665.
1-(3-(3,4-Dichlorobenzyloxy)benzyl)guanidinium 2,2,2-trifluoroacetate (9e): A white solid was obtained (99% yield). 1H NMR (400 MHz, DMSO-d6): δ 4.39 (2H, d, J = 5.1 Hz, CH2), 5.19 (2H, s, CH2), 6.93–7.05 (3H, m, ArH), 7.38 (1H, td, J = 7.9, 1.9 Hz, ArH), 7.50 (1H, dd, J = 8.0, 1.9 Hz, ArH), 7.72 (1H, d, J = 8.1 Hz, ArH), 7.78 1H, (t, J = 1.9 Hz, ArH), and 8.02 (1H, br.s, NH). HRMS (ESI): Calcd. for C15H16Cl2N3O (M + H)+ 324.0670 and found 324.0667.
1-(3-(2,5-Dichlorobenzyloxy)benzyl)guanidinium 2,2,2-trifluoroacetate (9f): A white solid was obtained (98% yield). 1H NMR (400 MHz, DMSO-d6): δ 4.42 (2H, d, J = 6.0 Hz, CH2), 5.20 (2H, s, CH2), 6.95–7.08 (3H, m, 3 × ArH), 7.40 (1H, td, J = 8.0, 1.8 Hz, ArH), 7.55 (1H, dd, J = 8.0, 2.1 Hz, ArH), 7.63 (1H, d, J = 8.0 Hz, ArH), 7.72 (1H, t, J = 2.1 Hz, ArH), and 8.10 (1H, br.s, NH). HRMS (ESI): Calcd. for C15H16Cl2N3O (M + H)+ 324.0670 and found 324.0675.
1-(3-(2,3-Dichlorobenzyloxy)benzyl)guanidinium 2,2,2-trifluoroacetate (9g): A white solid was obtained (92% yield). 1H NMR (400 MHz, DMSO-d6): δ 4.40 (2H, d, J = 5.5 Hz, CH2), 5.25 (2H, s, CH2), 6.95–7.08 (3H, m, 3 × ArH), 7.40 (1H, td, J = 8.0, 1.8 Hz, ArH), 7.48 (1H, t, J = 8.0 Hz, ArH), 7.63 (1H, dd, J = 8.0, 1.8 Hz, ArH), 7.72 (1H, dd, J = 8.0, 1.8 Hz, ArH), and 8.05 (1H, br.s, NH). 13C NMR (100 MHz, DMSO-d6): δ 43.9, 67.3, 113.6, 113.9, 120.0, 128.5, 129.9, 130.3, 130.5, 132.0, 136.9, 139.0, 156.8, and 157.2. (HRMS (ESI): Calcd. for C15H16Cl2N3O (M + H)+ 324.0670 and found 324.0661.
1-(3-(2,3-Dichlorobenzyloxy)benzyl)guanidinium chloride (9g.HCl): Compound 9g (5 mg) was converted to the hydrogen chloride salt by dissolving in a HCl-methanol (0.5M, 2 mL) solution and concentrating in vacuo. A white solid was obtained (4 mg). HRMS (ESI): Calcd. for C15H16Cl2N3O (M + H)+ 324.0670 and found 324.0677.
1-(3-[4-(Trifluoromethyl)benzyloxy]benzyl)guanidinium 2,2,2-trifluoroacetate (9h): A white solid was obtained (97% yield). 1H NMR (400 MHz, DMSO-d6): δ 4.40 (d2H, J = 5.5 Hz, CH2), 5.25 (2H, s, CH2), 6.95–7.12 (2H, m, 2 × ArH), 7.38 (2H, m, 2 × ArH), 7.72 (2H, d, J = 8.2 Hz, 2 × ArH), 7.82 (2H, d, J = 8.2 Hz, 2 × ArH), and 8.05 (1H, br.s, NH). HRMS (ESI): Calcd. for C26H33F3N3O5 (M + H)+ 524.2372 and found 524.2360.
1-(3-[3-(Trifluoromethyl)benzyloxy]benzyl)guanidinium 2,2,2-trifluoroacetate (9i): An off-white solid was obtained (98% yield). 1H NMR (400 MHz, DMSO-d6): δ 4.40 (2H, d, J = 6.1 Hz, CH2), 5.27 (2H, s, CH2), 6.95–7.08 (3H, m, 3 × ArH), 7.13 (1H, td, J = 7.9, 1.7 Hz, ArH), 7.71 (1H, t, J = 7.9 Hz, ArH), 7.78 (1H, d, J = 8.0 Hz, ArH), 7.82 (1H, d, J = 8.0 Hz, ArH), 7.87 (1H, s, ArH), and 8.05 (1H, br.s, NH). HRMS (ESI): Calcd. for C26H33F3N3O5 (M + H)+ 524.2372 and found 524.2397.
1-(3-(4-Bromobenzyloxy)benzyl)guanidinium 2,2,2-trifluoroacetate (9j): A white solid was obtained (97% yield). 1H NMR (400 MHz, DMSO-d6): δ 4.39 (2H, d, J = 6.2 Hz, CH2), 5.14 (2H, s, CH2), 6.95–7.08 (3H, m, 3 × ArH), 7.37 (1H, t, J = 8.0 Hz, ArH), 7.46 (2H, d, J = 8.0 Hz, 2 × ArH), 7.65 (2H, d, J = 8.1 Hz, 2 × ArH), and 8.10 (1H, br.s, NH). HRMS (ESI): Calcd. for C15H16BrN3NaO (M + Na)+ 356.0374 and found 356.0375.
1-(3-(4-Fluorobenzyloxy)benzyl)guanidinium 2,2,2-trifluoroacetate (9k): A white solid was obtained (90% yield). 1H NMR (400 MHz, DMSO-d6): δ 4.40 (2H, d, J = 6.2 Hz, CH2), 5.11 (2H, s, CH2), 6.97–7.10 (3H, m, 3 × ArH), 7.35 (1H, td, J = 8.0, 1.5 Hz, ArH), 7.45 (2H, d, J = 8.2 Hz, 2 × ArH), 7.60 (2H, d, J = 8.2 Hz, 2 × ArH), and 8.07 (1H, br.s, NH). HRMS (ESI): Calcd. for C15H17FN3O (M + H)+ 274.1356 and found 274.1366.
1-(3-[2-Chloro-3-(trifluoromethyl)benzyloxy]benzyl)guanidinium 2,2,2-trifluoroacetate (9m): A white solid was obtained (99% yield). 1H NMR (400 MHz, DMSO-d6): δ 4.41 (2H, d, J = 6.1 Hz, CH2), 5.31 (2H, s, CH2), 6.96–7.08 (3H, m, 3 × ArH), 7.40 (1H, t, J = 8.0 Hz, ArH), 7.68 (1H, t, J = 8.1 Hz, ArH), 7.95 (1H, d, J = 7.9 Hz, ArH), 7.99 (1H, d, J = 7.9 Hz, ArH), and 8.12 (1H, br.s, NH). HRMS (ESI): Calcd. for C16H16ClF3N3O (M + H)+ 358.0934 and found 358.0979.
1-(3-[2-Chloro-3-(trifluorobenzyloxy)benzyl]guanidinium chloride (9m.HCl): Compound 9m (9 mg) was converted to the hydrogen chloride salt by dissolving in a HCl-methanol (0.5 M, 2 mL) solution and concentrating in vacuo. A white solid was obtained (7 mg). HRMS (ESI): Calcd. for C16H16ClF3N3O (M + H)+ 358.0934 and found 358.0897.
1-(3-[3-Chlorobenzyloxy]-4-methoxybenzyl)guanidinium 2,2,2-trifluoroacetate (9n): A white solid was obtained (97% yield). 1H NMR (400 MHz, DMSO-d6): δ 4.36 (2H, d, J = 6.0 Hz, CH2), 5.27 (2H, s, CH2), 6.90–7.15 (3H, m, 3 × ArH), 7.39–7.45 (3H, m, 3 × ArH), 7.65 (1H, s, ArH), and 8.12 (1H, br.s, NH). HRMS (ESI): Calcd. for C16H18ClN3NaO2 (M + Na)+ 342.0985 and found 342.0977.
1-(3-[4-Chlorobenzyloxy]-4-methoxybenzyl)guanidinium 2,2,2-trifluoroacetate (9o): An off-white solid was obtained (95% yield). 1H NMR (400 MHz, DMSO-d6): δ 4.40 (2H, d, J = 6.0 Hz, CH2), 5.29 (2H, s, CH2), 6.87–7.02 (3H, m, 3 × ArH), 7.30–7.39 (4H, m, 4 × ArH), and 8.07 (1H, br.s, NH). HRMS (ESI): Calcd. for C16H18ClN3NaO2 (M + Na)+ 342.0985 and found 342.0994.
1-(3-(2,3-Dichlorobenzyloxy]-2,6-difluorobenzyl)guanidinium 2,2,2-trifluoroacetate (9p): A white solid was obtained (93% yield). 1H NMR (400 MHz, DMSO-d6): δ 4.50 (2H, s, CH2), 5.35 (2H, s, CH2), 7.30–7.40 (2H, m, 2 × ArH), 7.50 (1H, t, J = 7.9 Hz, ArH), 7.63 (1H, d, J = 7.9 Hz, ArH), 7.75 (1H, d, J = 8.0 Hz, ArH), and 7.96 (1H, br.s, NH). HRMS (ESI): Calcd. for C15H14Cl2F2N3O (M + H)+ 360.0482 and found 360.0493.
1-(2,6-Difluoro-3-[3-(trifluoromethyl)benzyloxy]benzyl)guanidinium 2,2,2-trifluoroacetate (9q): A white solid was obtained (98% yield). 1H NMR (400 MHz, DMSO-d6): δ 4.49 (2H, d, J = 5.3 Hz, CH2), 5.35 (2H, s, CH2), 7.18 (1H, dt, J = 7.9, 1.6 Hz, ArH), 7.32–7.40 (2H, m, 2 × ArH), 7.72 (1H, t, J = 8.1 Hz, ArH), 7.81 (1H, d, J = 7.9 Hz, ArH), 7.88 (1H, s, ArH), and 8.00 (1H, br.s, NH). HRMS (ESI): Calcd. for C16H15F5N3O (M + H)+ 360.1135 and found 360.1248.
tert-Butyl N-[{[(tert-butoxy)carbonyl]imino}({[(4-hydroxyphenyl)methyl]amino}) methyl]carbamate (15): In a solution of 4-(aminomethyl)phenol hydrochloride 14 (6.0 mmol) in DMF (20 mL), S-methyl-N,N’-bis(tert-butoxycarbonyl)isothiourea (1.6 g, 5.6 mmol) was added, followed by Et3N (2.0 mL). The mixture was stirred at r.t. overnight and partitioned between EtOAc (100 mL) and citric acid (50 mL, 5% in water). The organic layer was washed with brine, dried (MgSO4), and concentrated in vacuo to give an off-white solid. Purification by flash column chromatography (petrol ether to petrol ether/EtOAc 11:9) afforded 15 as a white solid (1.4 g, 68% yield). mp 137–138 °C. 1H NMR (400 MHz, CDCl3): δ 1.47 (9H, s, t-Bu), 1.49 (9H, s, t-Bu), 4.51 (2H, d, J = 5.2 Hz, CH2), 6.91 (2H, d, J = 8.1 Hz, 2 × ArH), 7.10 (2H, d, J = 8.1 Hz, 2 × ArH), 8.50 (1H, br.s, NH), and 11.5 (1H, s, NH). HRMS (ESI): Calcd. for C18H28N3O5 (M + H)+ 366.2029 and found 366.2037.
General Procedure: Benzylation of 4-(N,N′-di-Boc-guanydinomethyl)phenol (15): In a solution of 4-(N,N′-di-Boc-guanydinomethyl)phenol 15 (0.56 mmol) in acetone (8 mL), the substituted benzyl bromide (0.56 mmol) was added, followed by K2CO3 (96 mg, 0.7 mmol). The mixture was stirred at r.t. overnight and then partitioned between EtOAc (50 mL) and water (50 mL). The organic layer was washed with brine, dried (MgSO4), and concentrated in vacuo to give an off-white solid. Purification by flash column chromatography eluting with a gradient solvent (petrol ether to petrol ether/EtOAc 3:1) afforded 16a–e.
tert-Butyl N-[({[4-(benzyloxy)phenyl]methyl}amino)({[(tert-butoxy) carbonyl]-amino})methylidene]carbamate (16a): A white solid was obtained (82% yield), m.p. 115–116 °C. 1H NMR (400 MHz, CDCl3): δ 1.34 (9H, s, t-Bu), 1.49 (9H, s, t-Bu), 5.10 (2H, s, CH2), 5.15 (2H, br.s, CH2), 6.88 (2H, dt, J = 8.8, 2.0 Hz, 2 × ArH), 7.19 (2H, dt, J = 8.8, 2.1 Hz, 2 × ArH), 7.28–7.44 (5H, m, 5 × ArH), 9.30 (1H, br.s, NH), and 10.5 (1H, br.s, NH). HRMS (ESI): Calcd. for C25H33N3NaO5 (M + Na)+ 478.2318 and found 478.2329.
tert-Butyl N-[{[(tert-butoxy)carbonyl]amino}[({4-[(4-chlorophenyl)methoxy] phenyl}methyl)amino]methylidene]carbamate (16b): A white solid was obtained (79% yield), m.p. 120–121 °C. 1H NMR (400 MHz, CDCl3): δ 1.47 (9H, s, t-Bu), 1.52 (9H, s, t-Bu), 4.65 (2H, br.s, CH2), 5.01 (2H, s, CH2), 6.92 (2H, d, J = 8.2 Hz, 2 × ArH), 7.25 (2H, d, J = 8.2 Hz, 2 × ArH), 7.35 (4H, s, 4 × ArH), 8.75 (1H, br.s, NH), and 11.54 (1H, br.s, NH).
tert-Butyl N-[{[(tert-butoxy)carbonyl]amino}[({4-[(3-chlorophenyl)methoxy] phenyl}methyl)amino]methylidene]carbamate (16c): A white solid was obtained (67% yield), m.p. 120–122 °C. 1H NMR (400 MHz, CDCl3): δ 1.47 (9H, s, t-Bu), 1.52 (9H, s, t-Bu), 4.60 (2H, br.s, CH2), 5.02 (2H, s, CH2), 6.93 (2H, dd, J = 7.1, 2.0 Hz, 2 × ArH), 7.20–7.30 (5H, m, 5 × ArH), 7.43 (1H, s, ArH), 8.62 (1H, br.s, NH), and 11.5 (1H, br.s, NH).
tert-Butyl N-[{[(tert-butoxy)carbonyl]amino}[({4-[(3,4-dichlorophenyl)methoxy] phenyl}methyl)amino]methylidene]carbamate (16d): A white solid was obtained (79% yield), m.p. 156–158 °C. 1H NMR (400 MHz, CDCl3): δ 1.47 (9H, s, t-Bu), 1.52 (9H, s, t-Bu), 4.63 (2H, br.s, CH2), 5.00 (2H, s, CH2), 7.22 (1H, d, J = 7.9 Hz, ArH), 7.43 (1H, d, J = 8.1 Hz, ArH), 7.51 (1H, d, J = 1.6 Hz, ArH), 6.91 (2H, d, J = 7.9 Hz, 2 × ArH), 8.80 (1H, br.s, NH), and 11.5 (1H, br.s, NH).
tert-Butyl N-[{[(tert-butoxy)carbonyl]amino}[({4-[(2,3-dichlorophenyl)methoxy] phenyl}methyl)amino]methylidene]carbamate (16e): A white solid was obtained (89% yield), m.p. 99–101 °C. 1H NMR (400 MHz, CDCl3): δ 1.48 (9H, s, t-Bu), 1.53 (9H, s, t-Bu), 4.73 (2H, br.s, CH2), 5.16 (2H, s, CH2), 6.95 (2H, d, J = 8.1 Hz, 2 × ArH), 7.25–7.31 (3H, m, 3 × ArH), 7.43 (1H, d, J = 8.2 Hz, ArH), 7.47 (1H, d, J = 8.2 Hz, ArH), 8.90 (1H, br.s, NH), and 11.6 (1H, br.s, NH).
General Procedure: Synthesis of benzyl guanidine derivatives (9r–v): In a solution of the para-substituted N,N′-di-Boc-(4-guanidinomethyl)benzene (100 mg) (16a–e) in CH2Cl2 (2 mL), TFA (1 mL) was added. The mixture was shaken at r.t. overnight and then evaporated to dryness. Et2O (1 mL) was added, and the precipitate was collected, washed with ether, and dried in vacuo to give 9r–v as a white or off-white solid.
1-(4-(Benzyloxy)benzyl)guanidinium 2,2,2-trifluoroacetate (9r): A off-white solid was obtained (96% yield). 1H NMR (400 MHz, CD3OD): δ 4.38 (2H, br.s, CH2), 5.16 (2H, s, CH2), 7.06 (2H, d, J = 8.3 Hz, 2 × ArH), 7.32 (2H, d, J = 8.3 Hz, 2 × ArH), 7.36–7.43 (3H, m, 3 × ArH), and 7.50 (2H, d, J = 8.2 Hz, 2 × ArH). HRMS (ESI): Calcd. for C15H18N3O (M + H)+ 256.1450 and found 256.1539.
1-(4-(4-Chlorobenzyloxy)benzyl)guanidinium 2,2,2-trifluoroacetate (9s): A white solid was obtained (87% yield). 1H NMR (400 MHz, DMSO-d6): δ 4.55 (2H, br.s, CH2), 5.17 (2H, s, CH2), 6.94 (2H, d, J = 8.2 Hz, 2 × ArH), 7.27 (2H, d, J = 8.1 Hz, 2 × ArH), 7.37–7.41 (4H, m, 4 × ArH), and 8.15 (br s, 1H, NH). HRMS (ESI): Calcd. for C15H17ClN3O (M + H)+ 290.1060 and found 290.1066.
1-(4-(3-Chlorobenzyloxy)benzyl)guanidinium 2,2,2-trifluoroacetate (9t): A white solid was obtained (93% yield). 1H NMR (400 MHz, DMSO-d6): δ 4.33 (2H, br.s, CH2), 5.19 (2H, s, CH2), 7.08 (3H, m, 3 × ArH), 7.30 (2H, m, 2 × ArH), 7.40–7.50 (3H, m, 3 × ArH), and 7.95 (1H, br.s, NH). HRMS (ESI): Calcd. for C15H17ClN3O (M + H)+ 290.1060 and found 290.1071.
1-(4-(3,4-Dichlorobenzyloxy)benzyl)guanidinium 2,2,2-trifluoroacetate (9u): A white solid was obtained (93% yield). 1H NMR (400 MHz, DMSO-d6): δ 4.33 (2H, d, J = 6.0 Hz, CH2), 5.19 (2H, s, CH2), 7.09 (2H, d, J = 8.6 Hz, 2 × ArH), 7.31 (2H, d, J = 8.5 Hz, 2 × ArH), 7.50 (1H, dd, J = 7.8, 2.1 Hz, 2 × ArH), 7.72 (1H, d, J = 7.7 Hz, ArH), 7.77 (1H, d, J = 2.0 Hz, ArH), and 7.87 (1H, br.s, NH). HRMS (ESI): Calcd. for C15H15Cl2N3O (M + H)+ 324.0670 and found 324.0658.
1-(4-(2,3-Dichlorobenzyloxy)benzyl)guanidinium 2,2,2-trifluoroacetate (9v): A white solid was obtained (98% yield). 1H NMR (400 MHz, DMSO-d6): δ 4.38 (2H, br.s, CH2), 5.35 (2H, s, CH2), 7.14 (2H, d, J = 8.2 Hz, 2 × ArH), 7.32 (2H, d, J = 8.3 Hz, 2 × ArH), 7.46 (1H, t, J = 7.9 Hz, ArH), 7.63 (1H, d, J = 8.1 Hz, ArH), 7.72 (1H, d, J = 7.9 Hz, ArH), and 8.05 (1H, br.s, NH). 13C NMR (DMSO-d6): δ 43.5, 67.3, 114.9, 128.4, 128.4, 128.9, 130.2, 130.5, 132.0, 137.0, 156.8, and 157.5. HRMS (ESI): Calcd. for C15H15Cl2N3O (M + H)+ 324.0670 and found 324.0720.
