
Docking and Molecular Dynamic Investigations of Phenylspirodrimanes as Cannabinoid Receptor-2 Agonists

 ,
,  , and
, and 
Abstract
:1. Introduction
2. Results and Discussion
2.1. AI (Artificial Intelligence)-Based Target Prediction for Phenylspirodrimanes Derivatives
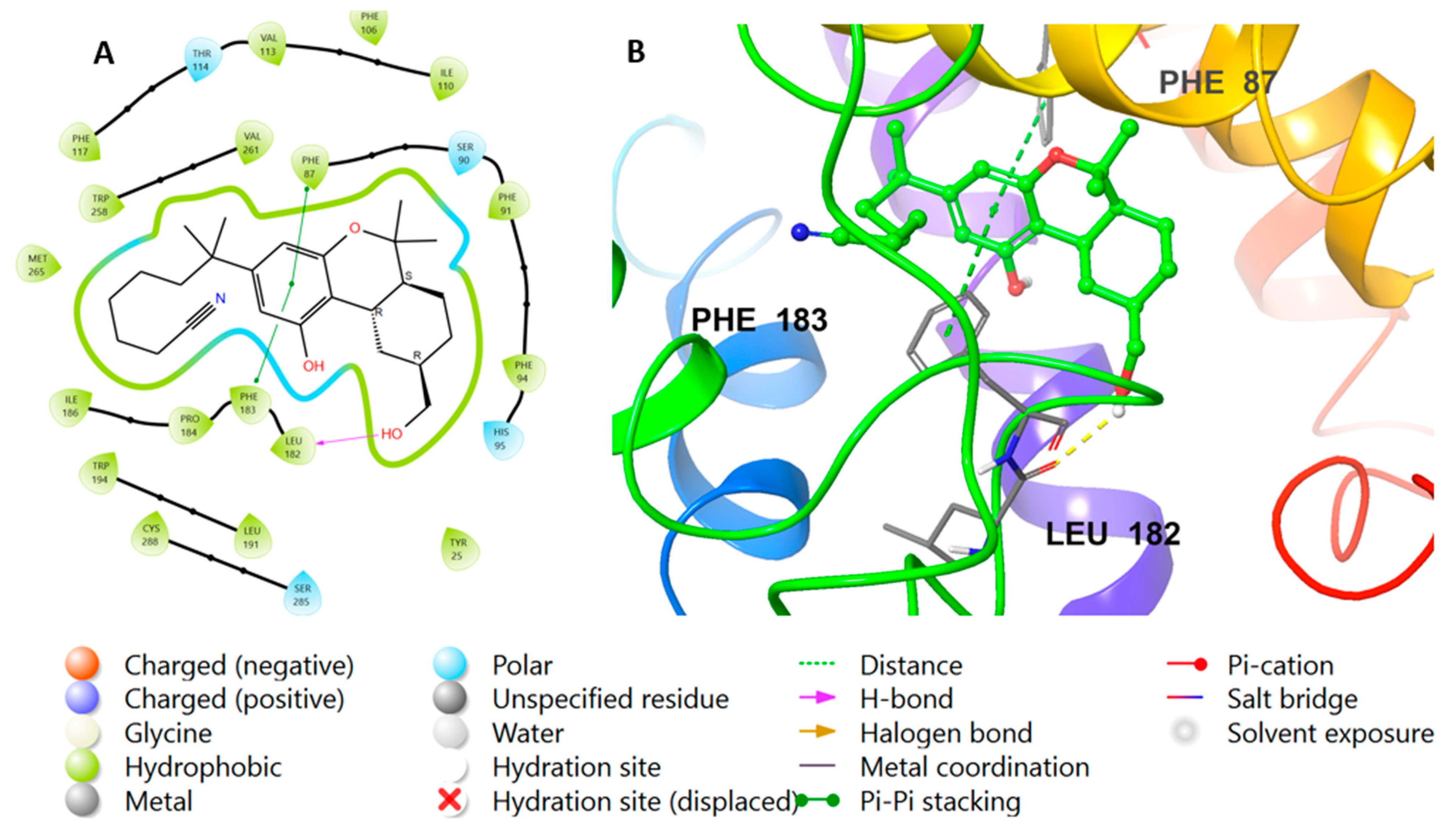
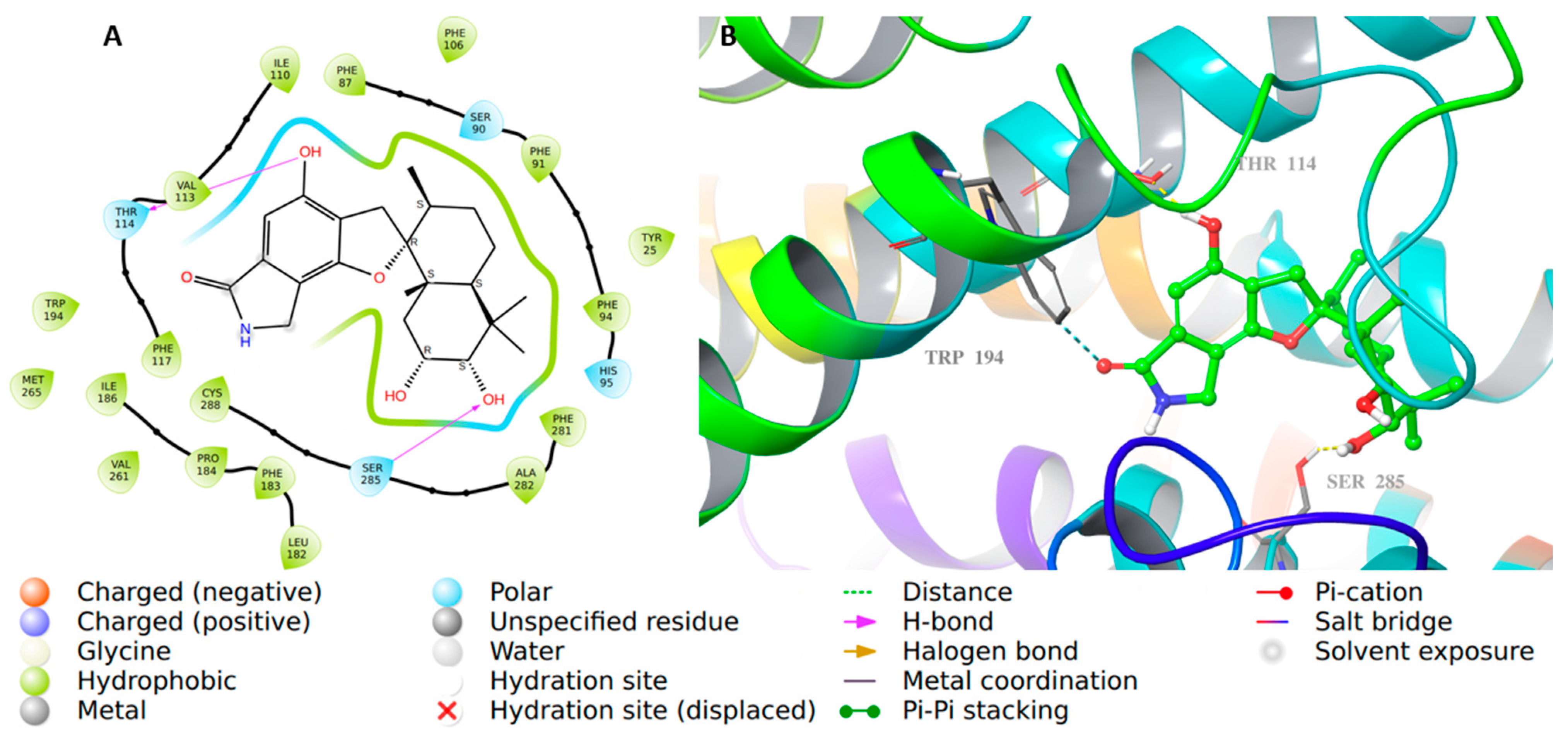
2.2. Molecular Docking Studies
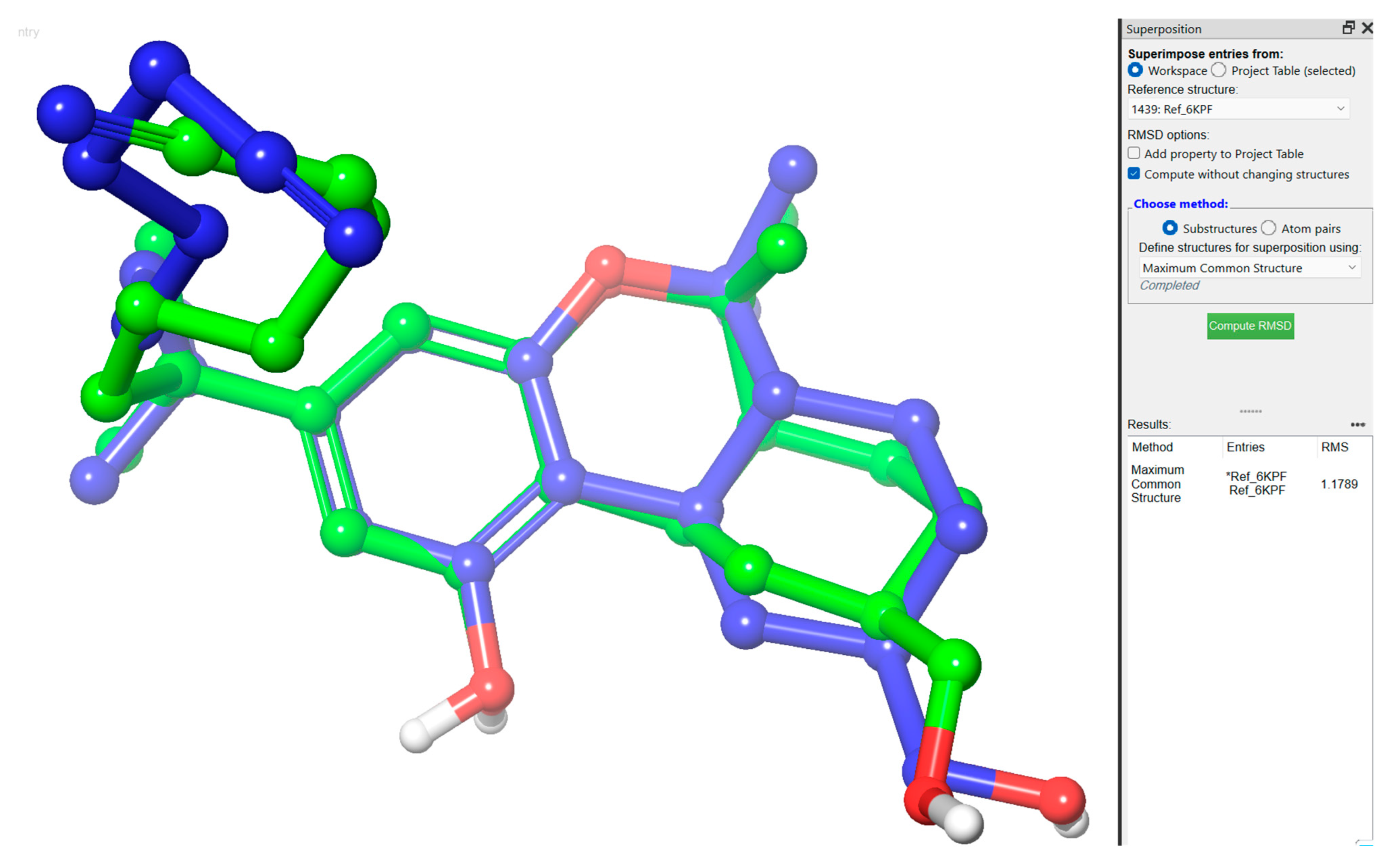
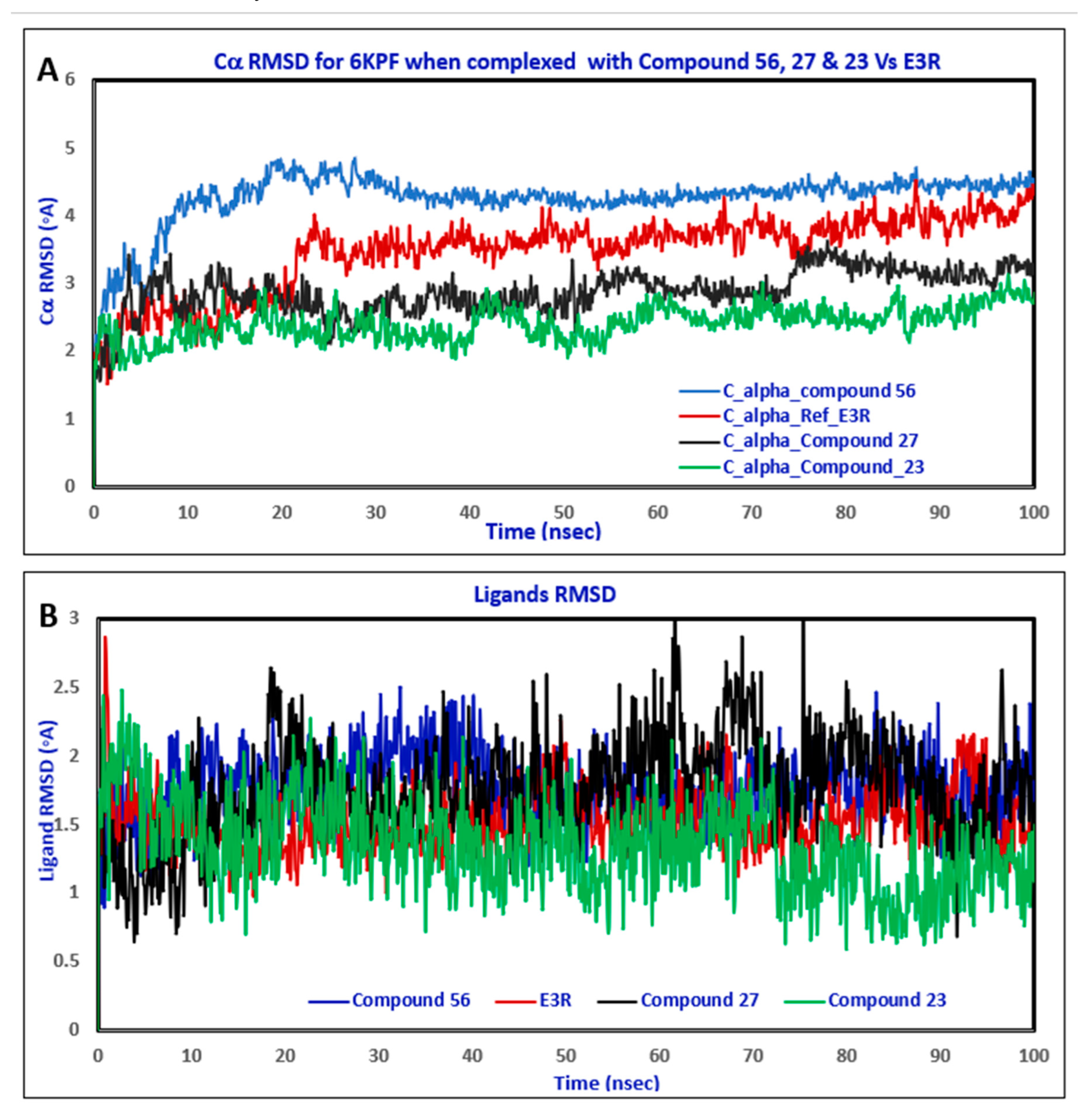
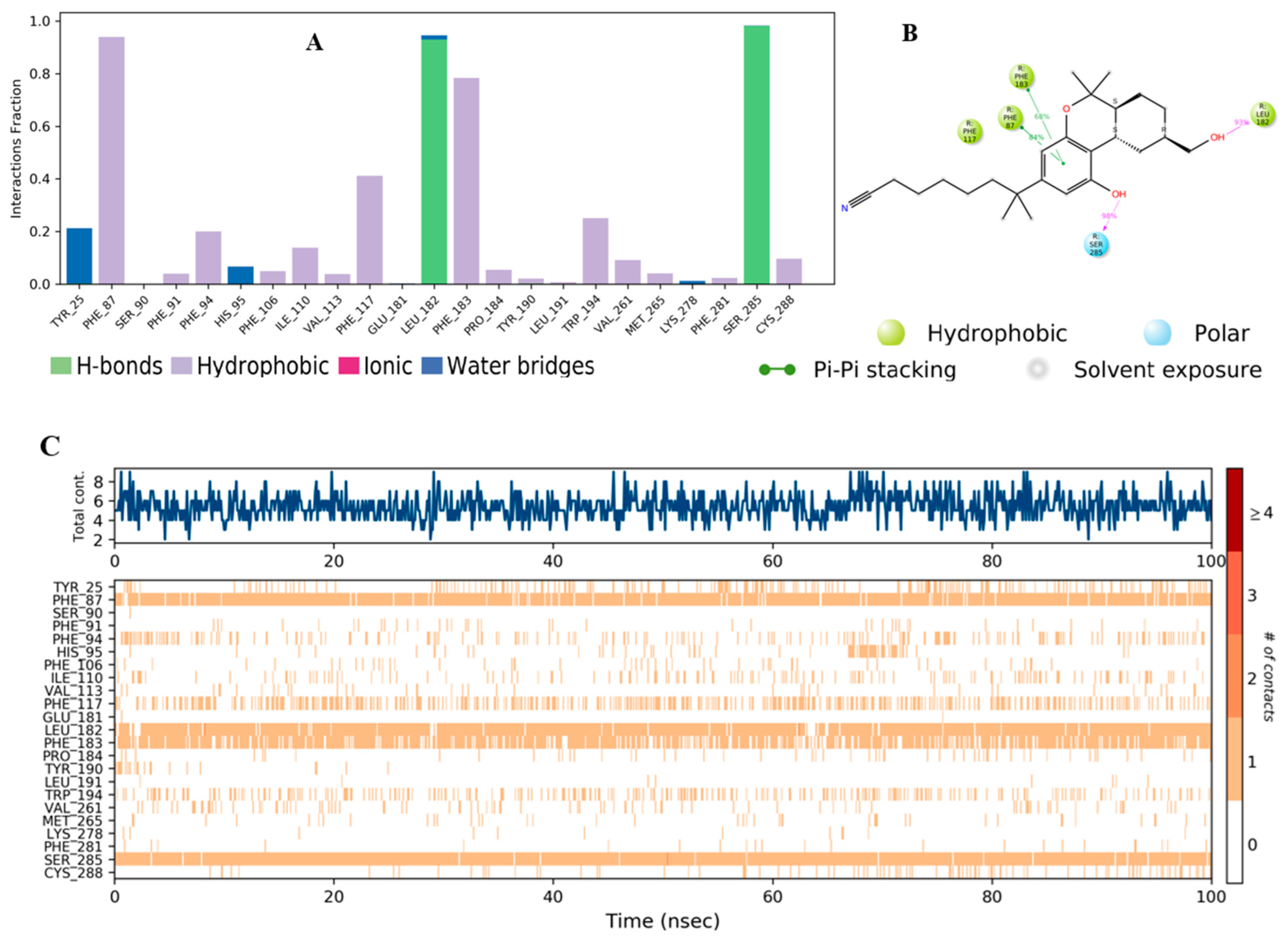
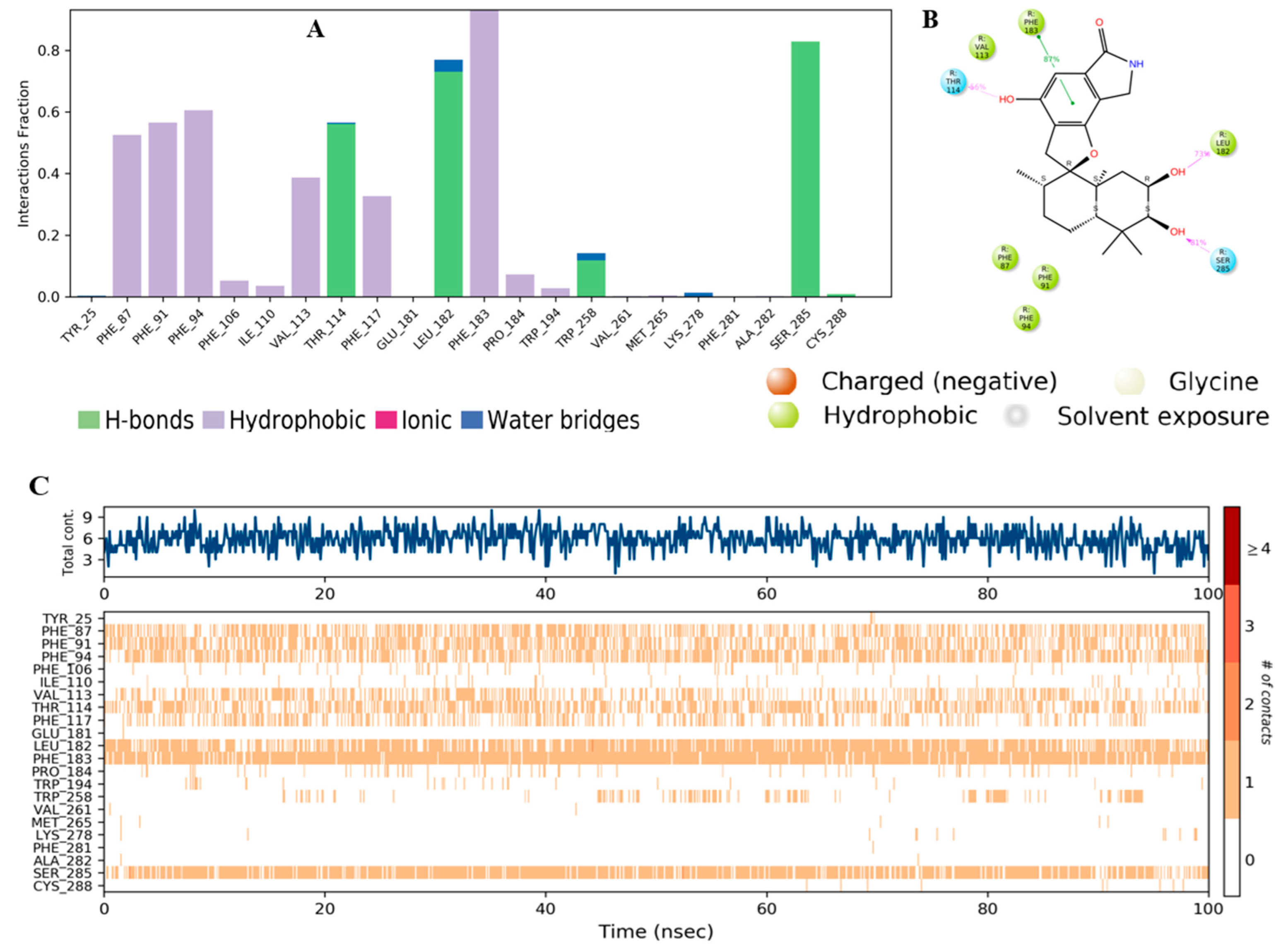
2.3. Molecular Dynamic (MD) Simulation
2.4. In Silico ADME Properties of Selected Ligand
3. Materials and Methods
3.1. Target Prediction
3.2. Ligand and Protein Preparation
3.3. Grid Generation and Molecular Docking
3.4. MD Simulation
3.5. ADMET Properties Prediction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guzmán, M. Cannabinoids: Potential Anticancer Agents. Nat. Rev. Cancer 2003, 3, 745–755. [Google Scholar] [CrossRef]
- Hua, T.; Li, X.; Wu, L.; Iliopoulos-Tsoutsouvas, C.; Wang, Y.; Wu, M.; Shen, L.; Brust, C.A.; Nikas, S.P.; Song, F.; et al. Activation and Signaling Mechanism Revealed by Cannabinoid Receptor-Gi Complex Structures. Cell 2020, 180, 655–665.e18. [Google Scholar] [CrossRef]
- Bie, B.; Wu, J.; Foss, J.F.; Naguib, M. An Overview of the Cannabinoid Type 2 Receptor System and Its Therapeutic Potential. Curr. Opin. Anaesthesiol. 2018, 31, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Lunn, C.A.; Reich, E.P.; Fine, J.S.; Lavey, B.; Kozlowski, J.A.; Hipkin, R.W.; Lundell, D.J.; Bober, L. Biology and Therapeutic Potential of Cannabinoid CB2 Receptor Inverse Agonists. Br. J. Pharmacol. 2008, 153, 226–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, P.; Roy, K.K.; Liu, H.; Ma, G.; Pettaway, S.; Alsharif, W.F.; Gadepalli, R.S.; Rimoldi, J.M.; McCurdy, C.R.; Cutler, S.J.; et al. Structure-Based Identification of Potent Natural Product Chemotypes as Cannabinoid Receptor 1 Inverse Agonists. Molecules 2018, 23, 2630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munro, S.; Thomas, K.L.; Abu-Shaar, M. Molecular Characterization of a Peripheral Receptor for Cannabinoids. Nature 1993, 365, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Romano, J.D.; Tatonetti, N.P. Informatics and Computational Methods in Natural Product Drug Discovery: A Review and Perspectives. Front. Genet. 2019, 10, 368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shankar, A.; Sharma, K.K. Fungal Secondary Metabolites in Food and Pharmaceuticals in the Era of Multi-Omics. Appl. Microbiol. Biotechnol. 2022, 106, 3465–3488. [Google Scholar] [CrossRef]