General procedure: Synthesis of Boc-protected aminobenzyl guanidine derivatives (18, 22): 3-Aminobenzylamine 17 or 4-aminobenzylamine 21 (10 mmol) was dissolved in DMF (8 mL). S-methyl-N,N’-bis(tert-butoxycarbonyl)isothiourea (10.5 mmol) and Et3N (20 mmol) were added successively at 0 °C. The reaction mixture was stirred for 18 h at r.t. and then evaporated at 70 °C. Water (180 mL) and brine (20 mL) were added, and the mixture was extracted with Et2O (2 × 200 mL). The organic layer was dried (Na2SO4), filtered, and concentrated in vacuo. Purification by flash column chromatography (CH2Cl2, 100%) gave 18 and 22.
tert-Butyl N-[(1E)-{[(3-aminophenyl)methyl]amino}({[(tert-butoxy)carbonyl] imino})methyl]carbamate (18): 2.85 g, 78%, white foam. 1H NMR (400 MHz, DMSO-d6): δ 1.44 (9H, s, t-Bu), 1.53 (9H, s, t-Bu), 4.41 (2H, d, J = 5.6 Hz, CH2), 5.14 (2H, br.s, NH2), 6.46 (1H, d, J = 7.2 Hz, ArH), 6.50 (1H, s, ArH), 6.51 (1H, d, J = 7.2 Hz, ArH), 7.02 (1H, t, J = 8.0 Hz, ArH), 8.56 (1H, t, J = 5.6 Hz, NH), and 11.60 (1H, br.s, NH). HRMS (ESI): Calcd. for C18H29N4O4 (M + H)+ 365.2183 and found 365.2189.
tert-Butyl N-[(1E)-{[(4-aminophenyl)methyl]amino}({[(tert-butoxy)carbonyl] imino})methyl]carbamate (22): 2.98 g, 81%, white foam. 1H NMR (400 MHz, DMSO-d6): δ 1.45 (9H, s, t-Bu), 1.50 (9H, s, t-Bu), 4.34 (2H, d, J = 5.6 Hz, CH2), 5.10 (2H, br.s, NH2), 6.58 (2H, d, J = 7.6 Hz, 2 × ArH), 7.03 (2H, d, J = 8.0 Hz, 2 × ArH), 8.41 (1H, t, J = 5.6 Hz, NH), and 11.55 (1H, br.s, NH). HRMS (ESI): Calcd. for C18H29N4O4 (M + H)+ 365.2183 and found 365.2187.
General Procedure: Synthesis of Boc-protected sulphonamide derivatives (19a–d, 23a–d): Compound 18 or 22 (0.2 mmol) was dissolved in CH2Cl2 (1.2 mL) and pyridine (0.8 mL) at 0 °C. The corresponding benzene sulphonyl chloride (0.22 mmol) was added, and the reaction mixture was stirred for 2 h at 0 °C. Water (40 mL) and brine (10 mL) were added, and the mixture was extracted with Et2O (2 × 50 mL). The organic layer was dried (Na2SO4), filtered, and concentrated to dryness. The residue was then co-evaporated with toluene (2 × 5 mL) and CH2Cl2 (2 × 5 mL) to give 19a–d or 23a–d.
tert-Butyl N-[(1E)-{[(tert-butoxy)carbonyl]imino}({[3-(3-chlorobenzene sulfonamido)phenyl]methyl}amino)methyl]carbamate (19a): 99 mg, 92%, white foam. 1H NMR (400 MHz, CDCl3): δ 1.47 (9H, s, t-Bu), 1.48 (9H, s, t-Bu), 4.51 (2H, s, CH2), 6.98–7.06 (3H, m, 3 × ArH), 7.18 (1H, t, J = 8.0 Hz, ArH), 7.34 (1H, t, J = 7.8 Hz, ArH), 7.46 (1H, ddd, J = 8.0, 2.0, 0.8 Hz, ArH), 7.54 (1H, br.s, NH), 7.64 (1H, ddd, J = 7.8, 1.6, 1.2 Hz, ArH), 7.79 (1H, t, J = 1.8 Hz, ArH), 8.60 (1H, br.s, NH), and 11.52 (1H, br.s, NH). HRMS (ESI): Calcd. for C24H32ClN4O6S (M + H)+ 539.1726 and found 539.1737.
tert-Butyl N-[(1E)-{[(tert-butoxy)carbonyl]imino}({[3-(4-chlorobenzene sulfonamido)phenyl]methyl}amino)methyl]carbamate (19b): 98 mg, 91%, foam. 1H NMR (400 MHz, CDCl3): δ 1.47 (9H, s, t-Bu), 1.48 (9H, s, t-Bu), 4.53 (2H, s, CH2), 7.00 (1H, d, J = 2.4 Hz, ArH), 7.02 (1H, d, J = 2.4 Hz, ArH), 7.07 (1H, s, ArH), 7.17 (1H, t, J = 7.8 Hz, ArH), 7.35 (2H, dt, J = 8.8, 2.2 Hz, 2 × ArH), 7.65 (1H, br.s, NH), 7.70 (2H, dt, J = 8.8, 2.2 Hz, 2 × ArH), 8.61 (1H, br.s, NH), and 11.52 (1H, br.s, NH). HRMS (ESI): Calcd. for C24H32ClN4O6S (M + H)+ 539.1726 and found 539.1738.
tert-Butyl N-[(1E)-{[(tert-butoxy)carbonyl]imino}({[3-(2,3-dichlorobenzene sulfonamido)phenyl]methyl}amino)methyl]carbamate (19c): 107 mg, 93%, yellow foam. 1H NMR (400 MHz, CDCl3): δ 1.48 (9H, s, t-Bu), 1.49 (9H, s, t-Bu), 4.55 (2H, s, CH2), 6.98–7.04 (2H, m, 2 × ArH), 7.08 (1H, s, ArH), 7.17 (1H, t, J = 7.8 Hz, ArH), 7.27 (1H, t, J = 8.0 Hz, ArH), 7.46 (1H, br.s, NH), 7.60 (1H, d, J = 8.0 Hz, ArH), 7.97 (1H, d, J = 8.0 Hz, ArH), 8.61 (1H, br.s, NH), and 11.52 (1H, br.s, NH). HRMS (ESI): Calcd. for C24H31Cl2N4O6S (M + H)+ 573.1336 and found 573.1349.
tert-Butyl N-[(1E)-{[(tert-butoxy)carbonyl]imino}[({3-[3-(trifluoromethyl) benzenesulfonamido]phenyl}methyl)amino]methyl]carbamate (19d): 107 mg, 93%, colourless glass. 1H NMR (400 MHz, CDCl3): δ 1.47 (18H, s, 2 × t-Bu), 4.52 (2H, s, CH2), 6.99–7.07 (3H, m, 3 × ArH), 7.18 (1H, t, J = 7.8 Hz, ArH), 7.55 (1H, t, J = 7.8 Hz, ArH), 7.68 (1H, br.s, NH), 7.75 (1H, d, J = 7.6 Hz, ArH), 7.95 (1H, d, J = 7.6 Hz, ArH), 8.05 (1H, s, ArH), 8.62 (1H, br.s, NH), and 11.51 (1H, br.s, NH). HRMS (ESI): Calcd. for C25H32F3N4O6S (M + H)+ 573.1989 and found 573.1998.
tert-Butyl N-[(1E)-{[(tert-butoxy)carbonyl]imino}({[4-(3-chloro benzenesulfonamido)phenyl]methyl}amino)methyl]carbamate (23a): 105 mg, 97%, pale yellow foam. 1H NMR (400 MHz, CDCl3): δ 1.46 (9H, s, t-Bu), 1.47 (9H, s, t-Bu), 4.54 (2H, s, CH2), 7.07 (2H, d, J = 8.0 Hz, 2 × ArH), 7.13 (2H, d, J = 8.4 Hz, 2 × ArH), 7.34 (1H, t, J = 8.0 Hz, ArH), 7.47 (1H, dd, J = 8.0, 1.2 Hz, ArH), 7.65 (1H, d, J = 7.6 Hz, ArH), 7.73 (1H, br.s, NH), 7.78 (1H, s, ArH), 8.61 (1H, br.s, NH), and 11.50 (1H, br.s, NH). HRMS (ESI): Calcd. for C24H32ClN4O6S (M + H)+ 539.1726 and found 539.1739.
tert-Butyl N-({[(tert-butoxy)carbonyl]imino}({[4-(4-chlorobenzenesulfonamido) phenyl]methyl}amino)methyl)carbamate (23b): 103 mg, 95%, white foam. 1H NMR (400 MHz, CDCl3): δ 1.46 (9H, s, t-Bu), 1.47 (9H, s, t-Bu), 4.55 (2H, s, CH2), 7.06 (2H, d, J = 8.4 Hz, 2 × ArH), 7.14 (2H, d, J = 8.4 Hz, 2 × ArH), 7.38 (2H, dt, J = 8.8, 2.2 Hz, 2 × ArH), 7.60 (1H, br.s, NH), 7.71 (2H, dd, J = 8.8, 2.2 Hz, 2 × ArH), 8.58 (1H, br.s, NH), and 11.51 (1H, br.s, NH). HRMS (ESI): Calcd. for C24H32ClN4O6S (M + H)+ 539.1726 and found 539.1741.
tert-Butyl N-({[(tert-butoxy)carbonyl]imino}({[4-(2,3-dichlorobenzene sulfonamido)phenyl]methyl}amino)methyl)carbamate (23c): 113 mg, 98%, yellow foam. 1H NMR (400 MHz, CDCl3): δ 1.45 (9H, s, t-Bu), 1.47 (9H, s, t-Bu), 4.51 (2H, s, CH2), 7.08 (2H, d, J = 8.8 Hz, 2 × ArH), 7.13 (2H, d, J = 8.4 Hz, 2 × ArH), 7.27 (1H, t, J = 8.2 Hz, ArH), 7.49 (1H, br.s, NH), 7.61 (1H, dd, J = 8.0, 1.6 Hz, ArH), 7.93 (1H, dd, J = 8.0, 1.6 Hz, ArH), 8.51 (1H, br.s, NH), and 11.50 (1H, br.s, NH). HRMS (ESI): Calcd. for C24H31Cl2N4O6S (M + H)+ 573.1336 and found 573.1347.
tert-Butyl N-[(1E)-{[(tert-butoxy)carbonyl]imino}[({4-[3-(trifluoromethyl) benzenesulfonamido]phenyl}methyl)amino]methyl]carbamate (23d): 111 mg, 97%, pale yellow foam. 1H NMR (400 MHz, CDCl3): δ 1.47 (9H, s, t-Bu), 1.48 (9H, s, t-Bu), 4.57 (2H, s, CH2), 7.07 (2H, d, J = 8.4 Hz, 2 × ArH), 7.13 (2H, d, J = 8.4 Hz, 2 × ArH), 7.56 (1H, t, J = 7.8 Hz, ArH), 7.69 (1H, br.s, NH), 7.76 (1H, d, J = 8.0 Hz, ArH), 7.96 (1H, d, J = 8.0 Hz, ArH), 8.04 (1H, s, ArH), 8.66 (1H, br.s, NH), and 11.50 (1H, br.s, NH). HRMS (ESI): Calcd. for C25H32F3N4O6S (M + H)+ 573.1989 and found 573.1996.
General Procedure: Synthesis of Boc-protected amide derivatives (19e, 23e): Compound 18 or 22 (0.5 mmol) and K2CO3 (1.0 mmol) were placed in an oven-dried 50 mL glass tube. Acetone (2.0 mL) and benzoyl chloride (0.75 mmol) were added successively, and the reaction mixture was stirred for 18 h at 80 °C. Water (80 mL) and brine (20 mL) were added, and the mixture was extracted with Et2O (2 × 100 mL). The organic layer was dried (Na2SO4), filtered, and concentrated to dryness. Purification by flash column chromatography (petrol ether to petrol ether/EtOAc 4:1) gave 19e or 23e.
tert-Butyl N-[(1E)-{[(3-benzamidophenyl)methyl]amino}({[(tert-butoxy) carbonyl]-imino})methyl]carbamate (19e): 45 mg, 19%, colourless glass. 1H NMR (400 MHz, CDCl3): δ 1.47 (9H, s, t-Bu), 1.48 (9H, s, t-Bu), 4.59 (2H, d, J = 5.2 Hz, CH2), 7.01 (1H, d, J = 7.6 Hz, ArH), 7.30 (1H, t, J = 8.0 Hz, ArH), 7.42–7.49 (3H, m, 3 × ArH), 7.53 (1H, t, J = 7.6 Hz, CH), 7.70 (1H, t, J = 8.0 Hz, ArH), 7.87–7.93 (2H, m, 2 × ArH), 8.19 (1H, s, NH), 8.70 (1H, s, NH), and 11.53 (1H, br.s, NH). HRMS (ESI): Calcd. for C25H33N4O5 (M + H)+ 469.2445 and found 469.2452.
tert-Butyl N-[(1E)-{[(4-benzamidophenyl)methyl]amino}({[(tert-butoxy) carbonyl]-imino})methyl]carbamate (23e): 27 mg, 11%, colourless glass. 1H NMR (400 MHz, CDCl3): δ 1.47 (9H, s, t-Bu), 1.48 (9H, s, t-Bu), 4.58 (2H, d, J = 5.2 Hz, CH2), 7.24 (2H, d, J = 8.4 Hz, 2 × ArH), 7.45 (2H, t, J = 7.4 Hz, 2 × ArH), 7.52 (1H, t, J = 7.2 Hz, ArH), 7.61 (2H, d, J = 8.8 Hz, 2 × ArH), 7.87 (2H, d, J = 8.4 Hz, 2 × ArH), 8.14 (1H, s, NH), 8.63 (1H, s, NH), and 11.53 (1H, br.s, NH). HRMS (ESI): Calcd. for C25H33N4O5 (M + H)+ 469.2445 and found 469.2451.
General procedure: Synthesis of Amino((arylsulfonamido)benzyl) amino) meth-animinium 2,2,2-trifluoroacetate derivatives (20a–d, 24a–d): Compound 19a–d, 23a–d (0.1 mmol) was dissolved in CH2Cl2 (0.8 mL) and then TFA (0.2 mL) was added. The reaction mixture was stirred for 2 h at r.t. and concentrated to dryness to give 20a–d and 24a–d.
Amino((3-((3-chlorophenyl)sulfonamido)benzyl)amino)methaniminium 2,2,2-trifluoroacetate (20a): 89 mg, 98%, glass. 1H NMR (400 MHz, CD3OD): δ 4.37 (2H, s, CH2), 7.07 (1H, d, J = 8.0 Hz, ArH), 7.13 (1H, d, J = 7.6 Hz, ArH), 7.23 (1H, s, ArH), 7.33 (1H, t, J = 7.8 Hz, ArH), 7.54 (1H, t, J = 8.0 Hz, ArH), 7.65 (1H, d, J = 8.0 Hz, ArH), 7.75 (1H, d, J = 8.0 Hz, ArH), and 7.81 (1H, s ArH). HRMS (ESI): Calcd. for C14H16ClN4O2S (M + H)+ 339.0677 and found 339.0681.
Amino((3-((4-chlorophenyl)sulfonamido)benzyl)amino)methaniminium 2,2,2-trifluoroacetate (20b): 88 mg, 97%, glass. 1H NMR (400 MHz, CD3OD): δ 4.38 (2H, s, CH2), 7.07 (1H, d, J = 8.0 Hz, ArH), 7.11 (1H, d, J = 7.6 Hz, ArH), 7.23 (1H, s, ArH), 7.31 (1H, t, J = 8.0 Hz, ArH), 7.54 (2H, d, J = 8.4 Hz, 2 × ArH), and 7.81 (2H, d, J = 8.4 Hz, 2 × ArH). HRMS (ESI): Calcd. for C14H16ClN4O2S (M + H)+ 339.0677 and found 339.0685.
Amino((3-((2,3-dichlorophenyl)sulfonamido)benzyl)amino)methaniminium 2,2,2-trifluoroacetate (20c): 96 mg, 98%, glass. 1H NMR (400 MHz, CD3OD): δ 4.39 (2H, s, CH2), 7.06 (1H, d, J = 7.6 Hz, ArH), 7.13 (1H, d, J = 8.0 Hz, ArH), 7.22 (1H, s, ArH), 7.30 (1H, t, J = 8.0 Hz, ArH), 7.49 (1H, dt, J = 8.2, 0.8 Hz, ArH), 7.81 (1H, d, J = 8.0 Hz, ArH), and 8.11 (1H, d, J = 8.0 Hz, ArH). HRMS (ESI): Calcd. for C14H15Cl2N4O2S (M + H)+ 373.0287 and found 373.0295.
Amino((3-((3-(trifluoromethyl)phenyl)sulfonamido)benzyl)amino) methaniminium 2,2,2-trifluoroacetate (20d): 95 mg, 97%, glass. 1H NMR (400 MHz, CD3OD): δ 4.42 (2H, s, CH2), 7.07 (1H, d, J = 8.0 Hz, ArH), 7.14 (1H, d, J = 7.6 Hz, ArH), 7.24 (1H, s, ArH), 7.32 (1H, t, J = 8.0 Hz, ArH), 7.77 (1H, d, J = 8.2 Hz, ArH), 7.96 (1H, d, J = 7.6 Hz, ArH), and 8.07 (2H, s, 2 × ArH). HRMS (ESI): Calcd. for C15H16F3N4O2S (M + H)+ 373.0941 and found 373.0946.
Amino((4-((3-chlorophenyl)sulfonamido)benzyl)amino)methaniminium 2,2,2-trifluoroacetate (24a): 90 mg, 99%, glass. 1H NMR (400 MHz, CD3OD): δ 4.38 (2H, s, CH2), 7.17–7.22 (2H, m, 2 × ArH), 7.24–7.31 (2H, m, 2 × ArH), 7.53 (1H, t, J = 7.4 Hz, ArH), 7.60–7.66 (1H, m, ArH), 7.71–7.77 (1H, m, ArH), and 7.77–7.81 (1H, m, ArH). HRMS (ESI): Calcd. for C14H16ClN4O2S (M + H)+ 339.0677 and found 339.0685.
Amino((4-((4-chlorophenyl)sulfonamido)benzyl)amino)methaniminium 2,2,2-trifluoroacetate (24b): 89 mg, 98%, glass, 1H NMR (400 MHz, CD3OD): δ 4.38 (2H, s, CH2), 7.20 (2H, d, J = 8.4 Hz, 2 × ArH), 7.27 (2H, d, J = 8.4 Hz, 2 × ArH), 7.55 (2H, d, J = 8.4 Hz, 2 × ArH), and 7.80 (2H, d, J = 8.4 Hz, 2 × ArH). HRMS (ESI): Calcd. for C14H16ClN4O2S (M + H)+ 339.0677 and found 339.0686.
Amino((4-((2,3-dichlorophenyl)sulfonamido)benzyl)amino)methaniminium 2,2,2-trifluoroacetate (24c): 97 mg, >99%, glass. 1H NMR (400 MHz, CD3OD): δ 4.34 (2H, s, CH2), 7.21–7.28 (4H, m, 4 × ArH), 7.46 (1H, t, J = 8.0 Hz, ArH), 7.81 (1H, dd, J = 8.0, 1.6 Hz, ArH), and 8.10 (1H, dd, J = 8.0, 1.6 Hz, ArH). HRMS (ESI): Calcd. for C14H15Cl2N4O2S (M + H)+ 373.0287 and found 373.0296.
Amino((4-((3-trifluoromethyl)phenyl)sulfonamido)benzyl)amino) methaniminium 2,2,2-trifluoroacetate (24d): 95 mg, 97%, glass. 1H NMR (400 MHz, CD3OD): δ 4.38 (2H, s, CH2), 7.20 (2H, d, J = 8.8 Hz, 2 × ArH), 7.29 (2H, d, J = 8.4 Hz, 2 × ArH), 7.76 (1H, t, J = 8.2 Hz, ArH), 7.95 (1H, d, J = 8.0 Hz, ArH), and 8.05–8.11 (2H, m, 2 × ArH). HRMS (ESI): Calcd. for C15H16F3N4O2S (M + H)+ 373.0941 and found 373.0945.
General procedure: Synthesis of amino((benzamidobenzyl)amino) methaniminium 2,2,2-trifluoroacetate derivatives (20e, 24e): Compound 19e or 23e (0.1 mmol) was dissolved in CH2Cl2 (0.8 mL) and then TFA (0.2 mL) was added. The reaction mixture was stirred for 2 h at 0 °C. The mixture was concentrated to dryness to give 20a or 24e.
Amino((3-benzamidobenzyl)amino)methaniminium 2,2,2-trifluoroacetate (20e): 38 mg, 99%, pale yellow glass. 1H NMR (400 MHz, DMSO-d6): δ 4.44 (2H, s, CH2), 7.10 (1H, d, J = 8.0 Hz, ArH), 7.42 (1H, t, J = 8.0 Hz, ArH), 7.56–7.62 (3H, m, 3 × ArH), 7.65 (1H, d, J = 7.2 Hz, ArH), 7.90 (1H, s, ArH), 8.00 (2H, d, J = 7.2 Hz, 2 × ArH), and 8.01 (1H, s, NH). HRMS (ESI): Calcd. for C15H17N4O (M + H)+ 269.1397 and found: 269.1401.