- Ibrahim, S.R.M.; Altyar, A.E.; Mohamed, S.G.A.; Mohamed, G.A. Genus Thielavia: Phytochemicals, Industrial Importance and Biological Relevance. Nat. Prod. Res. 2022, 36, 5108–5123. [Google Scholar] [CrossRef]
- Ibrahim, S.R.M.; Sirwi, A.; Eid, B.G.; Mohamed, S.G.A.; Mohamed, G.A. Bright Side of Fusarium Oxysporum: Secondary Metabolites Bioactivities and Industrial Relevance in Biotechnology and Nanotechnology. J. Fungi 2021, 7, 943. [Google Scholar] [CrossRef]
- Ibrahim, S.R.M.; Sirwi, A.; Eid, B.G.; Mohamed, S.G.A.; Mohamed, G.A. Fungal Depsides-Naturally Inspiring Molecules: Biosynthesis, Structural Characterization, and Biological Activities. Metabolites 2021, 11, 683. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, G.A.; Ibrahim, S.R.M. Untapped Potential of Marine-Associated Cladosporium Species: An Overview on Secondary Metabolites, Biotechnological Relevance, and Biological Activities. Mar. Drugs 2021, 19, 645. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, G.A.; Ibrahim, S.R.M.; El-Agamy, D.S.; Elsaed, W.M.; Sirwi, A.; Asfour, H.Z.; Koshak, A.E.; Elhady, S.S. Terretonin as a New Protective Agent against Sepsis-Induced Acute Lung Injury: Impact on SIRT1/Nrf2/NF-ΚBp65/NLRP3 Signaling. Biology 2021, 10, 1219. [Google Scholar] [CrossRef] [PubMed]
- El-Agamy, D.S.; Ibrahim, S.R.M.; Ahmed, N.; Khoshhal, S.; Abo-Haded, H.M.; Elkablawy, M.A.; Aljuhani, N.; Mohamed, G.A. Aspernolide F, as a New Cardioprotective Butyrolactone against Doxorubicin-Induced Cardiotoxicity. Int. Immunopharmacol. 2019, 72, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, S.R.M.; Mohamed, G.A.; Khedr, A.I.M. γ-Butyrolactones from Aspergillus Species: Structures, Biosynthesis, and Biological Activities. Nat. Prod. Commun. 