Amino((4-benzamidobenzyl)amino)methaniminium 2,2,2-trifluoroacetate (24e): 38 mg, 99%, pale yellow glass. 1H NMR (400 MHz, DMSO-d6): δ 4.39 (2H, s, CH2), 7.35 (2H, d, J = 8.4 Hz, 2 × ArH), 7.59 (2H, t, J = 7.2 Hz, 2 × ArH), 7.66 (1H, t, J = 7.2 Hz, ArH), 7.84 (2H, d, J = 8.4 Hz, 2 × ArH), 8.00 (2H, d, J = 8.0 Hz, 2 × ArH), and 8.01 (1H, s, NH). HRMS (ESI): Calcd. for C15H17N4O (M + H)+ 269.1397 and found: 269.1402.
tert-Butyl 7-hydroxy-1,2,3,4-dihydroisoquinoline-2-carboxylate (26a): In a solution of 25a (349 mg, 2.34 mmol) in THF/water (5 mL/1 mL), Boc2O (545 mg, 2.5 mmol) and Et3N (0.4 mL, 2.8 mmol) were added. The mixture was stirred at r.t. for 16 h and partitioned between EtOAc (50 mL) and water (50 mL). The organic layer was washed with brine, dried (MgSO4), and concentrated in vacuo. Purification by flash column chromatography (petrol ether/EtOAc 7:3) gave a white solid (475 mg, 81% yield), m.p. 130–131 °C. 1H NMR (400 MHz, CDCl3): δ 1.49 (9H, s, t-Bu), 2.74 (2H, t, J = 5.9 Hz, CH2), 3.62 (2H, t, J = 6.0 Hz, CH2), 4.51 (2H, s, CH2), 6.62–6.65 (2H, m, 2 × ArH), and 6.98 (1H, d, J = 8.2 Hz, ArH).
tert-Butyl 7-(2,3-dichlorobenzyloxy)-1,2,3,4-tetrahydroisoquinoline-2-carboxylate (27a): In a solution of 26a (370 mg, 1.48 mmol) in acetone (15 mL), 2,3-dichlorobenzyl bromide (437 mg, 1.6 mmol) was added, followed by K2CO3 (262 mg, 1.9 mmol). The mixture was stirred at r.t. overnight and partitioned between EtOAc (30 mL) and water (30 mL). The organic layer was washed with brine, dried (MgSO4), and concentrated in vacuo. Purification by flash column chromatography (petrol ether to petrol ether/EtOAc 3:1) afforded 27a as clear oil (500 mg, 83%). 1H NMR (400 MHz, CDCl3): δ 1.48 (9H, s, t-Bu), 2.76 (2H, t, J = 5.7 Hz, CH2), 3.62 (2H, t, J = 5.7 Hz, CH2), 4.52 (2H, s), 5.15 (2H, s), 6.75–6.79 (2H, m, 2 × ArH), 7.05 (1H, d, J = 8.4 Hz, ArH), 7.23 (1H, t, J = 7.9 Hz, ArH), and 7.45 (2H, m, 2 × ArH). HRMS (ESI): Calcd. for C21H23Cl2NNaO3 (M + Na)+ 430.0953 and found 430.0970.
tert-Butyl N-[{[(tert-butoxy)carbonyl]imino}({7-(2,3-dichlorobenzyloxy)-1,2,3,4-tetrahydroisoquinolin-2-yl})methyl]carbamate (28a): In a solution of 27a (450 mg, 1.1 mmol) in CH2Cl2 (63 mL), TFA (2 mL) was added. The mixture was shaken at r.t. for 10 h and evaporated in vacuo to give to a yellow residue. The crude product was dissolved in DMF (5 mL) and Et3N (0.6 mL). S-Methyl-N,N’-bis(tert-butoxycarbonyl)isothiourea (390 mg, 1.32 mmol) was added, followed by HgCl2 (200 mg, 2.37 mmol). The mixture was stirred at r.t. for 16 h, diluted with EtOAc (50 mL), and filtered through Celite. The organic layer was washed with brine, dried (MgSO4), and concentrated in vacuo. Purification by flash column chromatography (petrol ether to petrol ether/EtOAc 3:2) afforded 28a as a foamy powder solid (340 mg, 56% yield), m.p. 59–61 °C. 1H NMR (400 MHz, CDCl3): δ 1.50 (18H, s, 2 × t-Bu), 2.98 (2H, t, J = 5.7 Hz, CH2), 3.80 (2H, t, J = 5.7 Hz, CH2), 4.75 (2H, s), 5.12 (2H, s), 6.70 (1H, br.s, ArH), 6.81 (1H, dd, J = 8.0, 1.9 Hz, ArH), 7.05 (1H, d, J = 8.0 Hz, ArH), 7.22 (1H, d, J = 8.0 Hz, ArH), 7.40–7.46 (2H m, 2 × ArH), and 10.2 (1H, s, NH). HRMS (ESI): Calcd. for C27H34Cl2N3O5 (M + H)+ 550.1876 and found 550.1876.
7-(2,3-Dichlorobenzyloxy)-1,2,3,4-tetrahydroisoquinoline-2-carboximidamide 2,2,2-trifluoroacetate (29a): The compound was synthesised as described for 28a. A white solid (30 mg, 82%) was obtained. 1H NMR (400 MHz, DMSO-d6): δ 2.89 (2H, t, J = 5.5 Hz, CH2), 3.50 (2H, t, J = 5.2 Hz, CH2), 4.51 (2H, s), 5.20 (2H, s), 6.85 (1H, d, J = 1.7 Hz, ArH), 6.95 (1H, dd, J = 7.8, 1.5 Hz, ArH), 7.25 (1H, d, J = 7.7 Hz, ArH), 7.45 (1H, t, J = 7.9 Hz, ArH), 7.60 (1H, d, J = 8.0 Hz, ArH), and 7.70 (1H, d, J = 8.0 Hz, ArH). HRMS (ESI): Calcd. for C17H18Cl2N3O (M + H)+ 350.0827 and found 350.0821.
tert-Butyl 5-hydroxy-1,2,3,4-tetrahydroisoquinoline-2-carboxylate (26b): A solution of 25b (1.0 g, 90% purity, 6.2 mmol) in AcOH (20 mL) was reacted over H2 (1 atm) and PtO2 (85 mg) at r.t. for 48 h. The reaction mixture was then filtered through Celite and concentrated in vacuo. The residue was dissolved in acetone (3 mL) and diluted with Et2O (3 mL). The precipitate was collected and dried in vacuo. The crude product (700 mg, 4.7 mmol) was suspended in THF/water (10 mL/2 mL). Boc2O (1.1 g, 5.0 mmol) and Et3N (1.5 mL, 10 mmol) were added. The mixture was stirred at r.t. for 16 h and partitioned between EtOAc (50 mL) and water (50 mL). The organic layer was washed with brine, dried (MgSO4), and concentrated in vacuo. Purification by flash column chromatography (petrol ether/EtOAc 7:3) gave 26b as a white solid (490 mg, 42% yield), m.p. 156–158 °C. 1H NMR (400 MHz, CDCl3): δ 1.48 (s, 9H, t-Bu), 2.75 (2H, t, J = 6.0 Hz, CH2), 3.65 (2H, t, J = 6.1 Hz, CH2), 4.55 (2H, s, CH2), 6.63 (1H, d, J = 7.8 Hz, ArH), 6.68 (1H, d, J = 7.9 Hz, ArH), and 7.03 (1H, d, J = 7.8 Hz, ArH). HRMS (ESI): Calcd. for C14H19NNaO3 (M + Na)+ 272.1263 and found 272.1244.
tert-Butyl 5-(2,3-dichlorobenzyloxy)-1,2,3,4-tetrahydroisoquinoline-2-carboxylate (27b): The compound was synthesised as described for 27a. A white solid (480 mg, 79%) was obtained, m.p. 138–139 °C. 1H NMR (400 MHz, CDCl3): δ 1.49 (9H, s, t-Bu), 2.86 (2H, t, J = 5.8 Hz, CH2), 3.67 (2H, t, J = 5.9 Hz, CH2), 4.58 (2H, s), 5.16 (2H, s), 6.74–6.77 (2H, m, 2 × ArH), 7.14 (1H, t, J = 8.2 Hz, ArH), 7.22–7.26 (2H, m, 2 × ArH), 7.44 (1H, d, J = 8.7 Hz, ArH), and 7.49 (1H, d, J = 9.0 Hz, ArH). HRMS (ESI): Calcd. for C21H23Cl2NNaO3 (M + Na)+ 430.0953 and found 430.0917.
tert-Butyl N-[{[(tert-butoxy)carbonyl]amino}({5-(2,3-dichlorobenzyloxy)-1,2,3,4-tetrahydroisoquinolin-2-yl})methylidene]carbamate (28b): The compound was synthesised as described for 28a. A white solid (280 mg, 59%) was obtained, m.p. 129–130 °C. 1H NMR (400 MHz, CDCl3): δ 1.47 (9H, s, t-Bu), 1.51 (9H, s, t-Bu), 2.91 (2H, t, J = 5.5 Hz, CH2), 3.71 (2H, t, J = 5.5 Hz, CH2), 4.62 (2H, s), 5.21 (2H, s), 6.79–6.82 (2H, m, 2 × ArH), 7.18 (1H, t, J = 8.3 Hz, ArH)), 7.31–7.34 (2H, m, 2 × ArH), 7.46 (1H, d, J = 8.5 Hz, ArH), and 7.52 (1H, d, J = 8.3 Hz, ArH). HRMS (ESI): Calcd. for C27H34Cl2N3O5 (M + H)+ 550.1876 and found 550.1882.
5-(2,3-Dichlorobenzyloxy)-1,2,3,4-tetrahydroisoquinoline-2-carboximidamide hydrochloride (29b): The compound in TFA salt form (100 mg) was synthesised as described for 28a. The TFA salt was converted to hydrochloride 29b using HCl (0.5M in MeOH). 1H NMR (400 MHz, DMSO-d6): δ 2.85 (2H, t, J = 5.5 Hz, CH2), 3.52 (2H, t, J = 5.2 Hz, CH2), 4.60 (2H, s), 5.35 (2H, s), 6.82 (1H, d, J = 7.8 Hz, ArH), 7.05 (1H, d, J = 7.9 Hz, ArH), 7.28 (1H, t, J = 8.0 Hz, ArH), 7.47–7.51 (1H, m, ArH), 7.65 (1H, d, J = 8.3 Hz, ArH), and 7.73 (1H, d, J = 8.5 Hz, ArH). HRMS (ESI): Calcd. for C17H18Cl2N3O (M + H)+ 350.0827 and found 350.0899.
tert-Butyl N-[(7-bromo-1,2,3,4-tetrahydroisoquinolin-2-yl)({[(tert-butoxy) carbon yl]amino})methylidene]carbamate (31): In a solution of 7-bromo-1,2,3,4-tetrahydroisoquinoline 30 (318 mg, 1.5 mmol) in DMF (5 mL), S-methyl-N,N’-bis(tert-butoxycarbonyl)isothiourea (436 mg, 1.5 mmol) was added, followed by Et3N (0.5 mL). The mixture was stirred at r.t. for 4 h and partitioned between EtOAc (100 mL) and citric acid (50 mL, 5% in water). The organic layer was washed with brine, dried (MgSO4), filtered, and concentrated in vacuo. Purification by flash column chromatography (petrol ether to petrol ether/EtOAc 3:1) afforded 31 as a white solid (500 mg, 73% yield), m.p. 131–132 °C. 1H NMR (400 MHz, CDCl3): δ 1.50 (18H, s, 2 × t-Bu), 2.90 (2H, t, J = 5.5 Hz, CH2), 3.87 (2H, t, J = 5.7 Hz, CH2), 4.70 (2H, s, CH2), 7.01 (1H, d, J = 8.0 Hz, ArH), 7.23 (1H, br.s, ArH), 7.28 (1H, dd, J = 8.2, 1.9 Hz, ArH), and 10.3 (1H, s, NH). HRMS (ESI): Calcd. for C20H29BrN3O4 (M + H)+ 454.1341 and found 454.1356.
tert-Butyl N-[7-(4-tert-butylphenyl)-1,2,3,4-tetrahydroisoquinoline-2-carboximido yl]carbamate (32): In a solution of 31 (400 mg, 0.88 mmol) in dioxane (6 mL) and water (2 mL), 4-tert-butylphenylboronic acid (188 mg, 1.05 mmol) and K2CO3 (242 mg, 1.76 mmol) were added. The mixture was degassed under vacuum for 1 min and Pd(PPh3)4 (20 mg) was added. The reaction mixture was stirred at 100 °C under N2 for 4 h. After cooling to r.t., the mixture was partitioned between EtOAc (50 mL) and water (50 mL). The organic layer was washed with brine, dried (MgSO4), filtered, and concentrated in vacuo. Purification by flash column chromatography (CH2Cl2/MeOH (9:1) gave 32 as a clear oil (190 mg, 53% yield). 1H NMR (400 MHz, CDCl3): δ 1.35 (9H, s, t-Bu), 1.51 (9H, s, t-Bu), 2.90 (2H, t, J = 5.3 Hz, CH2), 3.75 (2H, t, J = 5.2 Hz, CH2), 4.73 (2H, s, CH2), 7.18 (1H, d, J = 8.3 Hz, ArH), 7.35 (1H, br.s, ArH), 7.41 (1H, dd, J = 8.1, 1.5 Hz, ArH), 7.46–7.50 (4H, m, 4 × ArH), and 10.1 (1H, s, NH). HRMS (ESI): Calcd. for C25H34N3O2 (M + H)+ 408.2651 and found 408.2687.
N-[7-(4-tert-Butylphenyl)-1,2,3,4-tetrahydroisoquinoline-2-carboximidoyl]cabam-ate 2,2,2-trifluoroacetate (33): The compound was synthesised as described for 28a. A white solid (35 mg, 77%) was obtained, m.p. 218–219 °C. 1H NMR (400 MHz, CD3OD): δ 1.35 (9H, s, t-Bu), 3.00 (2H, t, J = 5.5 Hz, CH2), 3.70 (2H, t, J = 5.5 Hz, CH2), 4.70 (2H, s, CH2), 7.39 (1H, d, J = 8.2 Hz, ArH), 7.46 (1H, d, J = 1.6 Hz, ArH), and 7.50–7.65 (5H, m, 5 × ArH). HRMS (ESI): Calcd. for C20H26N3 (M + H)+ 308.2127 and found 308.2131.
General Procedure: Guanylation of O-benzylhydroxylamines (34a–c): In a solution of the substituted amine (1.5 mmol) in DMF (5 mL), S-methyl-N,N’-bis(tert-butoxycarbonyl)isothiourea (285 mg, 0.98 mmol) was added, followed by Et3N (0.6 mL). The mixture was stirred at r.t. overnight and then partitioned between EtOAc (100) ml and brine (50 mL). The organic layer was washed with brine, dried (MgSO4), and concentrated in vacuo. Purification by flash column chromatography (petrol ether to petrol ether/EtOAc 11:9) afforded 35a–c.
tert-Butyl N-[{[(tert-butoxy)carbonyl]amino}({[(3-phenylphenyl)methoxy]amino}) methylidene]carbamate (35a): A foamy powder (190 mg, 69%) was obtained. 1H NMR (400 MHz, CDCl3): δ 1.46 (9H, s, t-Bu), 1.49 (9H, s, t-Bu), 5.13 (2H, s, CH2), 7.33–7.46 (5H, m, 5 × ArH), 7.56–7.63 (3H, m, 3 × ArH), 7.64–7.68 (1H, m, ArH), 7.73 (s, 1H, NH), and 9.16 (s, 1H, NH).
tert-Butyl N-[{[(tert-butoxy)carbonyl]amino}({[(3-phenoxyphenyl)methoxy] amino})methylidene]carbamate (35b): A foamy powder (185 mg, 65%) was obtained. 1H NMR (400 MHz, CDCl3): δ 1.36 (18H, s, 2 × t-Bu), 5.28 (2H, s, CH2), 7.41–7.55 (5H, m, 4 × ArH, NH), 7.59 (1H, dt, J = 8.1, 1.5 Hz, ArH), 7.69–7.74 (2H, m, 2 × ArH), 7.76 (1H, s, NH), 7.81 (1H, d, J = 8.1 Hz, ArH), and 7.82 (1H, d, J = 8.2 Hz, ArH).
tert-Butyl N-[{[(tert-butoxy)carbonyl]amino}({[3-(4-tert-butylphenyl)phenyl]meth- oxy}amino)methylidene]carbamate (35c): A foamy powder (200 mg, 80%) was obtained. 1H NMR (400 MHz, CDCl3): δ 1.36 (9H, s, t-Bu), 1.49 (9H, s, t-Bu), 1.50 (9H, s, t-Bu), 5.12 (2H, s, CH2), 7.38–7.45 (4H, m, 4 × ArH), 7.52–7.58 (3H, m, 3 × ArH), 7.62–7.66 (1H, m, ArH), 7.72 (br.s, 1H, NH), and 9.18 (1H, s, NH).
General Procedure: Synthesis of benzyloxy guanidine derivatives (36a–c): In a solution of the substituted N,N′-di-Boc-guanidino derivative (35a–c) (0.3 mmol) in CH2Cl2 (2 mL), TFA (1 mL) was added. The mixture was shaken at r.t. overnight and then evaporated to dryness. Et2O (1 mL) was added, and the precipitate was collected, washed with diethyl ether, and dried in vacuo to give 36a–c as a white or off-white solid.
1-[(3-Phenylphenyl)methoxy]guanidinium 2,2,2-trifluoroacetate (36a): An off-white solid (65 mg, 92%) was obtained. 1H NMR (400 MHz, DMSO-d6): δ 5.00 (2H, s, CH2), 7.43–7.57 (4H, m, 4 × ArH), 7.72 (1H, t, J = 1.9 Hz, ArH), 7.70–7.76 (4H, m, 4 × ArH), 7.83 (1H, br.s, NH), and 11.2 (1H, s, NH). HRMS (ESI): Calcd. for C14H16N3O (M + H)+ 242.1293 and found 242.1282.
1-[(3-Phenoxyphenyl)methoxy]guanidinium 2,2,2-trifluoroacetate (36b): A white solid (95 mg, 97%) was obtained. 1H NMR (400 MHz, CDCl3): δ 4.76 (2H, s, CH2), 6.96–7.05 (4H, m, 4 × ArH), 7.05 (1H, dt, J = 8.5, 1.9 Hz, ArH), 7.12 (1H, td, J = 8.9, 2.1 Hz, ArH), 7.30–7.35 (3H, m, 3 × ArH), and 10.9 (1H, s, NH). HRMS (ESI): Calcd. for C14H15N3NaO2 (M + Na)+ 280.1062 and found 280.1066.
1-{[3-(4-tert-Butylphenyl)phenyl]methoxy}guanidinium 2,2,2-trifluoroacetate (36c): A white solid (75 mg, 77%) was obtained. 1H NMR (400 MHz, CD3OD): δ 1.43 (9H, s, t-Bu), 5.12 (2H, s, CH2), 7.46 (1H, dt, J = 8.9, 1.7 Hz, ArH), 7.52–7.56 (3H, m, 3 × ArH), 7.63–7.66 (2H, m, 2 × ArH), 7.72 (1H, dt, J = 8.8, 2.0 Hz, ArH), and 7.76–7.79 (1H, m, ArH). HRMS (ESI): Calcd. for C18H24N3O (M + H)+ 298.1919 and found 298.1938.
General Procedure: Synthesis of 3-benzyloxybenzaldehyde derivatives (38a–t): The 3-hydroxybenzaldehyde derivative 37a–c (2 mmol) and K2CO3 (4 mmol) were placed in a round-bottom flask. DMF (3 mL) was added. Then, the corresponding benzyl halide (2.2 mmol) was added and the reaction mixture was stirred at r.t. for 18 h. Water (125 mL) and brine (25 mL) were added, and the mixture was extracted with Et2O (2 × 100 mL) or CH2Cl2 (for 38q; 2 × 100 mL). The combined organic layers were dried (NaCl), filtered, and concentrated in vacuo. Crystallisation from Et2O or pentane/Et2O gave the corresponding aldehyde derivatives 38a–c and 38h–t. Aldehyde derivatives 38d–g were purified by flash column chromatography.
3-(2,3-Dichlorobenzyloxy)-benzaldehyde (38a): 445 mg, 79%, white solid. 1H NMR (400 MHz, CDCl3): δ 5.22 (2H, s, CH2), 7.21–7.28 (3H, m, 3 × ArH), 7.42–7.55 (5H, m, 5 × ArH), and 9.99 (1H, s, CH=O). 13C NMR (100 MHz, CDCl3): δ 67.8, 113.5, 122.1, 124.2, 126.8, 127.6, 130.0, 130.4, 130.9, 133.4, 136.6, 138.1, 159.0, and 192.1. HRMS (ESI): Calcd. for C14H11Cl2O2 (M + H)+ 281.0131 and found 281.0142.
3-(2-Chloro-3-(trifluoromethyl)benzyloxy)-benzaldehyde (38b): 474 mg, 75%, white solid. 1H NMR (400 MHz, CDCl3): δ 5.27 (2H, s, CH2), 7.24–7.29 (1H, m, ArH), 7.42 (1H, t, J = 8.0 Hz, ArH), 7.46–7.54 (3H, m, 3 × ArH), 7.70 (1H, d, J = 7.6 Hz, ArH), 7.79 (1H, d, J = 7.6 Hz, ArH), and 9.99 (1H, s, CH=O). HRMS (ESI): Calcd. for C15H11ClF3O2 (M + H)+ 315.0394 and found 315.0403.
3-(4-Chlorobenzyloxy)-benzaldehyde (38c): 430 mg, 87%, white solid, m.p. 52–54 °C. 1H NMR (400 MHz, CDCl3): δ 5.08 (2H, s, CH2), 7.23 (1H, dt, J = 7.6, 2.0 Hz, ArH), 7.34–7.40 (4H, m, 4 × ArH), 7.43–7.51 (3H, m, 3 × ArH), and 9.97 (1H, s, CH=O). HRMS (ESI): Calcd. for C14H12ClO2 (M + H)+ 247.0520 and found 247.0531.
3-(4-(Trifluoromethyl)benzyloxy)-benzaldehyde (38d): Purification by flash column chromatography (petrol ether to petrol ether/EtOAc 9:1) gave 38d (501 mg, 89%, colourless oil). 1H NMR (400 MHz, CDCl3): δ 5.18 (2H, s, CH2), 7.23–7.28 (1H, m, ArH), 7.44–7.52 (3H, m, 3 × ArH), 7.56 (2H, d, J = 8.0 Hz, 2 × ArH), 7.66 (2H, d, J = 8.0 Hz, 2 × ArH), and 9.97 (1H, d, J = 1.2 Hz, CH=O). HRMS (ESI): Calcd. for C15H12F3O2 (M + H)+ 281.0784 and found 281.0789.
3-(3-Chlorobenzyloxy)-benzaldehyde (38e): Purification by flash column chromatography (petrol ether to petrol ether/EtOAc 9:1) gave 38e (424 mg, 86%, colourless oil). 1H NMR (400 MHz, CDCl3): δ 5.09 (2H, s, CH2), 7.22–7.26 (1H, m, ArH), 7.29–7.33 (3H, m, 3 × ArH), 7.44–7.51 (4H, m, 4 × ArH), and 9.97 (1H, s, CH=O). HRMS (ESI): Calcd. for C14H12ClO2 (M + H)+ 247.0520 and found 247.0525.
3-(3-(Trifluoromethyl)benzyloxy)-benzaldehyde (38f): Purification by flash column chromatography (petrol ether to petrol ether/EtOAc 9:1) gave 38f (478 mg, 85%, colourless oil). 1H NMR (400 MHz, CDCl3): δ 5.17 (2H, s, CH2), 7.24–7.28 (1H, m, ArH), 7.45–7.51 (3H, m, 3 × ArH), 7.53 (1H, d, J = 8.0 Hz, ArH), 7.61 (1H, d, J = 8.8 Hz, ArH), 7.63 (1H, d, J = 8.0 Hz, ArH), 7.72 (1H, s, ArH), and 9.98 (1H, s, CH=O). HRMS (ESI) calcd. for C15H12F3O2+ (M + H)+ 281.0784 and found 281.0790.
3-(2,3-Dichlorobenzyloxy)-4-methoxybenzaldehyde (38g): Purification by flash column chromatography (CH2Cl2 to CH2Cl2/EtOAc 9:1) gave 38g (436 mg, 87%). m.p. 117–119 °C. 1H NMR (400 MHz, CDCl3): δ 3.98 (3H, s, OCH3), 5.28 (2H, s, CH2), 7.08 (1H, d, J = 8.3 Hz, ArH), 7.23 (1H, d, J = 8.1 Hz, ArH), 7.43 (2H, d, J = 8.2 Hz, 2 × ArH), 7.52 (2H, dd, J = 8.0, 1.1 Hz, 2 × ArH), and 9.88 (1H, s, CH=O). HRMS (ESI): Calcd. for C15H13Cl2O3 (M + H)+ 311.0242 and found 311.0237.
2-Chloro-3-(2-chloro-3-methoxybenzyloxy)-benzaldehyde (38h): 597 mg, 96%, white solid, m.p. 124–126 °C. 1H NMR (400 MHz, CDCl3): δ 3.92 (3H, s, OCH3), 5.28 (2H, s, CH2), 6.92 (1H, dd, J = 7.0, 2.6 Hz, ArH), 7.19 (1H, dd, J = 8.0, 1.2 Hz, ArH), 7.24–7.33 (3H, m, 3 × ArH), 7.54 (1H, dd, J = 7.8, 1.4 Hz, ArH), and 10.53 (1H, d, J = 0.4 Hz, CH=O). HRMS (ESI): Calcd. for C15H13Cl2O3 (M + H)+ 311.0236 and found 311.0232.
2-Chloro-3-benzyloxybenzaldehyde (38i): 425 mg, 86%, white solid, m.p. 103–105 °C. 1H NMR (400 MHz, CDCl3): δ 5.20 (2H, s, CH2), 7.19 (1H, dd, J = 8.0, 1.2 Hz, ArH), 7.29 (1H, t, J = 7.8 Hz, ArH), 7.35 (1H, d, J = 6.8 Hz, ArH), 7.41 (2H, t, J = 7.4 Hz, 2 × ArH), 7.47 (2H, d, J = 7.2 Hz, 2 × ArH), 7.53 (1H, dd, J = 7.8, 1.4 Hz, ArH), and 10.54 (1H, s, CH=O). HRMS (ESI): Calcd. for C14H12ClO2 (M + H)+ 247.0520 and found 247.0528.
2-Chloro-3-(4-chlorobenzyloxy)-benzaldehyde (38j): 522 mg, 93%, white solid, m.p. 94–96 °C. 1H NMR (400 MHz, CDCl3): δ 5.15 (2H, s, CH2), 7.16 (1H, dd, J = 8.0, 1.6 Hz, ArH), 7.30 (1H, t, J = 8.0 Hz, ArH), 7.35–7.43 (4H, m, 4 × ArH), 7.54 (1H, dd, J = 7.8, 1.4 Hz, ArH), and 10.53 (1H, d, J = 0.8 Hz, CH=O). HRMS (ESI): Calcd. for C14H11Cl2O2 (M + H)+ 281.0131 and found 281.0127.
2-Chloro-3-(2,3-dichlorobenzyloxy)-benzaldehyde (38k): 600 mg, 95%, white solid, m.p. 150–152 °C. 1H NMR (400 MHz, CDCl3): δ 5.27 (2H, s, CH2), 7.20 (1H, d, J = 8.0 Hz, ArH), 7.28 (1H, t, J = 8.0 Hz, ArH), 7.34 (1H, t, J = 8.0 Hz, ArH), 7.46 (1H, d, J = 8.0 Hz, ArH), 7.57 (1H, d, J = 8.0 Hz, ArH), 7.61 (1H, d, J = 8.0 Hz, ArH), and 10.54 (1H, s, CH=O). HRMS (ESI): Calcd. for C14H10Cl3O2 (M + H)+ 314.9741 and found 314.9748.
2-Chloro-3-(2-chloro-3-(trifluoromethyl)benzyloxy)-benzaldehyde (38m): 632 mg, 90%, white solid, m.p. 128–131 °C. 1H NMR (400 MHz, CDCl3): δ 5.31 (2H, s, CH2), 7.23 (1H, dd, J = 8.2, 1.4 Hz, ArH), 7.36 (1H, t, J = 8.0 Hz, ArH), 7.47 (1H, t, J = 7.8 Hz, ArH), 7.59 (1H, dd, J = 7.8, 1.4 Hz, ArH), 7.71 (1H, d, J = 8.0 Hz, ArH), 7.94 (1H, d, J = 7.6 Hz, ArH), and 10.55 (1H, d, J = 0.8 Hz, CH=O). HRMS (ESI): Calcd. for C15H10Cl2F3O2 (M + H)+ 349.0004 and found 349.0011.
2-Chloro-3-(2,4-dichlorobenzyloxy)-benzaldehyde (38n): 580 mg, 92%, white solid, m.p. 115–116 °C. 1H NMR (400 MHz, CDCl3): δ 5.22 (2H, s, CH2), 7.20 (1H, dd, J = 8.2, 1.4 Hz, ArH), 7.30–7.36 (2H, m, 2 × ArH), 7.43 (1H, d, J = 2.4 Hz, ArH), 7.57 (1H, dd, J = 7.8, 1.4 Hz, ArH), 7.62 (1H, d, J = 8.4 Hz, ArH), and 10.54 (1H, d, J = 0.8 Hz, CH=O). HRMS (ESI): Calcd. for C14H10Cl3O2 (M + H)+ 314.9741 and found 314.9748.
2-Chloro-3-(2,5-dichlorobenzyloxy)-benzaldehyde (38o): 535 mg, 85%, white solid, m.p. 132–134 °C. 1H NMR (400 MHz, CDCl3): δ 5.22 (2H, s, CH2), 7.21 (1H, dd, J = 8.4, 1.2 Hz, ArH), 7.27 (1H, dd, J = 8.4, 2.4 Hz, ArH), 7.34 (1H, d, J = 8.8 Hz, ArH), 7.35 (1H, t, J = 8.0 Hz, ArH), 7.58 (1H, dd, J = 8.0, 1.6 Hz, ArH), 7.69 (1H, d, J = 2.4 Hz, ArH), and 10.55 (1H, s, CH=O). HRMS (ESI): Calcd. for C14H10Cl3O2 (M + H)+ 314.9741 and found 314.9745.
2-Chloro-3-(3,4-dichlorobenzyloxy)-benzaldehyde (38p): 610 mg, 97%, white solid, m.p. 140–142 °C. 1H NMR (400 MHz, CDCl3): δ 5.13 (2H, s, CH2), 7.15 (1H, dd, J = 8.2, 1.4 Hz, ArH), 7.29–7.34 (2H, m, 2 × ArH), 7.48 (1H, d, J = 8.4 Hz, ArH), 7.56 (1H, dd, J = 8.0, 1.2 Hz, ArH), 7.58 (1H, s, ArH), and 10.53 (1H, s, CH=O). HRMS (ESI): Calcd. for C14H10Cl3O2 (M + H)+ 314.9741 and found 314.9744.
2-Chloro-3-(2,3,5-trichlorobenzyloxy)-benzaldehyde (38q): 610 mg, 87%, white solid, m.p. 158–161 °C. 1H NMR (400 MHz, CDCl3): δ 5.22 (2H, s, CH2), 7.20 (1H, dd, J = 8.2, 1.4 Hz, ArH), 7.36 (1H, t, J = 8.0 Hz, ArH), 7.48 (1H, d, J = 2.4 Hz, ArH), 7.60 (1H, dd, J = 8.0, 1.2 Hz, ArH), 7.64 (1H, d, J = 2.4 Hz, ArH), and 10.55 (1H, d, J = 0.4 Hz, CH=O). HRMS (ESI): Calcd. for C14H9Cl4O2 (M + H)+ 348.9351 and found 348.9365.
2-Chloro-3-(3-(trifluoromethyl)benzyloxy)-benzaldehyde (38r): 617 mg, 98%, white solid, m.p. 83–85 °C. 1H NMR (400 MHz, CDCl3): δ 5.23 (2H, s, CH2), 7.19 (1H, dd, J = 8.0, 1.2 Hz, ArH), 7.33 (1H, t, J = 8.0 Hz, ArH), 7.54 (1H, t, J = 7.8 Hz, ArH), 7.57 (1H, dd, J = 7.8, 1.4 Hz, ArH), 7.62 (1H, d, J = 8.0 Hz, ArH), 7.69 (1H, d, J = 7.6 Hz, ArH), 7.74 (1H, s, ArH), and 10.54 (1H, d, J = 0.4 Hz, CH=O). HRMS (ESI): Calcd. for C15H11ClF3O2 (M + H)+ 315.0394 and found 315.0401.
2-Chloro-3-(4-(trifluoromethyl)benzyloxy)-benzaldehyde (38s): 605 mg, 96%, white solid, m.p. 82–84 °C. 1H NMR (400 MHz, CDCl3): δ 5.25 (2H, s, CH2), 7.18 (1H, dd, J = 7.8, 1.4 Hz, ArH), 7.32 (1H, dt, J = 8.0, 0.8 Hz, ArH), 7.56 (1H, dd, J = 7.8, 1.4 Hz, ArH), 7.60 (2H, d, J = 8.0 Hz, 2 × ArH), 7.67 (2H, d, J = 7.6 Hz, 2 × ArH), and 10.54 (1H, s, CH=O). HRMS (ESI): Calcd. for C15H11ClF3O2 (M + H)+ 315.0394 and found 315.0398.
2-Chloro-3-(3-chlorobenzyloxy)-benzaldehyde (38t): 418 mg, 74%, white solid. 1H NMR (400 MHz, CDCl3): δ 5.16 (2H, s, CH2), 7.17 (1H, dd, J = 8.2, 1.4 Hz, ArH), 7.31 (1H, t, J = 8.0 Hz, ArH), 7.31–7.37 (3H, m, 3 × ArH), 7.46–7.48 (1H, m, ArH), 7.56 (1H, dd, J = 7.8, 1.4 Hz, ArH), and 10.54 (1H, d, J = 0.8 Hz, CH=O). HRMS (ESI): Calcd. for C14H11Cl2O2 (M + H)+ 281.0131 and found 281.0139.
(E)-Amino(2-(3-(2,3-dichlorobenzyloxy)benzylidene)hydrazineyl)methaniminium acetate (10a): A mixture of 38a (228 mg, 0.81 mmol) and N-aminoguanidine bicarbonate (110 mg, 0.81 mmol) in MeOH-AcOH (3 mL/0.2 mL) was refluxed under N2 for 4 h, cooled to r.t., and concentrated in vacuo. CH2Cl2 (1 mL) was added, and the precipitate was collected, washed with CH2Cl2, and dried in vacuo to give 10a as a white solid (170 mg, 53%, m.p. 159–160 °C). 1H NMR (400 MHz, DMSO-d6): δ 1.87 (3H, s, CH3CO2), 5.29 (2H, s, CH2), 7.03 (1H, d, J = 8.9 Hz, ArH), 7.07 (4H, br.s, 2 × NH2), 7.48 (t, J = 7.9 Hz, 1H, ArH), 7.53–7.55 (2H, m, 2 × ArH), 7.66–7.72 (2H, m, 2 × ArH), 7.58 (1H, s, ArH), and 8.08 (1H, s, CH=N). HRMS (ESI): Calcd. for C15H15Cl2N4O (M + H)+ 337.0623 and found 337.0693.
(E)-Amino(2-(3-(2-chloro-3-(trifluoromethyl)benzyloxy)benzylidene)hydrazine-yl)-methaniminium acetate (10b): A mixture of 38b (320 mg, 1.02 mmol) and N-aminoguanidine bicarbonate (138 mg, 1.02 mmol) in MeOH-AcOH (4 mL/0.2 mL) was refluxed under N2 for 6 h, cooled to r.t., and concentrated in vacuo. EtOAc (3 mL) was added, and the precipitate was collected, washed with EtOAc, and dried in vacuo to give 10b as a white solid (283 mg, 75%, m.p. 190–192 °C). 1H NMR (400 MHz, DMSO-d6): δ 1.82 (3H, s, CH3CO2), 5.25 (2H, s, CH2), 6.95 (4H, br.s, 2 × NH2), 7.02 (1H, dd, J = 7.8, 1.9 Hz, ArH), 7.26–7.33 (2H, m, 2 × ArH), 7.50 (1H, s, ArH), 7.58 (1H, t, J = 7.8 Hz, ArH), 7.86 (1H, d, J = 8.0 Hz, ArH), 7.91 (1H, d, J = 7.8 Hz, ArH), and 7.99 (s, 1H, CH=N). HRMS (ESI): Calcd. for C16H15ClF3N4O (M + H)+ 371.0886 and found 371.0895.
(E)-Amino(2-(3-(2,3-dichlorobenzyloxy)-4-methoxybenzylidene)hydrazineyl)-methaniminium acetate (10g): A white solid was obtained (278 mg, 80%). 1H NMR (400 MHz, DMSO-d6): δ 1.89 (3H, s, CH3CO2), 3.85 (3H, s, OCH3), 5.30 (2H, s, CH2), 6.98 (4H, br.s, 2 × NH2), 7.07 (1H, d, J = 8.1 Hz, ArH), 7.28 (1H, d, J = 8.2 Hz, ArH), 7.50 (1H, t, J = 7.9 Hz, ArH), 7.65–7.75 (3H, m, 3 × ArH), and 8.19 (1H, s, CH=N). HRMS (ESI): Calcd. for C16H17Cl2N4O2 (M + H)+ 367.0729 and found 367.0701.
General procedure: Synthesis of (E)-amino(2-(3-(benzyloxy)benzylidene) hydrazineyl)methaniminium chloride derivatives (10c–f, 10h–t): 3-benzyloxybenzaldehyde derivatives 36c–t (0.2 mmol) and N-aminoguanidine bicarbonate (0.205 mmol) were placed in a 50 mL round-bottom flask. HCl (0.5M in MeOH, 2.0 mL) was added, and the reaction mixture was stirred at 80 °C for 0.5 h and then evaporated to dryness. Crystallisation from Et2O (~5–6 mL) with a very small portion of MeOH (~0.3–0.5 mL) gave the corresponding N-aminoguanidinium chloride or acetate salts 10c–f or 10h–t.
(E)-Amino(2-(3-(4-chlorobenzyloxy)benzylidene)hydrazineyl)methaniminium chloride (10c): 54 mg, 79%, white solid, m.p. 191–193 °C. 1H NMR (400 MHz, DMSO-d6): δ 5.21 (2H, s, CH2), 7.11–7.18 (1H, m, ArH), 7.38–7.45 (2H, m, 2 × ArH), 7.52 (2H, d, J = 8.4 Hz, 2 × ArH), 7.56 (2H, d, J = 8.4 Hz, 2 × ArH), 7.67 (1H, s, ArH), 7.86 (4H, s, br, 2 × NH2), 8.20 (1H, s, CH=N), and 11.97 (1H, s, N-NH). HRMS (ESI): Calcd. for C15H16ClN4O (M + H)+ 303.1007 and found 303.1013.
(E)-Amino(2-(3-(4-(trifluoromethyl)benzyloxy)benzylidene) hydrazineyl) methan-iminium chloride (10d): 58 mg, 77%, white solid, m.p. 176–179 °C. 1H NMR (400 MHz, DMSO-d6): δ 5.33 (2H, s, CH2), 7.14–7.20 (1H, m, ArH), 7.40–7.47 (2H, m, 2 × ArH), 7.67 (1H, s, ArH), 7.75 (2H, d, J = 8.0 Hz, 2 × ArH), 7.83 (2H, d, J = 8.4 Hz, 2 × ArH), 7.85 (4H, s, br, 2 × NH2), 8.20 (1H, s, CH=N), and 11.96 (1H, s, N-NH). HRMS (ESI): Calcd. for C16H16F3N4O (M + H)+ 337.1271 and found 337.1278.
(E)-Amino(2-(3-(3-chlorobenzyloxy)benzylidene)hydrazineyl)methaniminium chloride (10e): 69 mg, >99%, beige glass. 1H NMR (400 MHz, DMSO-d6): δ 5.22 (2H, s, CH2), 7.12–7.18 (1H, m, ArH), 7.42 (2H, d, J = 6.0 Hz, 2 × ArH), 7.45 (1H, dd, J = 5.0, 2.0 Hz, ArH), 7.49 (2H, d, J = 6.0 Hz, 2 × ArH), 7.59 (1H, s, ArH), 7.66 (1H, d, J = 2.8 Hz, ArH), 7.84 (4H, s, br, 2 × NH2), 8.21 (1H, s, CH=N), and 11.97 (1H, s, N-NH). HRMS (ESI): Calcd. for C15H16ClN4O (M + H)+ 303.1007 and found 303.1015.
(E)-Amino(2-(3-(4-(trifluoromethyl)benzyloxy)benzylidene)hydrazineyl)methan-iminium chloride (10f): 68 mg, 92%, pale yellow glass. 1H NMR (400 MHz, DMSO-d6): δ 5.31 (2H, s, CH2), 7.15–7.21 (1H, m, ArH), 7.41–7.45 (2H, m, 2 × ArH), 7.69 (1H, d, J = 2.8 Hz, ArH), 7.71 (1H, d, J = 8.4 Hz, ArH), 7.76 (1H, d, J = 8.0 Hz, ArH), 7.84 (1H, d, J = 7.6 Hz, ArH), 7.86 (4H, s, br, 2 × NH2), 7.89 (1H, s, ArH), 8.22 (1H, s, CH=N), and 11.97 (1H, s, N-NH). HRMS (ESI): Calcd. for C16H16F3N4O (M + H)+ 337.1271 and found 337.1282.
(E)-Amino(2-(2-chloro-3-(2-chloro-3-methoxybenzyloxy)benzylidene) hydrazine-yl)methaniminium chloride (10h): 68 mg, 84%, white solid, m.p. 258–260 °C. 1H NMR (400 MHz, DMSO-d6): δ 5.33 (2H, s, CH2), 7.23 (1H, dd, J = 8.0, 1.2 Hz, ArH), 7.28 (1H, dd, J = 7.6 Hz, 1.2, ArH), 7.37 (1H, dd, J = 8.0, 1.6 Hz, ArH), 7.42 (1H, d, J = 8.0 Hz, ArH), 7.43 (1H, t, J = 7.8 Hz, ArH), 7.93 (4H, s, br, 2 × NH2), 7.96 (1H, dd, J = 7.8, 1.4 Hz, ArH), 8.65 (1H, s, CH=N), and 12.26 (1H, s, N-NH). HRMS (ESI): Calcd. for C16H17Cl2N4O2 (M + H)+ 367.0723 and found 367.0734.