2017, 12, 1934578X1701200526. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, S.R.M.; Fadil, S.A.; Fadil, H.A.; Eshmawi, B.A.; Mohamed, S.G.A.; Mohamed, G.A. Fungal Naphthalenones; Promising Metabolites for Drug Discovery: Structures, Biosynthesis, Sources, and Pharmacological Potential. Toxins 2022, 14, 154. [Google Scholar] [CrossRef]
- Ibrahim, S.R.M.; Bagalagel, A.A.; Diri, R.M.; Noor, A.O.; Bakhsh, H.T.; Muhammad, Y.A.; Mohamed, G.A.; Omar, A.M. Exploring the Activity of Fungal Phenalenone Derivatives as Potential CK2 Inhibitors Using Computational Methods. J. Fungi 2022, 8, 443. [Google Scholar] [CrossRef]
- Omar, A.M.; Mohamed, G.A.; Ibrahim, S.R.M. Chaetomugilins and Chaetoviridins-Promising Natural Metabolites: Structures, Separation, Characterization, Biosynthesis, Bioactivities, Molecular Docking, and Molecular Dynamics. J. Fungi 2022, 8, 127. [Google Scholar] [CrossRef]
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; Orhan, I.E.; Banach, M.; Rollinger, J.M.; Barreca, D.; Weckwerth, W.; Bauer, R.; Bayer, E.A.; et al. Natural Products in Drug Discovery: Advances and Opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–216. [Google Scholar] [CrossRef]
- Wang, J.B.; St. Leger, R.J.; Wang, C. Advances in Genomics of Entomopathogenic Fungi. Adv. Genet. 2016, 94, 67–105. [Google Scholar] [CrossRef]
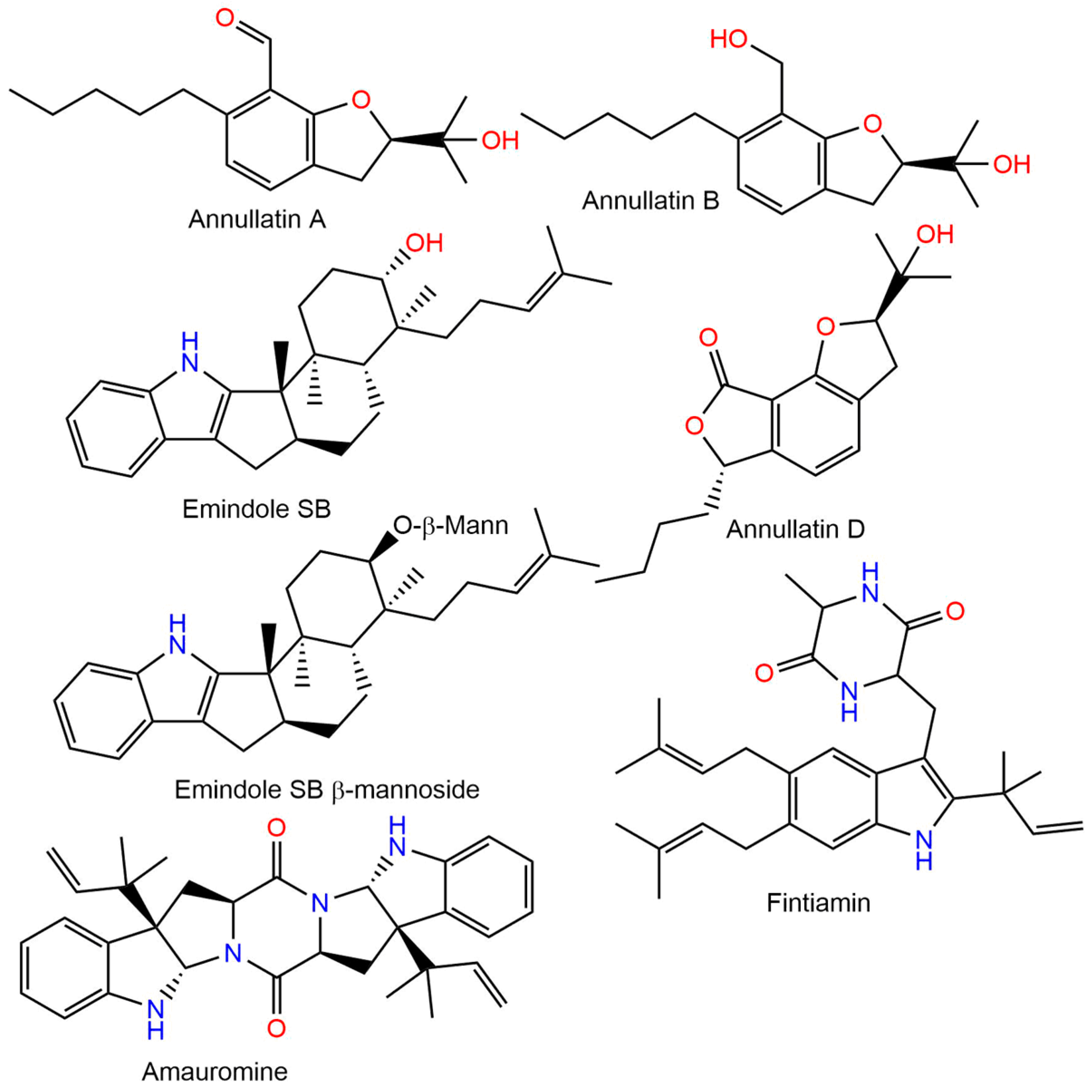
- Harms, H.; Rempel, V.; Kehraus, S.; Kaiser, M.; Hufendiek, P.; Müller, C.E.; König, G.M. Indoloditerpenes from a Marine-Derived Fungal Strain of Dichotomomyces Cejpii with Antagonistic Activity at GPR18 and Cannabinoid Receptors. J. Nat. Prod. 2014, 77, 673–677. [Google Scholar] [CrossRef] [PubMed]