(E)-Amino(2-(3-(benzyloxy)-2-chlorobenzylidene)hydrazineyl)methaniminium chloride (10i): 56 mg, 82%, white solid, m.p. 203–205 °C. 1H NMR (400 MHz, DMSO-d6): δ 5.30 (2H, s, CH2), 7.37 (1H, dd, J = 8.4, 2.0 Hz, ArH), 7.39–7.44 (2H, m, 2 × ArH), 7.47 (2H, t, J = 7.4 Hz, 2 × ArH), 7.53 (1H, s, ArH), 7.55 (1H, d, J = 1.6 Hz, ArH), 7.92 (4H, s, br, 2 × NH2), 7.94 (1H, dd, J = 7.6, 1.6 Hz, ArH), 8.65 (1H, s, CH=N), and 12.27 (1H, s, N-NH). HRMS (ESI): Calcd. for C15H16ClN4O (M + H)+ 303.1007 and found 303.1012.
(E)-Amino(2-(2-chloro-3-(4-chlorobenzyloxy)benzylidene)hydrazineyl)methan-iminium chloride (10j): 63 mg, 84%, white solid, m.p. 237–239 °C. 1H NMR (400 MHz, DMSO-d6): δ 5.30 (2H, s, CH2), 7.35 (1H, dd, J = 8.0, 1.2 Hz, ArH), 7.42 (1H, t, J = 8.0 Hz, ArH), 7.51–7.59 (4H, m, 4 × ArH), 7.93 (4H, s, br, 2 × NH2), 7.95 (1H, dd, J = 7.6, 1.6 Hz, ArH), 8.65 (1H, s, CH=N), and 12.32 (1H, s, N-NH). HRMS (ESI): Calcd. for C15H15Cl2N4O (M + H)+ 337.0617 and found 337.0625.
(E)-Amino(2-(2-chloro-3-(2,3-dichlorobenzyloxy)benzylidene)hydrazineyl) meth-animinium chloride (10k): 71 mg, 87%, white solid, m.p. 250–253 °C. 1H NMR (400 MHz, DMSO-d6): δ 5.38 (2H, s, CH2), 7.40 (1H, dd, J = 8.4, 1.6 Hz, ArH), 7.46 (1H, t, J = 7.6 Hz, ArH), 7.51 (1H, t, J = 8.0 Hz, ArH), 7.70 (1H, dd, J = 7.8, 1.4 Hz, ArH), 7.73 (1H, dd, J = 8.0, 1.2 Hz, ArH), 7.95 (4H, s, br, 2 × NH2), 7.98 (1H, dd, J = 7.6, 1.6 Hz, ArH), 8.65 (1H, s, CH=N), and 12.30 (1H, s, N-NH). HRMS (ESI): Calcd. for C15H14Cl3N4O (M + H)+ 371.0228 and found 371.0235.
(E)-Amino(2-(2-chloro-3-(2-chloro-3-(trifluoromethyl)benzyloxy)benzylidene) hydrazineyl)methaniminium chloride (10m): 73 mg, 82%, white solid, m.p. 274–277 °C. 1H NMR (400 MHz, DMSO-d6): δ 5.44 (2H, s, CH2), 7.42–7.48 (2H, m, 2 × ArH), 7.71 (1H, t, J = 7.8 Hz, ArH), 7.95 (1H, d, J = 8.0 Hz, ArH), 7.97 (4H, s, br, 2 × NH2), 7.99 (1H, dd, J = 7.0, 1.4 Hz, ArH), 8.04 (1H, d, J = 7.6 Hz, ArH), 8.66 (1H, s, CH=N), and 12.31 (1H, s, N-NH). 13C NMR (100 MHz, DMSO-d6): δ 67.6 (CH2), 115.5 (CH), 119.8 (CH), 122.3, 127.6 (CH), 127.8 (CH), 127.9 (CH), 132.0, 133.5 (CH), 136.7, 142.9 (CH), 153.5, and 155.2. HRMS (ESI): Calcd. for C16H14Cl2F3N4O (M + H)+ 405.0491 and found 405.0498.
(E)-Amino(2-(2-chloro-3-(2,4-dichlorobenzyloxy)benzylidene)hydrazineyl) meth-animinium chloride (10n): 73 mg, 89%, white solid, m.p. 268–270 °C. 1H NMR (400 MHz, DMSO-d6): δ 5.32 (2H, s, CH2), 7.40 (1H, dd, J = 8.4, 1.6 Hz, ArH), 7.44 (1H, t, J = 8.0 Hz, ArH), 7.58 (1H, dd, J = 8.4, 2.0 Hz, ArH), 7.73 (1H, d, J = 8.4 Hz, ArH), 7.77 (1H, d, J = 2.4 Hz, ArH), 7.95 (4H, s, br, 2 × NH2), 7.98 (1H, dd, J = 7.6, 1.6 Hz, ArH), 8.65 (1H, s, CH=N), and 12.34 (1H, s, N-NH). HRMS (ESI): Calcd. for C15H14Cl3N4O (M + H)+ 371.0228 and found 371.0239.
(E)-Amino(2-(2-chloro-3-(2,5-dichlorobenzyloxy)benzylidene)hydrazineyl) meth-animinium chloride (10o): 72 mg, 88%, white solid, m.p. 242–244 °C. 1H NMR (400 MHz, DMSO-d6): δ 5.33 (2H, s, CH2), 7.42 (1H, dd, J = 7.4, 1.6 Hz, ArH), 7.46 (1H, t, J = 7.6 Hz, ArH), 7.56 (1H, dd, J = 8.6, 2.2 Hz, ArH), 7.64 (1H, d, J = 8.8 Hz, ArH), 7.79 (1H, d, J = 2.4 Hz, ArH), 7.93 (4H, s, br, 2 × NH2), 7.99 (1H, dd, J = 7.2, 1.6 Hz, ArH), 8.65 (1H, s, CH=N), and 12.25 (1H, s, N-NH). HRMS (ESI): Calcd. for C15H14Cl3N4O (M + H)+ 371.0228 and found 371.0237.
(E)-Amino(2-(2-chloro-3-(3,4-dichlorobenzyloxy)benzylidene)hydrazineyl) meth-animinium chloride (10p): 69 mg, 84%, white solid, m.p. 230–233 °C. 1H NMR (400 MHz, DMSO-d6): δ 5.32 (2H, s, CH2), 7.35 (1H, dd, J = 8.4, 1.2 Hz, ArH), 7.43 (1H, t, J = 7.8 Hz, ArH), 7.53 (1H, dd, J = 8.0, 2.0 Hz, ArH), 7.75 (1H, d, J = 8.4 Hz, ArH), 7.81 (1H, d, J = 2.0 Hz, ArH), 7.92 (4H, s, br, 2 × NH2), 7.96 (1H, dd, J = 8.0, 1.2 Hz, ArH), 8.65 (1H, s, CH=N), and 12.25 (1H, s, N-NH). HRMS (ESI): Calcd. for C15H14Cl3N4O (M + H)+ 371.0228 and found 371.0239.
(E)-Amino(2-(2-chloro-3-(2,3,5-trichlorobenzyloxy)benzylidene)hydrazineyl) methaniminium chloride (10q): 52 mg, 59%, white solid, m.p. 273–276 °C. 1H NMR (400 MHz, DMSO-d6): δ 5.36 (2H, s, CH2), 7.42 (1H, dd, J = 7.4, 2.0 Hz, ArH), 7.46 (1H, t, J = 7.6 Hz, ArH), 7.77 (1H, d, J = 2.4 Hz, ArH), 7.96 (4H, s, br, 2 × NH2), 7.96 (1H, d, J = 2.8 Hz, ArH), 7.99 (1H, dd, J = 7.4, 1.8 Hz, ArH), 8.65 (1H, s, CH=N), and 12.32 (1H, s, N-NH). HRMS (ESI): Calcd. for C15H13Cl4N4O (M + H)+ 404.9838 and found 404.9851.
(E)-Amino(2-(2-chloro-3-(3-(trifluoromethyl)benzyloxy)benzylidene)hydrazineyl) methaniminium chloride (10r): 69 mg, 84%, white powder, m.p. 198–201 °C. 1H NMR (400 MHz, DMSO-d6): δ 5.41 (2H, s, CH2), 7.39 (1H, d, J = 8.4 Hz, ArH), 7.44 (1H, t, J = 7.8 Hz, ArH), 7.73 (1H, t, J = 7.6 Hz, ArH), 7.78 (1H, d, J = 8.0 Hz, ArH), 7.85 (1H, d, J = 7.2 Hz, ArH), 7.91 (1H, s, ArH), 7.94 (4H, s, br, 2 × NH2), 7.97 (1H, d, J = 7.6 Hz, ArH), 8.65 (1H, s, CH=N), and 12.33 (1H, s, N-NH). HRMS (ESI): Calcd. for C16H15ClF3N4O (M + H)+ 371.0881 and found 371.0891.
(E)-Amino(2-(2-chloro-3-(4-(trifluoromethyl)benzyloxy)benzylidene)hydrazineyl) methaniminium chloride (10s): 65 mg, 80%, white solid, m.p. 245–247 °C. 1H NMR (400 MHz, DMSO-d6): δ 5.43 (2H, s, CH2), 7.36 (1H, d, J = 8.0 Hz, ArH), 7.43 (1H, t, J = 8.0 Hz, ArH), 7.76 (2H, d, J = 8.0 Hz, 2 × ArH), 7.85 (2H, d, J = 7.6 Hz, 2 × ArH), 7.95 (4H, s, br, 2 × NH2), 7.96 (1H, d, J = 7.6 Hz, ArH), 8.66 (1H, s, CH=N), and 12.31 (1H, s, N-NH). HRMS (ESI): Calcd. for C16H15ClF3N4O (M + H)+ 371.0881 and found 371.0887.
(E)-Amino(2-(2-chloro-3-(3-chlorobenzyloxy)benzylidene)hydrazineyl) methaniminium chloride (10t): 75 mg, >99%, pale yellow glass. 1H NMR (400 MHz, DMSO-d6): δ 5.31 (2H, s, CH2), 7.35 (1H, dd, J = 8.0, 1.2 Hz, ArH), 7.41 (1H, t, J = 8.0 Hz, ArH), 7.43–7.48 (1H, m, ArH), 7.48–7.51 (2H, m, 2 × ArH), 7.59 (1H, s, ArH), 7.92 (4H, s, br, 2 × NH2), 7.94 (1H, dd, J = 8.0, 1.2 Hz, ArH), 8.65 (1H, s, CH=N), and 12.23 (1H, s, N-NH). HRMS (ESI): Calcd. for C15H15Cl2N4O (M + H)+ 337.0617 and found 337.0629.
General Procedure: Synthesis of 1-benzyl-1H-imidazole-2-carbaldehyde derivatives (40a–d) and 1-benzyl-1H-pyrrole-2-carbaldehyde derivatives (40e–h): Imidazole 2-carboxaldehyde 39a (1 mmol) or pyrrole 2-carboxaldehyde 39b (1 mmol) and K2CO3 (4 mmol) were placed in a 25 mL round-bottom flask. DMF (1 mL) was added. Then, the corresponding benzyl halide (1.2 mmol) was added and the reaction mixture was stirred at r.t. for 18 h. Water (80 mL) and brine (20 mL) were added, and the mixture was extracted with Et2O (100 mL). The organic layer was dried (NaCl), filtered, and concentrated in vacuo. All crude compounds 40a–d were purified by flash column chromatography.
1-(3-Chlorobenzyl)-1H-imidazole-2-carbaldehyde (40a): Purification by flash column chromatography (CH2Cl2 to CH2Cl2/EtOAc 9:1) gave 40a (177 mg, 80%, colourless oil). 1H NMR (400 MHz, CDCl3): δ 5.58 (2H, s, CH2), 7.05–7.08 (1H, m, ArH), 7.15 (2H, s, 2 × ArH), 7.25–7.28 (2H, m, 2 × ArH), 7.32 (1H, s, ArH), and 9.84 (1H, d, J = 0.5 Hz, CH=O). HRMS (ESI): Calcd. for C11H10ClN2O (M + H)+ 221.0476 and found 221.0469.
1-(4-Chlorobenzyl)-1H-imidazole-2-carbaldehyde (40b): Purification by flash column chromatography (CH2Cl2 to CH2Cl2/EtOAc 9:1) gave 40b (184 mg, 83%, beige solid). 1H NMR (400 MHz, CDCl3): δ 5.57 (2H, s, CH2), 7.11–7.13 (1H, m, ArH), 7.13–7.15 (2H, m, 2 × ArH), 7.30 (2H, dt, J = 8.4, 2.2 Hz, 2 × ArH), 7.32 (1H, s, ArH), and 9.86 (1H, s, CH=O). HRMS (ESI): Calcd. for C11H10ClN2O (M + H)+ 221.0476 and found 221.0471.
1-(3-(Trifluoromethyl)benzyl)-1H-imidazole-2-carbaldehyde (40c): Purification by flash column chromatography (CH2Cl2 to CH2Cl2/EtOAc 9:1) gave 40c (197 mg, 77%, colourless oil). 1H NMR (400 MHz, DMSO-d6): δ 5.75 (2H, s, CH2), 7.42 (1H, d, J = 0.8 Hz, ArH), 7.52 (1H, d, J = 8.0 Hz, ArH), 7.62–7.67 (2H, m, 2 × ArH), 7.70–7.74 (1H, m, ArH), 7.90 (1H, s, ArH), and 9.75 (1H, d, J = 0.8 Hz, CH=O). HRMS (ESI): Calcd. for C12H10F3N2O (M + H)+ 255.0740 and found 255.0747.
1-(4-(Trifluoromethyl)benzyl)-1H-imidazole-2-carbaldehyde (40d): Purification by flash column chromatography (CH2Cl2 to CH2Cl2/EtOAc 9:1) gave 40d (100 mg, 39%, beige solid). 1H NMR (400 MHz, CDCl3): δ 5.68 (2H, s, CH2), 7.19 (1H, s, ArH), 7.29 (2H, d, J = 8.0 Hz, 2 × ArH), 7.37 (1H, s, ArH), 7.60 (2H, d, J = 8.0 Hz, 2 × ArH), and 9.88 (1H, s, CH=O). HRMS (ESI): Calcd. for C12H10F3N2O (M + H)+ 255.0740 and found 255.0749.
1-(3-Chlorobenzyl)-1H-pyrrole-2-carbaldehyde (40e): Purification by flash column chromatography (petrol ether to petrol ether/EtOAc 9:1) gave 40e (165 mg, 75%, colourless oil). 1H NMR (400 MHz, CDCl3): δ 5.53 (2H, s, CH2), 6.29 (1H, dd, J = 3.8, 2.6 Hz, ArH), 6.96–6.99 (2H, m, 2 × ArH), 7.07–7.09 (1H, m, ArH), 7.21–7.23 (2H, m, 2 × ArH), 7.25–7.27 (1H, m, ArH), and 9.54 (1H, d, J = 0.8 Hz, CH=O). HRMS (ESI): Calcd. for C12H11ClNO (M + H)+ 220.0524 and found 220.0519.
1-(4-Chlorobenzyl)-1H-pyrrole-2-carbaldehyde (40f): Purification by flash column chromatography (petrol ether to petrol ether/EtOAc 9:1) gave 40f (185 mg, 84%, beige solid). 1H NMR (400 MHz, CDCl3): δ 5.51 (2H, s, CH2), 6.28 (1H, t, J = 3.2 Hz, ArH), 6.95–6.98 (2H, m, 2 × ArH), 7.07 (2H, dt, J = 8.4, 2.2 Hz, 2 × ArH), 7.26 (2H, dt, J = 8.4, 2.2 Hz, 2 × ArH), and 9.54 (1H, s, CH=O). HRMS (ESI): Calcd. for C12H11ClNO (M + H)+ 220.0524 and found 220.0521.
1-(3-(Trifluoromethyl)benzyl)-1H-pyrrole-2-carbaldehyde (40g): Purification by flash column chromatography (petrol ether to petrol ether/EtOAc 9:1) gave 40g (216 mg, 85%, beige-brown oil). 1H NMR (400 MHz, CDCl3): δ 5.61 (2H, s, CH2), 6.31 (1H, t, J = 3.2 Hz, ArH), 6.98–7.01 (2H, m, 2 × ArH), 7.30 (1H, d, J = 7.6 Hz, ArH), 7.37 (1H, s, ArH), 7.42 (1H, t, J = 7.8 Hz, ArH), 7.52 (1H, d, J = 8.0 Hz, ArH), and 9.55 (1H, s, CH=O). HRMS (ESI): Calcd. for C13H11F3NO (M + H)+ 254.0787 and found 254.0793.
1-(4-(Trifluoromethyl)benzyl)-1H-pyrrole-2-carbaldehyde (40h): Purification by flash column chromatography (petrol ether to petrol ether/EtOAc 9:1) gave 40h (82 mg, 32%, beige solid). 1H NMR (400 MHz, CDCl3): δ 5.61 (2H, s, CH2), 6.31 (1H, t, J = 3.0 Hz, ArH), 6.98–7.01 (2H, m, 2 × ArH), 7.21 (2H, d, J = 8.0 Hz, 2 × ArH), 7.55 (2H, d, J = 8.0 Hz, 2 × ArH), and 9.54 (1H, s, CH=O). HRMS (ESI): Calcd. for C13H11F3NO (M + H)+ 254.0787 and found 254.0795.
General Procedure: Synthesis of (E)-amino(2-((1-benzyl-1H-imidazol-2-yl)methylene)hydrazineyl)methaniminium chloride derivatives (41a–d) and (E)-Amino(2-((1-benzyl-1H-pyrrol-2-yl)methylene)hydrazineyl)methaniminium chloride derivatives (41e–h): Aldehyde derivatives (40a–h) (0.2 mmol) and N-aminoguanidine bicarbonate (0.24 mmol) were placed in a 50 mL round-bottom flask. HCl (0.5M in MeOH, 2.0 mL) was added, and the reaction mixture was stirred at 80 °C for 2 h and then evaporated to dryness.
(E)-Amino(2-((1-(3-chlorobenzyl)-1H-imidazol-2-yl)methylene)hydrazineyl) methaniminium chloride (41a): 87 mg, >99%, pale yellow glass. 1H NMR (400 MHz, DMSO-d6): δ 5.71 (2H, s, CH2), 7.35 (1H, dt, J = 4.4, 1.6 Hz, ArH), 7.46–7.50 (2H, m, 2 × ArH), 7.52 (1H, s, ArH), 7.88 (1H, d, J = 2.2 Hz, ArH), 7.94 (1H, d, J = 2.2 Hz, ArH), 8.35 (4H, s, br, 2 × NH2), 8.56 (1H, s, CH=N), and 12.85 (1H, s, br, N-NH). HRMS (ESI): Calcd. for C12H14ClN6 (M + H)+ 277.0963 and found 277.0956.
(E)-Amino(2-((1-(4-chlorobenzyl)-1H-imidazol-2-yl)methylene)hydrazineyl) methaniminium chloride (41b): 88 mg, >99%, pale yellow glass. 1H NMR (400 MHz, DMSO-d6): δ 5.70 (2H, s, CH2), 7.42 (2H, dt, J = 8.4, 2.2 Hz, 2 × ArH), 7.52 (2H, dt, J = 8.4, 2.2 Hz, 2 × ArH), 7.86 (1H, d, J = 1.8 Hz, ArH), 7.91 (1H, d, J = 1.8 Hz, ArH), 8.35 (4H, s, br, 2 × NH2), 8.53 (1H, s, CH=N), and 12.82 (1H, s, br, N-NH). HRMS (ESI): Calcd. for C12H14ClN6 (M + H)+ 277.0963 and found 277.0955.
(E)-Amino(2-((1-(3-(trifluoromethyl)benzyl)-1H-imidazol-2-yl)methylene) hydrazineyl)methaniminium chloride (41c): 94 mg, >99%, pale yellow glass. 1H NMR (400 MHz, DMSO-d6): δ 5.80 (2H, s, CH2), 7.65 (1H, d, J = 7.8 Hz, ArH), 7.70 (1H, t, J = 7.8 Hz, ArH), 7.80 (1H, d, J = 7.6 Hz, ArH), 7.85 (1H, s, ArH), 7.90 (1H, d, J = 1.8 Hz, ArH), 7.94 (1H, d, J = 1.8 Hz, ArH), 8.35 (4H, s, br, 2 × NH2), 8.59 (1H, s, CH=N), and 12.85 (1H, s, br, N-NH). HRMS (ESI): Calcd. for C13H14F3N6 (M + H)+ 311.1227 and found 311.1234.
(E)-Amino(2-((1-(4-(trifluoromethyl)benzyl)-1H-imidazol-2-yl)methylene) hydrazineyl)methaniminium chloride (41d): 95 mg, >99%, pale yellow glass. 1H NMR (400 MHz, DMSO-d6): δ 5.83 (2H, s, CH2), 7.58 (2H, d, J = 8.0 Hz, 2 × ArH), 7.82 (2H, d, J = 8.0 Hz, 2 × ArH), 7.88 (1H, d, J = 1.6 Hz, ArH), 7.94 (1H, d, J = 1.6 Hz, ArH), 8.29 (4H, s, br, 2 × NH2), 8.51 (1H, s, CH=N), and 12.73 (1H, s, br, N-NH). HRMS (ESI): Calcd. for C13H14F3N6 (M + H)+ 311.1227 and found 311.1235.
(E)-Amino(2-((1-(3-chlorobenzyl)-1H-pyrrol-2-yl)methylene)hydrazineyl) methan-iminium chloride (41e): 64 mg, >99%, purple-black glass. 1H NMR (400 MHz, DMSO-d6): δ 5.61 (2H, s, CH2), 6.29 (1H, dd, J = 3.8, 2.6 Hz, ArH), 6.77 (1H, dd, J = 3.8, 1.8 Hz, ArH), 7.01 (1H, dt, J = 7.2, 1.6 Hz, ArH), 7.07 (1H, t, J = 1.6 Hz, ArH), 7.26 (1H, dd, J = 2.4, 2.0 Hz, ArH), 7.35 (1H, dt, J = 8.0, 1.8 Hz, ArH), 7.39 (1H, t, J = 7.6 Hz, ArH), 7.59 (4H, s, br, 2 × NH2), 8.09 (1H, s, CH=N), and 11.55 (1H, s, N-NH). HRMS (ESI): Calcd. for C13H15ClN5 (M + H)+ 276.1010 and found 276.1006.
(E)-Amino(2-((1-(4-chlorobenzyl)-1H-pyrrol-2-yl)methylene)hydrazineyl) methan-iminium chloride (41f): 64 mg, >99%, purple-black glass. 1H NMR (400 MHz, DMSO-d6): δ 5.58 (2H, s, CH2), 6.27 (1H, dd, J = 3.8, 1.4 Hz, ArH), 6.76 (1H, dd, J = 3.8, 1.8 Hz, ArH), 7.07 (2H, d, J = 8.0 Hz, 2 × ArH), 7.25 (1H, dd, J = 6.4, 1.6 Hz, ArH), 7.42 (2H, d, J = 8.0 Hz, 2 × ArH), 7.49 (4H, s, br, 2 × NH2), 8.07 (1H, s, CH=N), and 11.54 (1H, s, N-NH). HRMS (ESI): Calcd. for C13H15ClN5 (M + H)+ 276.1010 and found 276.1004.
(E)-Amino(2-((1-(3-(trifluoromethyl)benzyl)-1H-pyrrol-2-yl)methylene) hydrazin-eyl)methaniminium chloride (41g): 81 mg, >99%, purple-black glass. 1H NMR (400 MHz, DMSO-d6): δ 5.72 (2H, s, CH2), 6.30 (1H, dd, J = 3.8, 2.6 Hz, ArH), 6.77 (1H, dd, J = 3.8, 1.8 Hz, ArH), 7.28–7.32 (2H, m, 2 × ArH), 7.44 (1H, s, ArH), 7.50 (4H, s, br, 2 × NH2), 7.60 (1H, t, J = 7.8 Hz, ArH), 7.65 (1H, d, J = 7.6 Hz, ArH), 8.08 (1H, s, CH=N), and 11.57 (1H, s, N-NH). HRMS (ESI): Calcd. for C14H15F3N5 (M + H)+ 310.1274 and found 310.1278.
(E)-Amino(2-((1-(4-(trifluoromethyl)benzyl)-1H-pyrrol-2-yl)methylene) hydrazin-eyl)methaniminium chloride (41h): 80 mg, >99%, purple-black glass. 1H NMR (400 MHz, DMSO-d6): δ 5.71 (2H, s, CH2), 6.31 (1H, t, J = 2.8 Hz, ArH), 6.78–6.79 (1H, m, ArH), 7.23 (2H, d, J = 8.0 Hz, 2 × ArH), 7.27 (1H, t, J = 2.6 Hz, ArH), 7.46 (4H, s, br, 2 × NH2), 7.73 (2H, d, J = 8.0 Hz, 2 × ArH), 8.07 (1H, s, CH=N), and 11.57 (1H, s, N-NH). HRMS (ESI): Calcd. for C14H15F3N5 (M + H)+ 310.1274 and found 310.1281.
tert-Butyl N-[{[(tert-butoxy)carbonyl]imino}[(4-hydroxyphenyl)amino] methyl]-carbamate (43): In a solution of 4-aminophenol 42 (273 mg, 2.5 mmol) in DMF (6 mL), S-methyl-N,N’-bis(tert-butoxycarbonyl)isothiourea (690 mg, 2.37 mmol) was added, followed by Et3N (0.7 mL) and HgCl2 (640 mg, 2.37 mmol). The mixture was stirred at r.t. overnight, and then diluted with EtOAc (30 mL) and filtered through Celite. The organic layer was washed with brine, dried (MgSO4), and concentrated in vacuo. Purification by flash chromatography (petrol ether to petrol ether/EtOAc 3:2) afforded 43 as a white solid (535 mg, 64% yield), m.p. 182–184 °C. 1H NMR (400 MHz, CDCl3): δ 1.44 (9H, s, t-Bu), 1.47 (9H, s, t-Bu), 6.56 (2H, br.s, 2 × ArH), 7.02 (3H, br.s, 2 × ArH and OH), 9.93 (1H, s, NH), and 11.6 (1H, s, NH). HRMS (ESI): Calcd. for C17H25N3NaO5 (M + Na)+ 374.1692 and found 374.1697.
General Procedure: Benzylation of 4-(N,N′-di-Boc-guanydino)phenol (43): In a solution of 4-(N,N′-di-Boc-guanydino)phenol 43 (110 mg, 0.53 mmol) in acetone (5 mL), the substituted benzyl bromide (0.37 mmol) was added, followed by K2CO3 (72 mg, 0.52 mmol). The mixture was stirred at r.t. overnight and partitioned between EtOAc (30 mL) and water (30 mL). The organic layer was washed with brine, dried (MgSO4), and concentrated in vacuo to give an off-white solid. Purification by flash column chromatography (petrol ether to petrol ether/EtOAc 3:1) afforded 44a–c.
tert-Butyl N-[{[4-(benzyloxy)phenyl]amino}({[(tert-butoxy)carbonyl]imino}) meth-yl]-carbamate (44a): A white solid was obtained (79% yield), m.p. 141–143 °C. 1H NMR (400 MHz, CDCl3): δ 1.48 (9H, s, t-Bu), 1.52 (9H, s, t-Bu), 5.05 (2H, s, OCH2), 6.92 (2H, d, J = 8.2 Hz, 2 × ArH), 7.30–7.42 (5H, m, 5 × ArH), 7.47 (2H, d, J = 8.3 Hz, 2 × ArH), 10.2 (1H, br.s, NH), and 11.6 (1H, s, NH). HRMS (ESI): Calcd. for C24H31N3NaO5 (M + Na)+ 464.2161 and found 464.2179.
tert-Butyl N-[{[(tert-butoxy)carbonyl]imino}({4-[(4-chlorophenyl)methoxy] phenyl}amino)methyl]carbamate (44b): A white solid was obtained (89% yield), m.p. 95–97 °C. 1H NMR (400 MHz, CDCl3): δ 1.48 (9H, s, t-Bu), 1.52 (9H, s, t-Bu), 5.00 (2H, s, OCH2), 6.91 (2H, dd, J = 7.8, 2.4 Hz, 2 × ArH), 7.30 (4H, s, 4 × ArH), 7.51 (2H, dd, J = 7.8, 2.4 Hz, 2 × ArH), 10.2 (1H, br.s, NH), and 11.6 (1H, s, NH).
tert-Butyl N-[{[3-(benzyloxy)phenyl]amino}({[(tert-butoxy)carbonyl]imino}) methyl]carbamate (44c): A white solid was obtained (62% yield), m.p. 79–83 °C. 1H NMR (400 MHz, CDCl3): δ 1.47 (9H, s, t-Bu), 1.51 (9H, s, t-Bu), 5.05 (2H, s, OCH2), 6.92 (2H, d, J = 8.4 Hz, 2 × ArH), 7.25–7.36 (4H, m, 4 × ArH), 7.40 (2H, d, J = 8.3 Hz, 2 × ArH), 10.8 (1H, br.s, NH), and 11.8 (1H, s, NH).
General Procedure: Synthesis of the phenyl guanidine derivatives (45a–c): In a solution of the para-substituted N,N′-di-Boc-(4-guanydino)benzene (100 mg) (44a–c) in CH2Cl2 (2 mL), TFA (1 mL) was added. The mixture was shaken at r.t. overnight and then evaporated to dryness. Et2O (1 mL) was added, and the precipitate was collected, washed with Et2O, and dried in vacuo to give 45a–c as a white or off-white solid.
1-[4-(Benzyloxy)phenyl]guanidinium 2,2,2-trifluoroacetate (45a): A white solid was obtained (95% yield). 1H NMR (400 MHz, CD3OD): δ 5.12 (2H, s, OCH2), 7.12 (2H, d, J = 8.1 Hz, 2 × ArH), 7.22 (2H, d, J = 8.1 Hz, 2 × ArH), 7.32–7.40 (3H, m, 3 × ArH), and 7.46 (2H, d, J = 8.3 Hz, 2 × ArH). HRMS (ESI): Calcd. for C14H16N3O (M + H)+ 242.1293 and found 242.1283.
1-[4-(4-Chlorobenzyloxy)phenyl]guanidinium 2,2,2-trifluoroacetate (45b): A white solid was obtained (88% yield). 1H NMR (400 MHz, CD3OD): δ 5.12 (2H, s, OCH2), 7.21–7.30 (4H, m, 4 × ArH), 7.37 (2H, d, J = 8.2 Hz, 2 × ArH), and 7.55 (2H, d, J = 8.2 Hz, 2 × ArH). HRMS (ESI): Calcd. for C14H15ClN3O (M + H)+ 276.0904 and found 276.0932.
1-[4-(3-Chlorobenzyloxy)phenyl}guanidinium 2,2,2-trifluoroacetate (45c): A white solid was obtained (92% yield). 1H NMR (400 MHz, CD3OD): δ 5.21 (s, 2H, OCH2), 7.32 (2H, d, J = 8.0 Hz, 2 × ArH), 7.38 (1H, s, ArH), 7.47–7.53 (3H, m, 3 × ArH), and 7.59 (2H, d, J = 8.3 Hz, 2 × ArH). HRMS (ESI) calcd. for C14H15ClN3O (M + H)+ 276.0904 and found 276.0911.
General Procedure: Synthesis of Boc-protected amide/sulphonamide derivative (47a-b): 4-Aminobenzylamine 21 (0.5 mmol) was dissolved in DMF (1.0 mL) and Et3N (2.0 mmol). Benzoyl chloride or benzenesulphonyl chloride (0.5 mmol) was added successively, and the reaction mixture was stirred for 2 h at 0 °C. HgCl2 (0.5 mmol) and S-methyl-N,N′-bis(tert-butoxycarbonyl) isothiourea (0.5 mmol) were added, and the reaction mixture was stirred at r.t. for 18 h. Et2O (100 mL) was added, and the mixture was filtered through a paper filter. The organic layer was washed with water (80 mL) and brine (20 mL), dried (NaCl), filtered, and concentrated to dryness. Crystallisation from Et2O gave 47a–b.
tert-Butyl N-[(1Z)-{[(tert-butoxy)carbonyl]imino}({4-[(phenylformamido)methyl] phenyl}amino)methyl]carbamate (47a): 113 mg, 48%, white solid, m.p. 152–156 °C, mixture of conformers as revealed by 1H NMR. Major conformer: 1H NMR (400 MHz, CDCl3): δ 1.45 (9H, s, t-Bu), 1.53 (9H, s, t-Bu), 4.55 (2H, d, J = 5.2 Hz, CH2), 6.64 (1H, br.s, NH), 7.25 (2H, d, J = 8.0 Hz, 2 × ArH), 7.39–7.52 (5H, m, 5 × ArH), 7.81 (2H, d, J = 8.0 Hz, 2 × ArH), 10.40 (1H, br.s, NH), and 11.66 (1H, br.s, NH). HRMS (ESI): Calcd. for C25H33N4O5 (M + H)+ 469.2445 and found 469.2451.
tert-Butyl N-[(1Z)-{[4-(benzenesulfonamidomethyl)phenyl]amino}({[(tert-butoxy)-carbonyl]imino})methyl]carbamate (47b): 128 mg, 50%, white solid, m.p. 154–158 °C. 1H NMR (400 MHz, CDCl3): δ 1.47 (9H, s, t-Bu), 1.53 (9H, s, t-Bu), 4.10 (2H, d, J = 6.0 Hz, CH2), 4.90 (1H, br.s, NH), 7.17 (2H, d, J = 8.0 Hz, 2 × ArH), 7.46 (2H, d, J = 8.4 Hz, 2 × ArH), 7.49–7.55 (2H, m, 2 × ArH), 7.56–7.62 (1H, m, ArH), 7.87 (1H, d, J = 1.6 Hz, ArH), 7.89 (1H, s, ArH), 10.51 (1H, br.s, NH), and 11.65 (1H, br.s, NH). HRMS (ESI): Calcd. for C24H33N4O6S (M + H)+ 505.2115 and found 505.2127.
Amino((4-(benzamidomethyl)phenyl)amino)methaniminium 2,2,2-trifluoroacet-ate (48a): Compound 47a (0.1 mmol) was dissolved in CH2Cl2 (0.8 mL) and TFA (0.2 mL) was added, and the reaction mixture was stirred for 2 h at 0 °C. The mixture was concentrated to dryness to give 48a as a pale yellow glass (38 mg, 99%). The product was a mixture of conformers as revealed by 1H NMR. Major conformer: 1H NMR (400 MHz, DMSO-d6): δ 4.54 (2H, s, CH2), 7.25 (2H, d, J = 8.0 Hz, 2 × ArH), 7.44 (4H, br.s, 2 × NH2), 7.49–7.67 (5H, m, 5 × ArH), 7.94 (2H, d, J = 7.2 Hz, 2 × ArH), 9.17 (1H, t, J = 5.8 Hz, NH), and 9.79 (1H, s, NH). HRMS (ESI): Calcd. for C15H17N4O (M + H)+ 269.1397 and found 269.1405.
Amino((4-(phenylsulfonamidomethyl)phenyl)amino)methaniminium 2,2,2-trifluoroacetate (48b): Compound 47b (0.1 mmol) was dissolved in CH2Cl2 (0.8 mL) and TFA (0.2 mL) was added, and the reaction mixture was stirred for 2 h at 0 °C. The mixture was concentrated to dryness to give 48b as a pale yellow glass (38 mg, 99%). 1H NMR (400 MHz, DMSO-d6): δ 4.03 (2H, d, J = 6.4 Hz, CH2), 7.21 (2H, d, J = 8.0 Hz, 2 × ArH), 7.37 (2H, d, J = 8.0 Hz, 2 × ArH), 7.45 (4H, br.s, 2 × NH2), 7.61–7.72 (3H, m, 3 × ArH), 7.87 (2H, d, J = 7.2 Hz, 2 × ArH), 8.27 (1H, t, J = 6.4 Hz, NH), and 9.78 (1H, br.s, NH). HRMS (ESI): Calcd. for C14H17N4O2S (M + H)+ 305.1067 and found 305.1078.
(3-[2,3-Dichlorobenzyloxy]benzyl)(methyl)amine (49a): In a solution of 38a (330 mg, 1.17 mmol) in MeOH (5 mL), MeNH2 (2.0 M in MeOH, 0.76 mL, 1.5 mmol) was added. The mixture was stirred at r.t. for 10 h and then cooled to 0 °C. NaBH4 (53 mg, 1.4 mmol) was added slowly. The mixture was stirred at r.t. for 4 h and then partitioned between NaOH (1M in water) and EtOAc. The organic layer was washed with brine, dried (MgSO4), and concentrated in vacuo to give 49a as a clear oil (340 mg, 98%). 1H NMR (400 MHz, CDCl3): δ 2.45 (3H, s, NCH3), 3.74 (2H, s, CH2), 5.15 (2H, s, OCH2), 6.85 (1H, dd, J = 8.0, 1.5 Hz, ArH), 6.92–7.00 (2H, m, 2 × ArH), 7.18–7.28 (2H, m, 2 × ArH), 7.40 (1H, d, J = 7.9 Hz, ArH), and 7.47 (1H, d, J = 7.9 Hz, ArH). HRMS (ESI): Calcd. for C15H16Cl2NO (M + H)+ 296.0609 and found 296.0680.
(3-[2,3-Dichlorobenzyloxy]benzyl)(2-methoxyethyl)amine (49b): In a solution of 38a (330 mg, 1.17 mmol) in MeOH (5 mL), 2-methoxyethan-1-amine (113 mg, 1.5 mmol) was added. The mixture was stirred at r.t. for 10 h and then cooled to 0°C. NaBH4 (57 mg, 1.5 mmol) was added slowly. The mixture was stirred at r.t. for 4 h and then partitioned between NaOH (1M in water) and EtOAc. The organic layer was washed with brine, dried (MgSO4), and concentrated in vacuo to give 49b as a clear oil (350 mg, 88%). 1H NMR (400 MHz, CDCl3): δ 2.80 (2H, t, J = 5.2 Hz, OCH2CH2N), 3.35 (3H, s, OCH3), 3.51 (2H, t, J = 5.3 Hz, OCH2CH2N), 3.80 (2H, s, CH2), 5.16 (2H, s, OCH2), 6.85 (1H, dd, J = 7.9, 2.6 Hz, ArH), 6.96 (1H, d, J = 7.5 Hz, ArH), 6.99 (1H, s, ArH), 7.20–7.26 (2H, m, 2 × ArH), 7.43 (1H, d, J = 8.0 Hz, ArH), and 7.48 (1H, d, J = 7.9 Hz, ArH). HRMS (ESI): Calcd. for C17H20Cl2NO2 (M + H)+ 340.0871 and found 340.0848.
tert-Butyl N-[{[(tert-butoxy)carbonyl]imino}[(3-(2,3-dichlorobenzyl) benzyl)(methyl)amino]methyl]carbamate (50a): In a solution of 49a (330 mg, 1.11 mmol) in DMF (5 mL), S-methyl-N,N’-bis(tert-butoxycarbonyl)isothiourea (389 mg, 1.3 mmol) was added, followed by Et3N (0.3 mL) and HgCl2 (200 mg). The mixture was stirred at r.t. overnight, and then diluted with EtOAc (50 mL) and filtered through Celite. The organic layer was washed with brine, dried (MgSO4), and concentrated in vacuo to give a colourless oil. Purification by flash column chromatography (petrol ether to petrol ether/EtOAc 4:1) afforded 50a as a white solid (350 mg, 59% yield), m.p. 127–129 °C. 1H NMR (400 MHz, CDCl3): δ 1.50 (18H, s, 2 × t-Bu), 2.90 (3H, s, NCH3), 4.70 (2H, s, CH2), 5.16 (2H, s, OCH2), 6.88–6.97 (2H, m, 2 × ArH), 7.22–7.28 (3H, m, 3 × ArH), 7.41 (1H, d, J = 7.9 Hz, ArH), 7.49 (1H, d, J = 7.9 Hz, ArH), and 10.2 (1H, s, NH). HRMS (ESI): Calcd. for C26H33Cl2N3NaO5 (M + Na)+ 560.1695 and found 560.1697.
tert-Butyl N-[{[(tert-butoxy)carbonyl]imino}[(3-(2,3-dichlorobenzyl)benzyl)(2-methoxyethyl)amino]methyl]carbamate (50b): In a solution of 49b (320 mg, 0.94 mmol) in DMF (3 mL), S-methyl-N,N’-bis(tert-butoxycarbonyl)isothiourea (330 mg, 1.13 mmol) was added, followed by Et3N (0.5 mL) and HgCl2 (306 mg). The mixture was stirred at r.t. overnight, and then diluted with EtOAc (50 mL) and filtered through Celite. The organic layer was washed with brine, dried (MgSO4), and concentrated in vacuo to give a colourless oil. Purification by flash column chromatography eluting with a gradient solvent (petrol ether to petrol ether/EtOAc 4:1) afforded 50b as a white solid (340 mg, 62% yield), m.p. 125–126 °C. 1H NMR (400 MHz, CDCl3): δ 1.50 (18H, s, 2 × t-Bu), 3.30 (3H, s, OCH3), 3.45 (2H, s, CH2), 3.65 (2H, br.s, OCH2CH2N), 4.89 (2H, br.s, OCH2CH2N), 5.20 (2H, s, OCH2), 6.86–6.97 (2H, m, 2 × ArH), 7.21–7.28 (3H, m, 3 × ArH), 7.43 (1H, d, J = 8.3 Hz, ArH), 7.49 (1H, d, J = 8.2 Hz, ArH), and 9.56 (1H, s, NH). HRMS (ESI): Calcd. for C28H38Cl2N3O6 (M + H)+ 582.2138 and found 582.2177.
1-(3-(2,3-Dichlorobenzyloxy)benzyl)-1-methylguanidinium chloride (51a): In a solution of 50a (126 mg, 0.23 mmol) in CH2Cl2 (1.5 mL), TFA (0.75 mL) was added. The mixture was shaken at r.t. overnight and then evaporated to dryness. Et2O (1 mL) was added, and the precipitate was collected, washed with Et2O, and dried in vacuo to give a white solid (69 mg, 87%). The solid (18 mg) was dissolved in HCl (0.5M in MeOH, 5 mL), and concentrated in vacuo to give 51a as a white solid (15 mg). 1H NMR (400 MHz, CDCl3): δ 2.96 (3H, s, NCH3), 4.56 (2H, br.s, CH2), 5.18 (2H, s, OCH2), 6.76–6.86 (2H, m, 2 × ArH), 6.98 (1H, d, J = 8.2 Hz, ArH), 7.18–7.36 (2H, m, 2 × ArH), and 7.49 (2H, br.s, 2 × ArH). HRMS (ESI): Calcd. for C16H18Cl2N3O (M + H)+ 338.0827 and found 338.0909.
1-(3-(2,3-Dichlorobenzyl)benzyl)-1-(2-methoxyethyl)guanidinium 2,2,2-trifluoroacetate (51b): In a solution of the intermediate 50b (98 mg, 0.17 mmol) in CH2Cl2 (1.5 mL), TFA (0.75 mL) was added. The mixture was shaken at r.t. overnight and then evaporated to dryness. Et2O (1 mL) was added, and the precipitate was collected, washed with Et2O, and dried in vacuo to give 51b as a white solid (70 mg, 86%). 1H NMR (400 MHz, CDCl3): δ 3.31 (3H, s, NCH3), 3.56 (4H, m, OCH2CH2N and CH2), 4.67 (2H, br.s, OCH2CH2N), 5.26 (2H, s, OCH2), 6.85–6.95 (2H, m, 2 × ArH), 7.06 (1H, dd, J = 7.9, 2.1 Hz, ArH), 7.37–7.50 (2H, m, 2 × ArH), 7.62 (1H, dd, J = 8.1, 1.8 Hz, ArH), and 7.73 (1H, dd, J = 8.1, 1.7 Hz, ArH). HRMS (ESI): Calcd. for C18H22Cl2N3O2 (M + H)+ 382.1089 and found 382.1195.
3-(2,3-Dichlorobenzyloxy)phenylmethanol (52): Compound 38a (1.3 g, 4.6 mmol) was dissolved in EtOH (20 mL) and THF (5 mL) and cooled to 0 °C. NaNH4 (176 mg, 4.6 mmol) was added portionwise and the mixture was stirred at r.t. overnight. Acetone (2 mL) was added, and the mixture was partitioned between EtOAc and a saturated NH4Cl solution. The organic layer was washed with brine, dried (MgSO4), and concentrated in vacuo to give 52 as a white solid (1.2 g, 92% yield), m.p. 79–80 °C. 1H NMR (400 MHz, CDCl3): δ 4.64 (2H, s, OCH2), 5.24 (2H, s, OCH2), 6.90 (1H, dd, J = 8.0, 1.9 Hz, ArH), 6.96–7.03 (2H, m, 2 × ArH), 7.20–7.31 (2H, m, 2 × ArH), 7.43 (1H, d, J = 8.2 Hz, ArH), and 7.49 (1H, d, J = 8.2 Hz, ArH).
1,2-Dichloro-3-[3-(chloromethyl)phenoxymethyl]benzene (53): In a solution of 52 (1.1 g, 3.9 mmol) in CH2Cl2 (20 mL), Et3N (1.1 mL, 7.8 mmol) was added, followed by methanesulfonyl chloride (0.6 mL, 7.8 mmol). The mixture was stirred at r.t. overnight and partitioned between CH2Cl2 and a saturated NaHCO3 solution. The organic layer was washed with brine, dried (MgSO4), and concentrated in vacuo to give 53 as a white solid (0.95 g, 91% yield), m.p. 91–95 °C. 1H NMR (400 MHz, CDCl3): δ 4.53 (2H, s, CH2), 5.17 (2H, s, OCH2), 6.92 (1H, dd, J = 8.1, 1.7 Hz, ArH), 7.00–7.05 (2H, m, 2 × ArH), 7.21–7.32 (2H, m, 2 × ArH), 7.44 (1H, d, J = 8.1 Hz, ArH), and 7.49 (1H, d, J = 8.1 Hz, ArH).
tert-Butyl N-[{[(tert-butoxy)carbonyl](3-(2,3-dichlorobenzyloxy) benzyl)amino}-(methylsulfanyl)methylidene]carbamate (54): In a solution of 53 (330 mg, 1.1 mmol) in CH2Cl2 (5 mL), KOH (112 mg, 2.2 mmol) in water (5 mL) and Bu4NHSO4 (34 mg, 0.1 mmol) were added. The mixture was stirred at r.t. overnight and partitioned between CH2Cl2 and water. The organic layer was washed with brine, dried (MgSO4), and concentrated in vacuo to give 54 as a clear oil (300 mg, 49% yield). 1H NMR (400 MHz, CDCl3): δ 1.39 (9H, s, t-Bu), 1.51 (9H, s, t-Bu), 2.40 (3H, s, SCH3), 4.75 (2H, s, CH2), 5.15 (2H, s, OCH2), 6.86 (1H, dd, J = 7.9, 1.7 Hz, ArH), 6.95–7.01 (2H, m, 2 × ArH), 7.18–7.26 (2H, m, 2 × ArH), 7.38 (1H, d, J = 8.2 Hz, ArH), and 7.47 (1H, d, J = 8.1 Hz, ArH). HRMS (ESI): Calcd. for C26H32Cl2N2NaO5S (M + Na)+ 577.1306 and found 577.1303.
tert-Butyl N-[{[(tert-butoxy)carbonyl](3-(2,3-dichlorobenzyl) benzyl)amino}(methylamino)methylidene]carbamate (55): In a solution of 54 (200 mg, 0.36 mmol) in DMF (1 mL), MeNH2 (2M in MeOH, 0.27 mL, 0.54 mmol) was added, followed by Et3N (0.2 mL) and HgCl2 (116 mg). The mixture was stirred at r.t. for 1.5 h, and then diluted with EtOAc (30 mL) and filtered through Celite. The organic layer was washed with brine, dried (MgSO4), and concentrated in vacuo to give a colourless oil. Purification by flash column chromatography (petrol ether to petrol ether/EtOAc 4:1) afforded 55 as a white solid (100 mg, 52% yield), m.p. 111–113 °C. 1H NMR (400 MHz, CDCl3): δ 1.46 (9H, s, t-Bu), 1.51 (9H, s, t-Bu), 2.52 (3H, s, NCH3), 4.57 (2H, s, CH2), 5.15 (2H, s, OCH2), 6.87–6.96 (3H, m, 3 × ArH), 7.19–7.26 (2H, m, 2 × ArH), 7.43 (1H, d, J = 7.9 Hz, ArH), 7.49 (1H, d, J = 8.0 Hz, ArH), and 9.87 (1H, s, NH).
3-(3-(2,3-Dichlorobenzyloxy)benzyl)-1-methylguanidinium 2,2,2-trifluoroacetate (56): In a solution of 55 (85 mg, 0.16 mmol) in CH2Cl2 (1 mL), TFA (0.5 mL) was added. The mixture was shaken at r.t. overnight and then evaporated to dryness. The residue was dissolved in MeOH and evaporated to dryness to give 56 as a foamy powder (60 mg, 87%). 1H NMR (400 MHz, CDCl3): δ 2.85 (3H, s, NCH3), 4.39 (2H, s, CH2), 5.19 (2H, s, OCH2), 6.88–6.95 (3H, m, 3 × ArH), 7.24–7.33 (2H, m, 2 × ArH), and 7.52 (2H, d, J = 7.6 Hz, 2 × ArH). HRMS (ESI): Calcd. for C16H18Cl2N3O (M + H)+ 338.0827 and found 338.0945.













{kind=link}
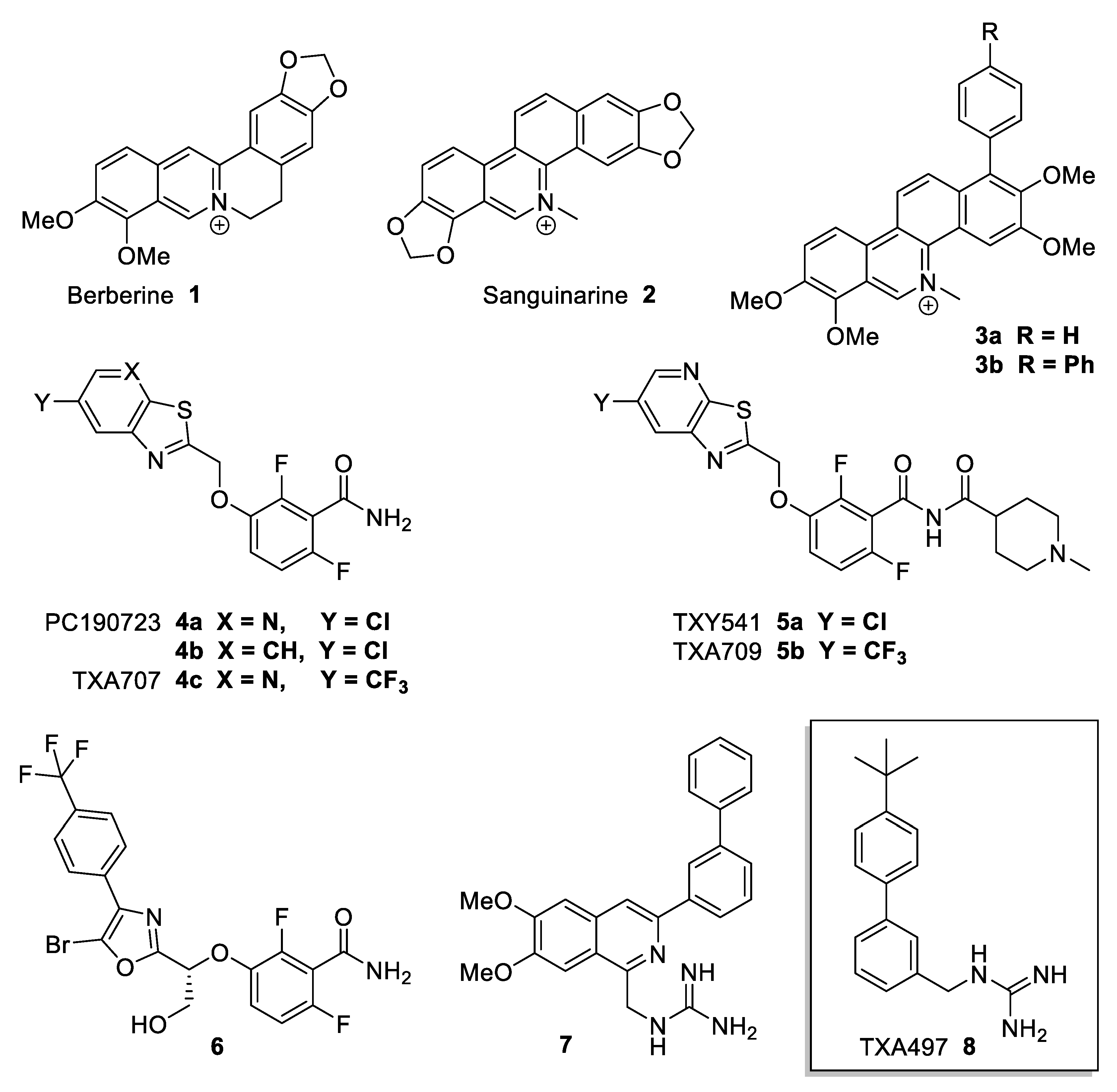{kind=link}
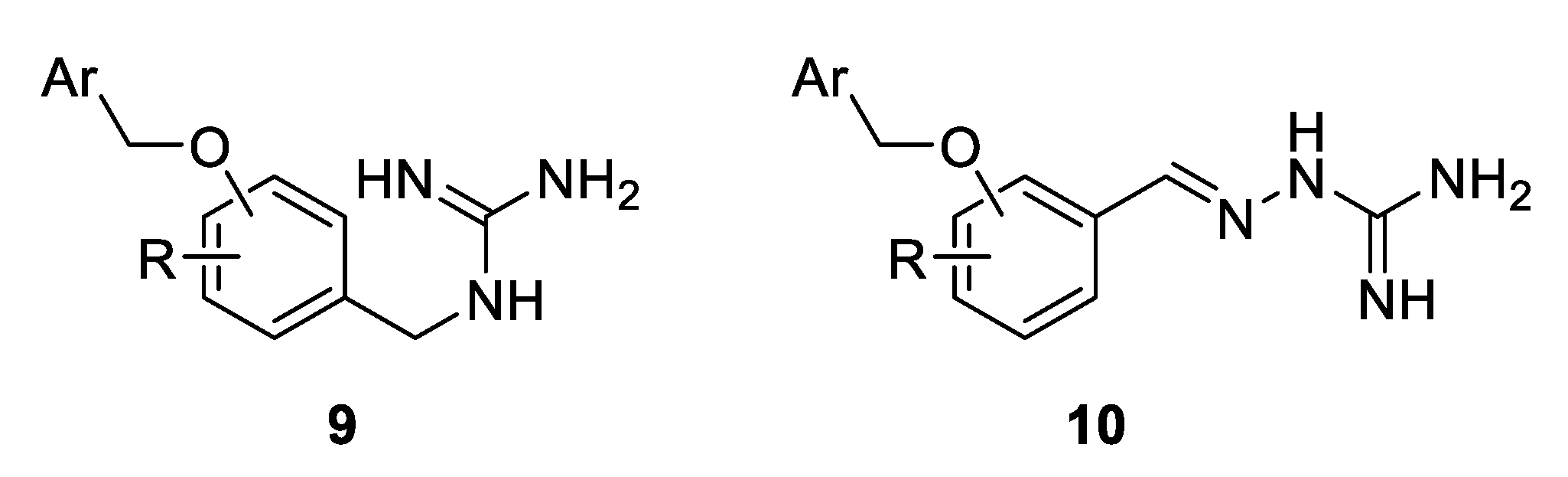{kind=link}
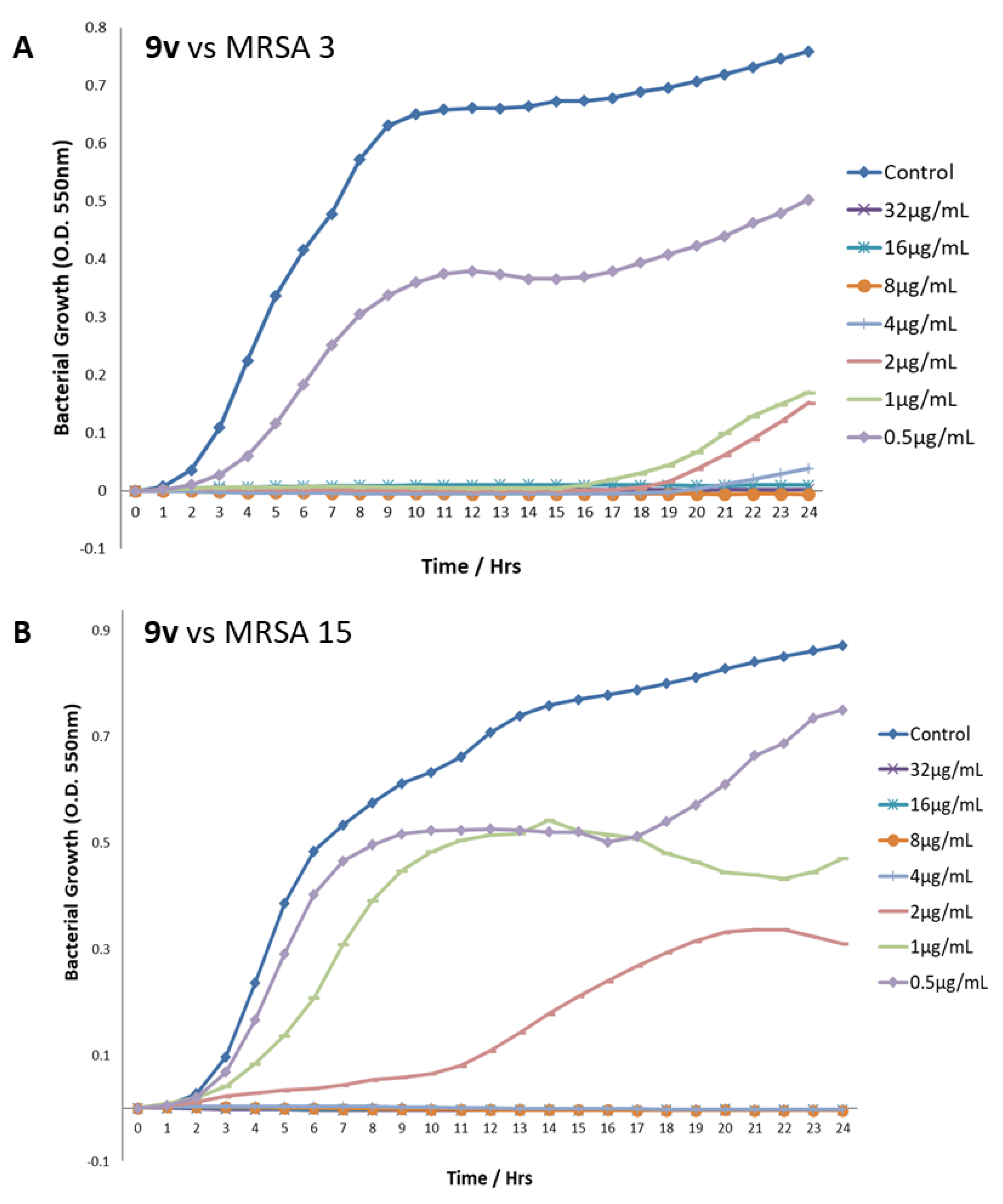{kind=link}
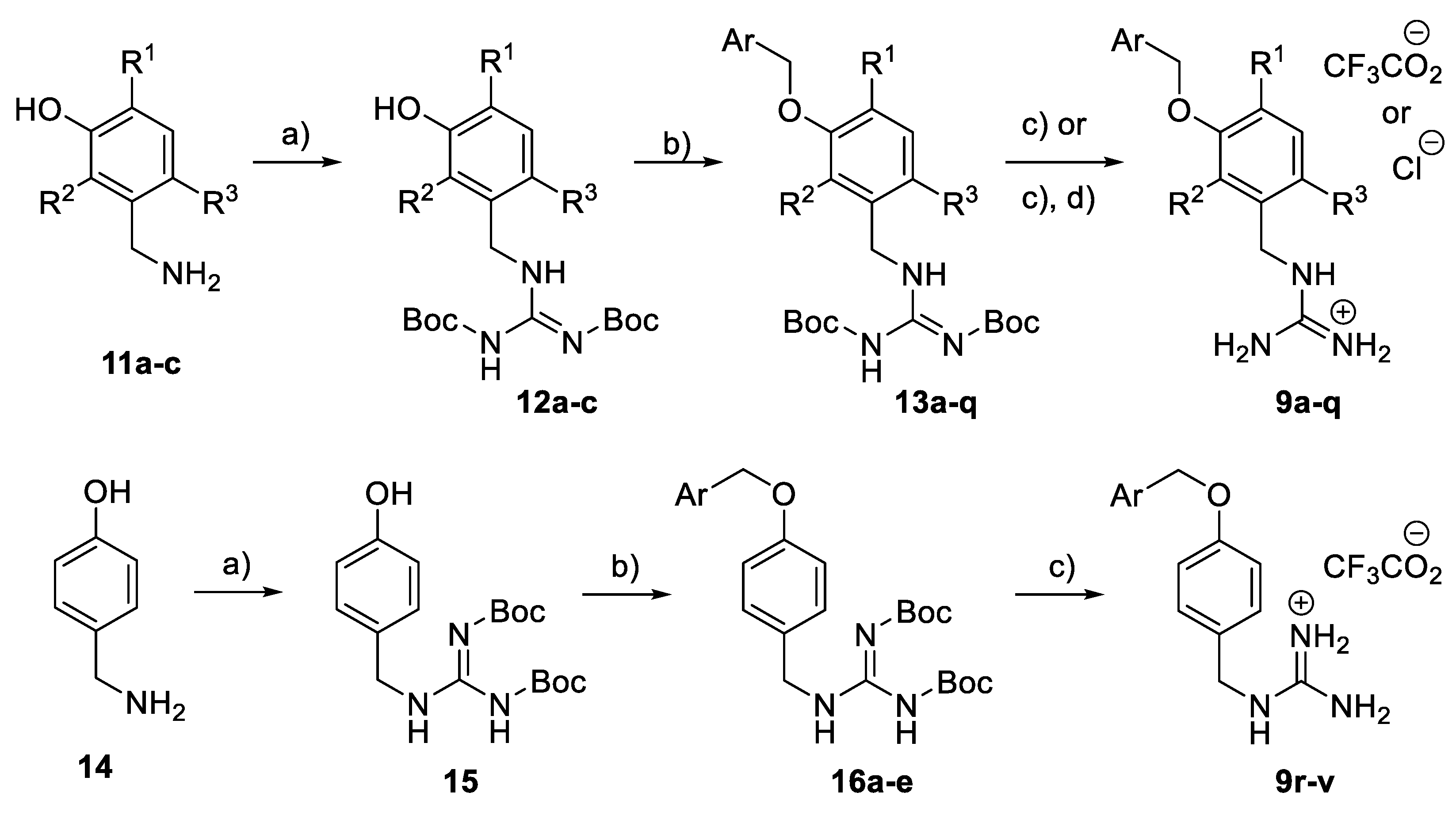{kind=link}
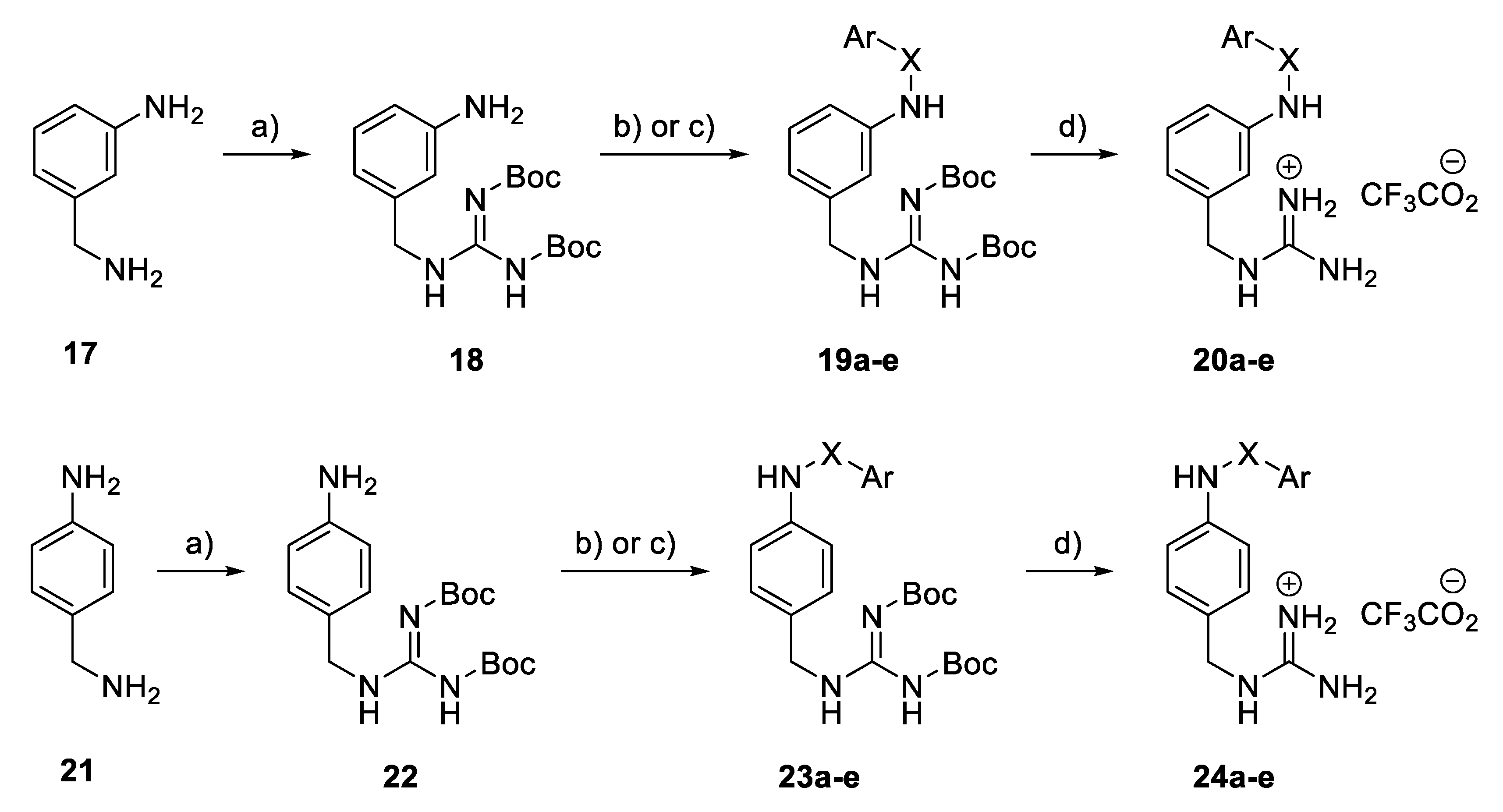{kind=link}
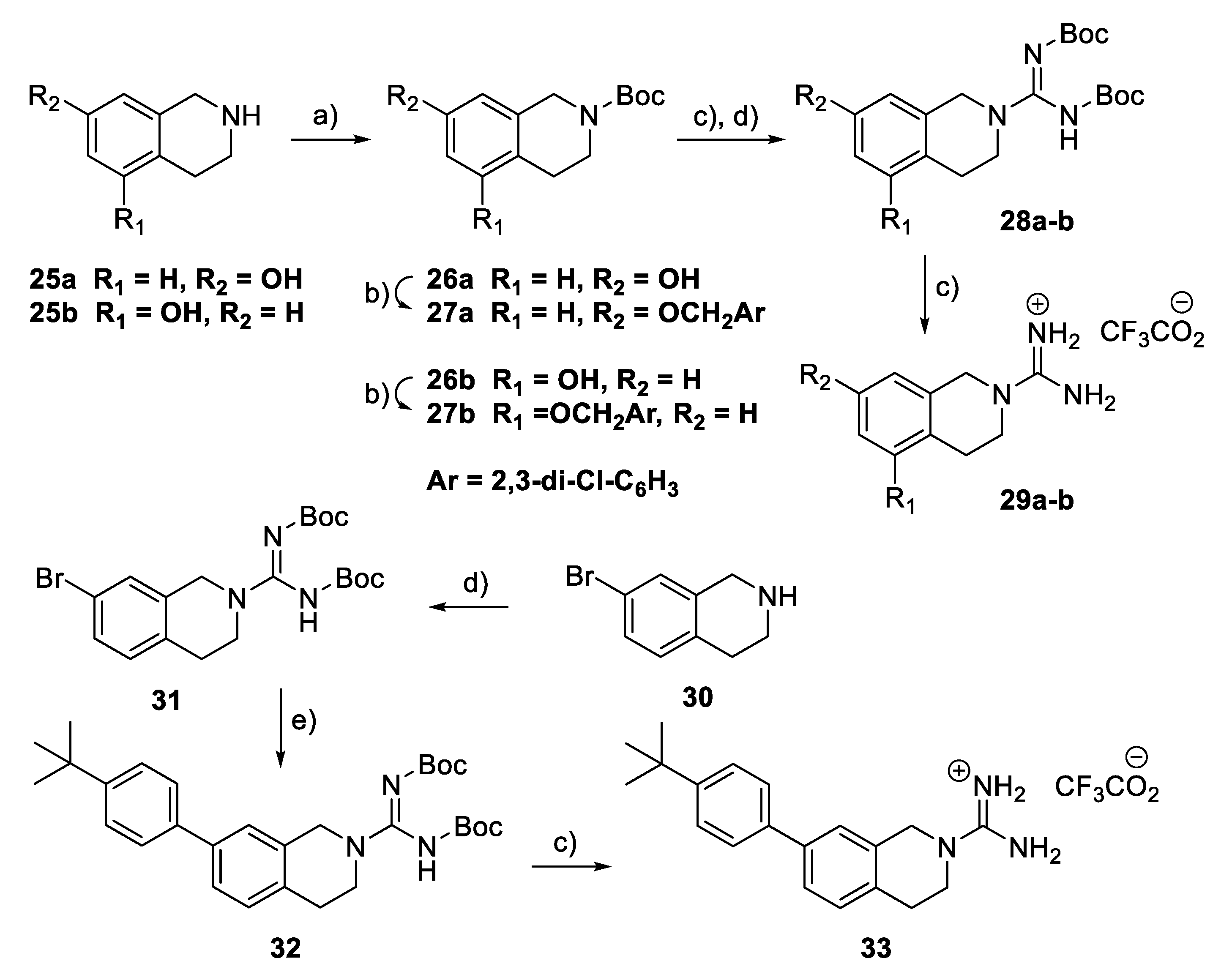{kind=link}
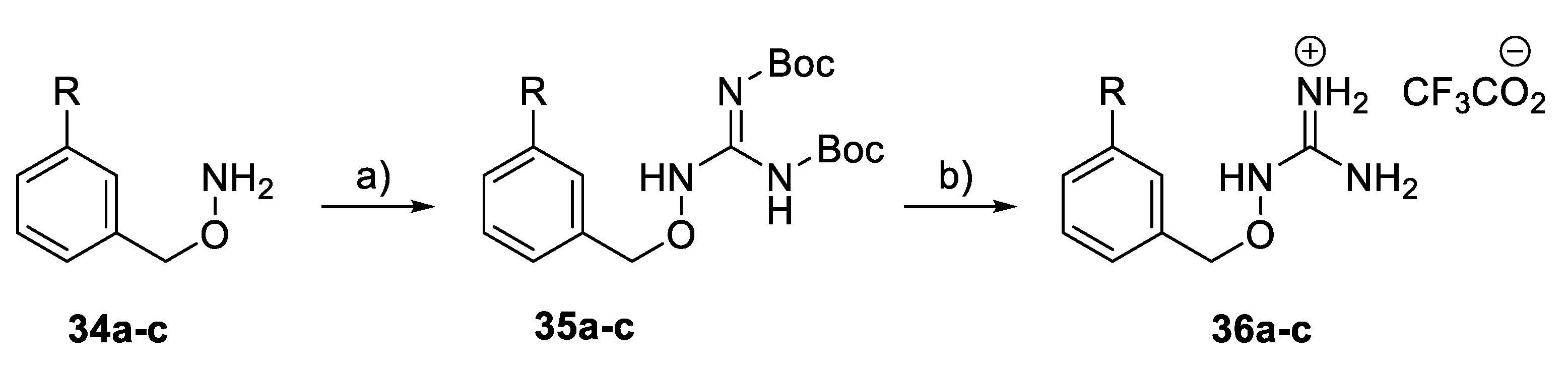{kind=link}
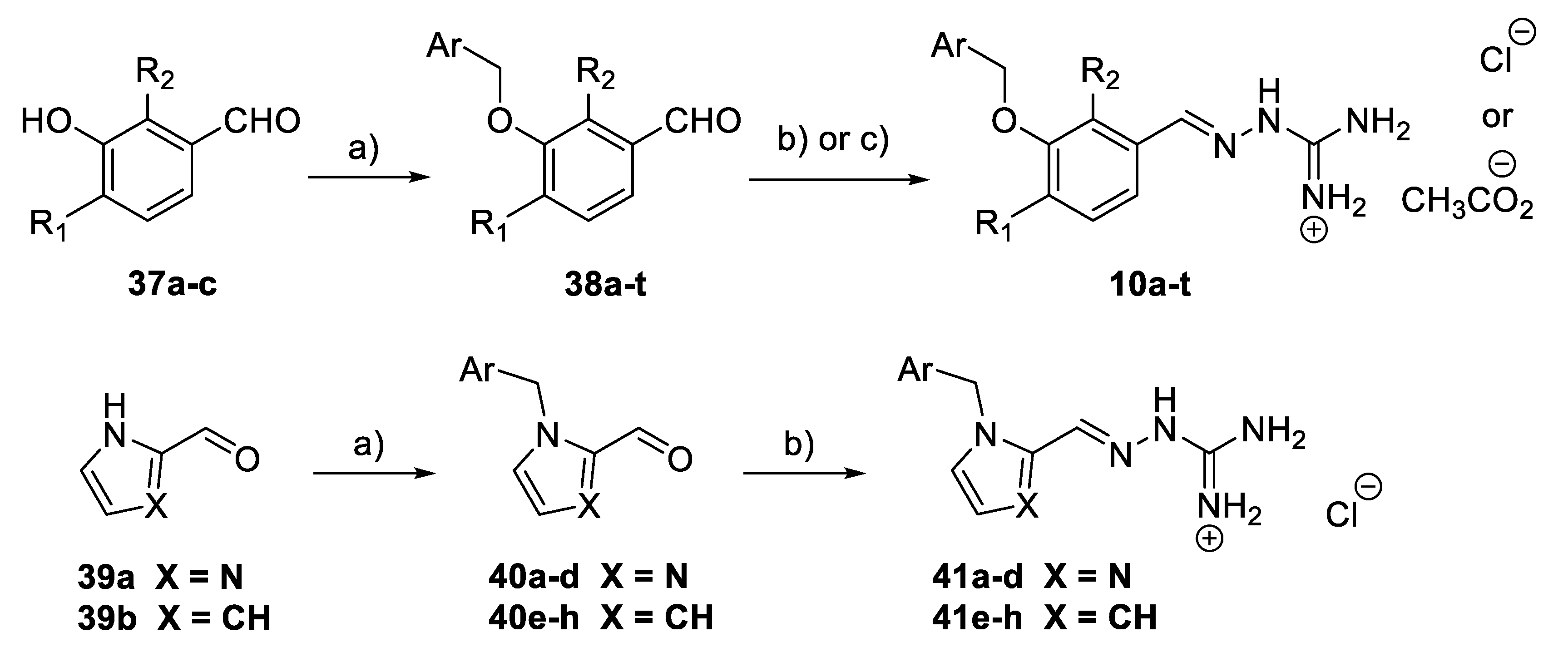{kind=link}
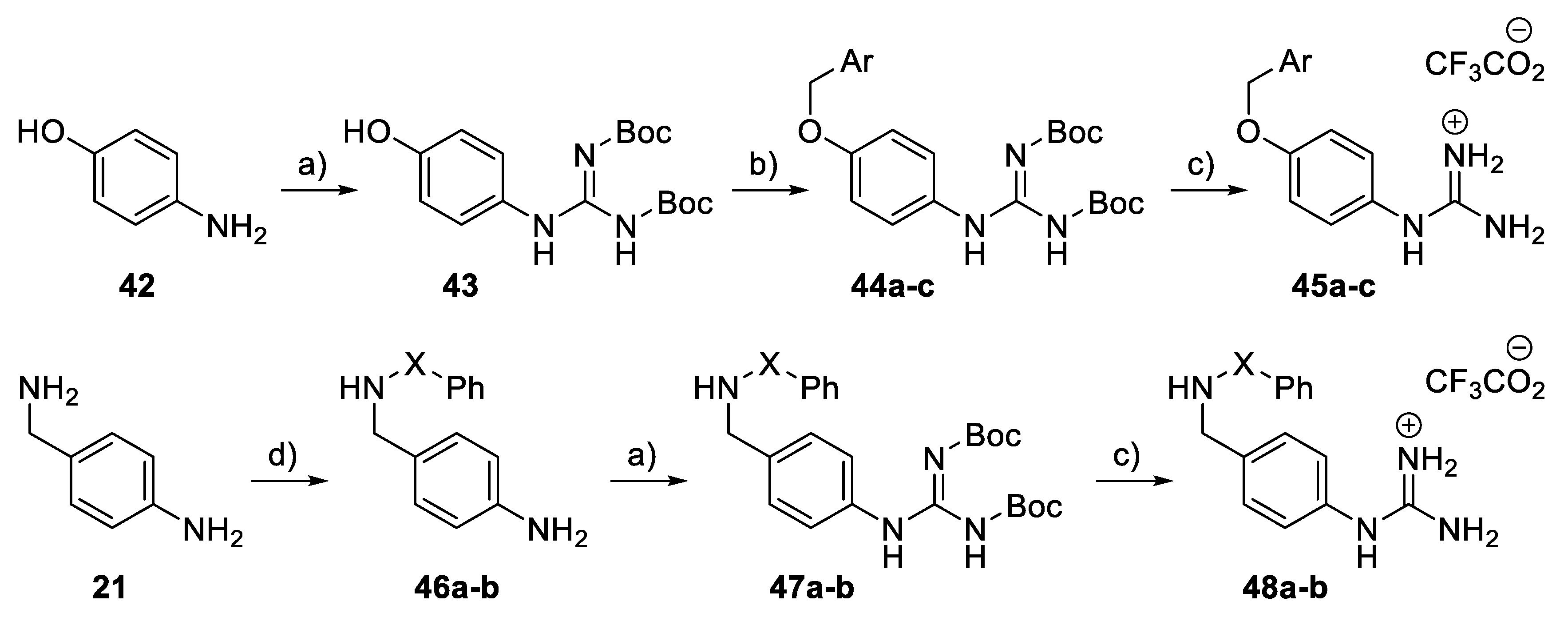{kind=link}
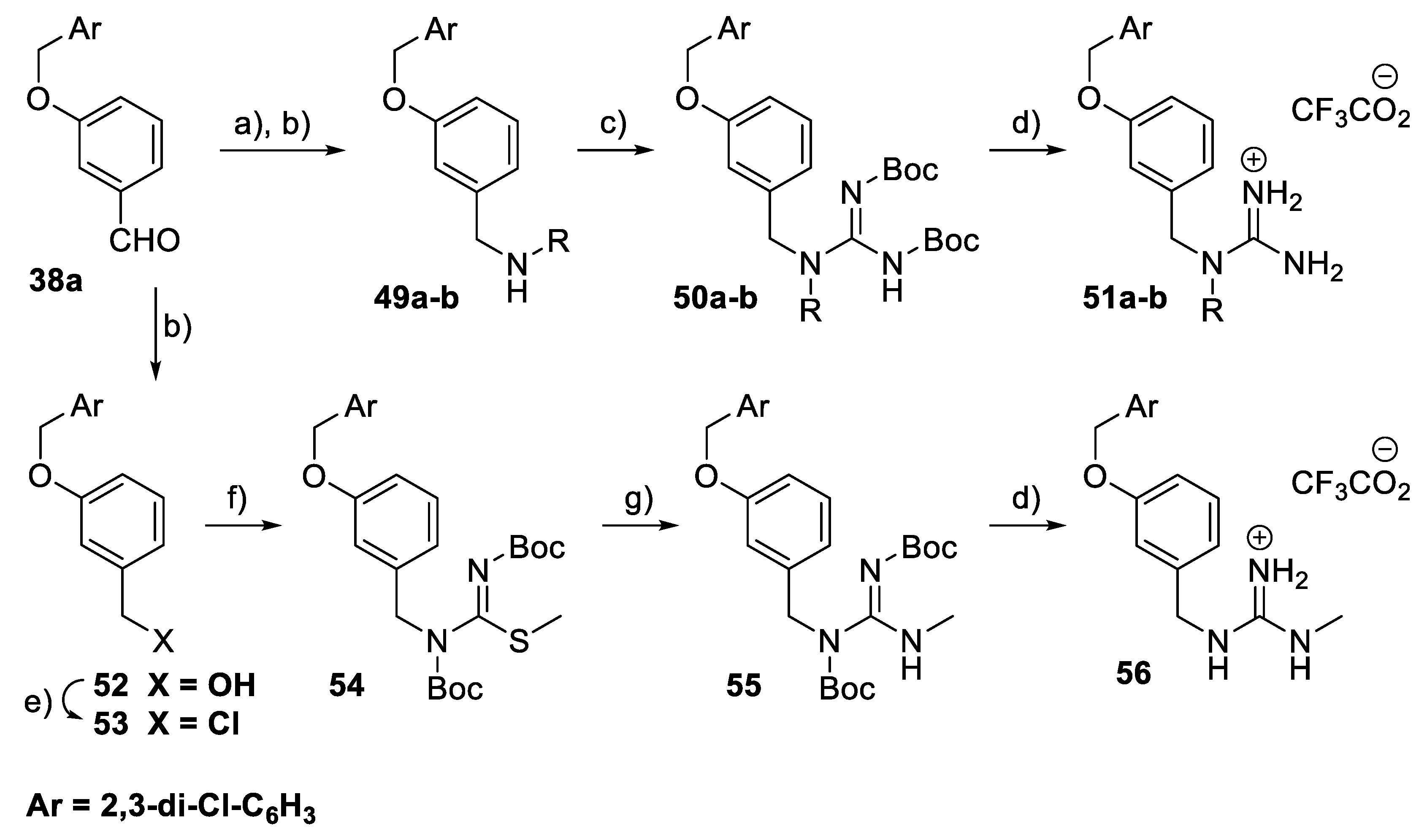{kind=link}