- Asai, T.; Luo, D.; Obara, Y.; Taniguchi, T.; Monde, K.; Yamashita, K.; Oshima, Y. Dihydrobenzofurans as Cannabinoid Receptor Ligands from Cordyceps Annullata, an Entomopathogenic Fungus Cultivated in the Presence of an HDAC Inhibitor. Tetrahedron Lett. 2012, 53, 2239–2243. [Google Scholar] [CrossRef]
- Elsebai, M.F.; Rempel, V.; Schnakenburg, G.; Kehraus, S.; Müller, C.E.; König, G.M. Identification of a Potent and Selective Cannabinoid CB1 Receptor Antagonist from Auxarthron Reticulatum. ACS Med. Chem. Lett. 2011, 2, 866–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elsebai, M.F.; Schoeder, C.T.; Müller, C.E. Fintiamin: A Diketopiperazine from the Marine Sponge-Derived Fungus Eurotium Sp. Arch. Pharm. 2021, 354, 2100206. [Google Scholar] [CrossRef] [PubMed]
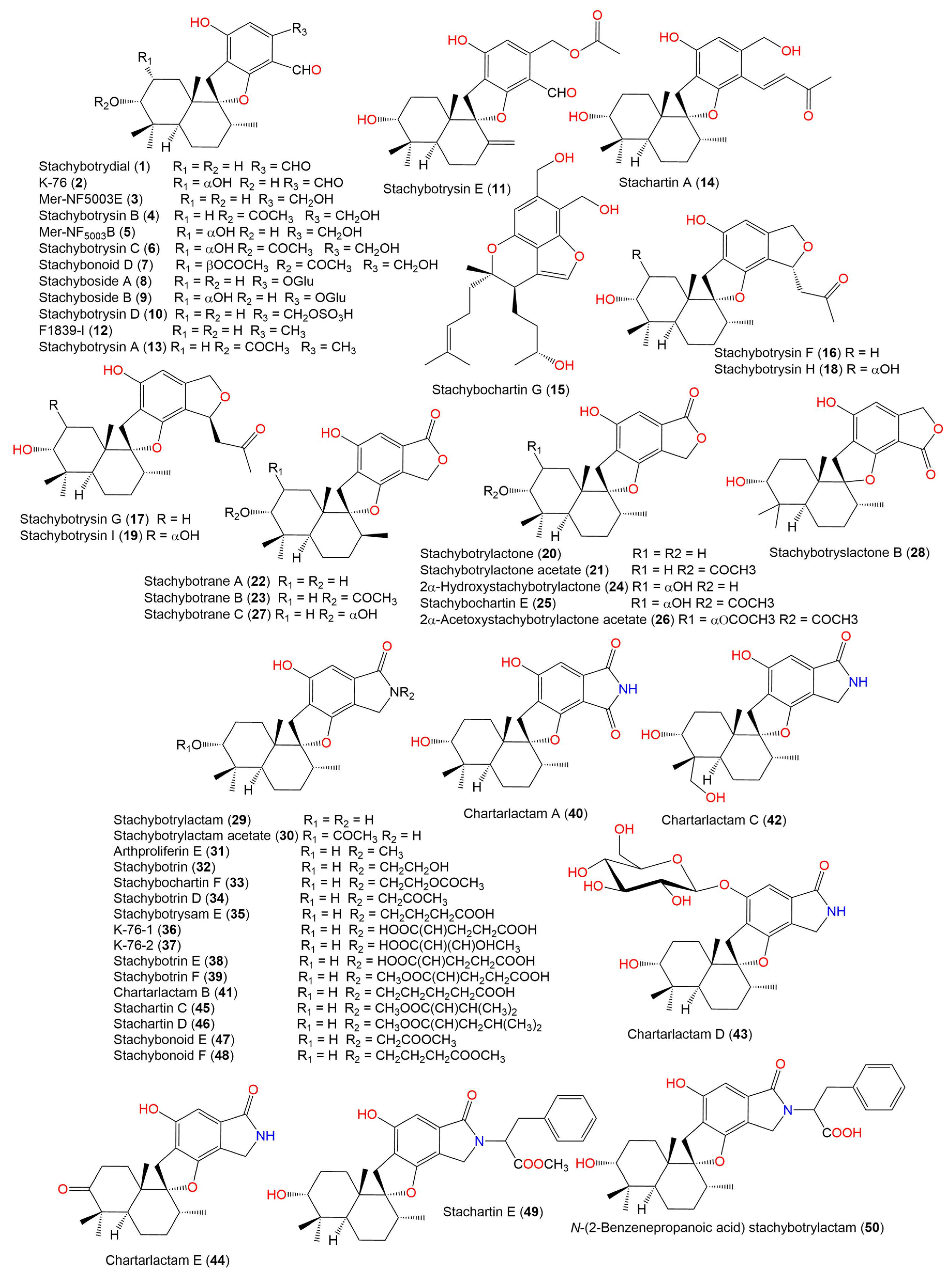
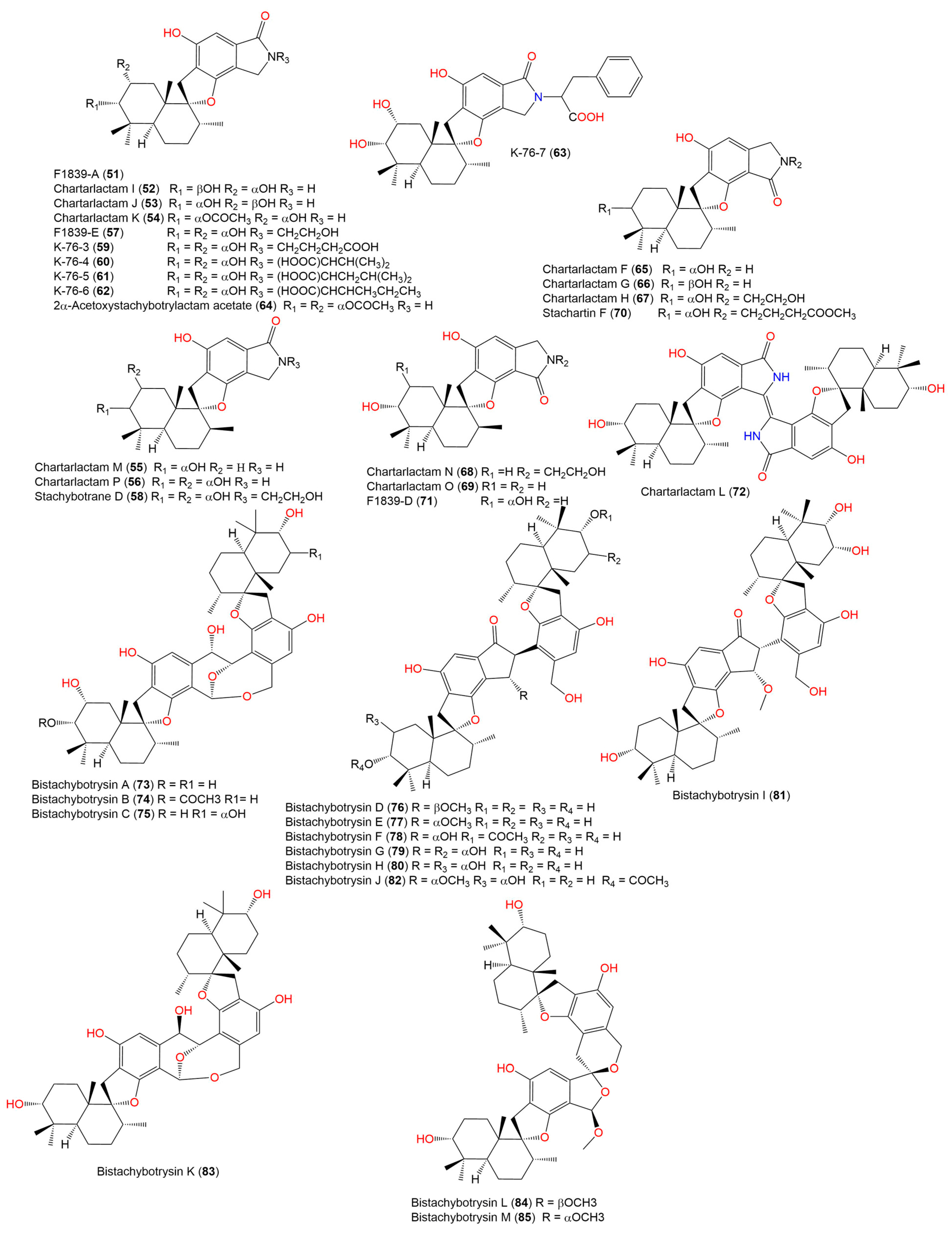
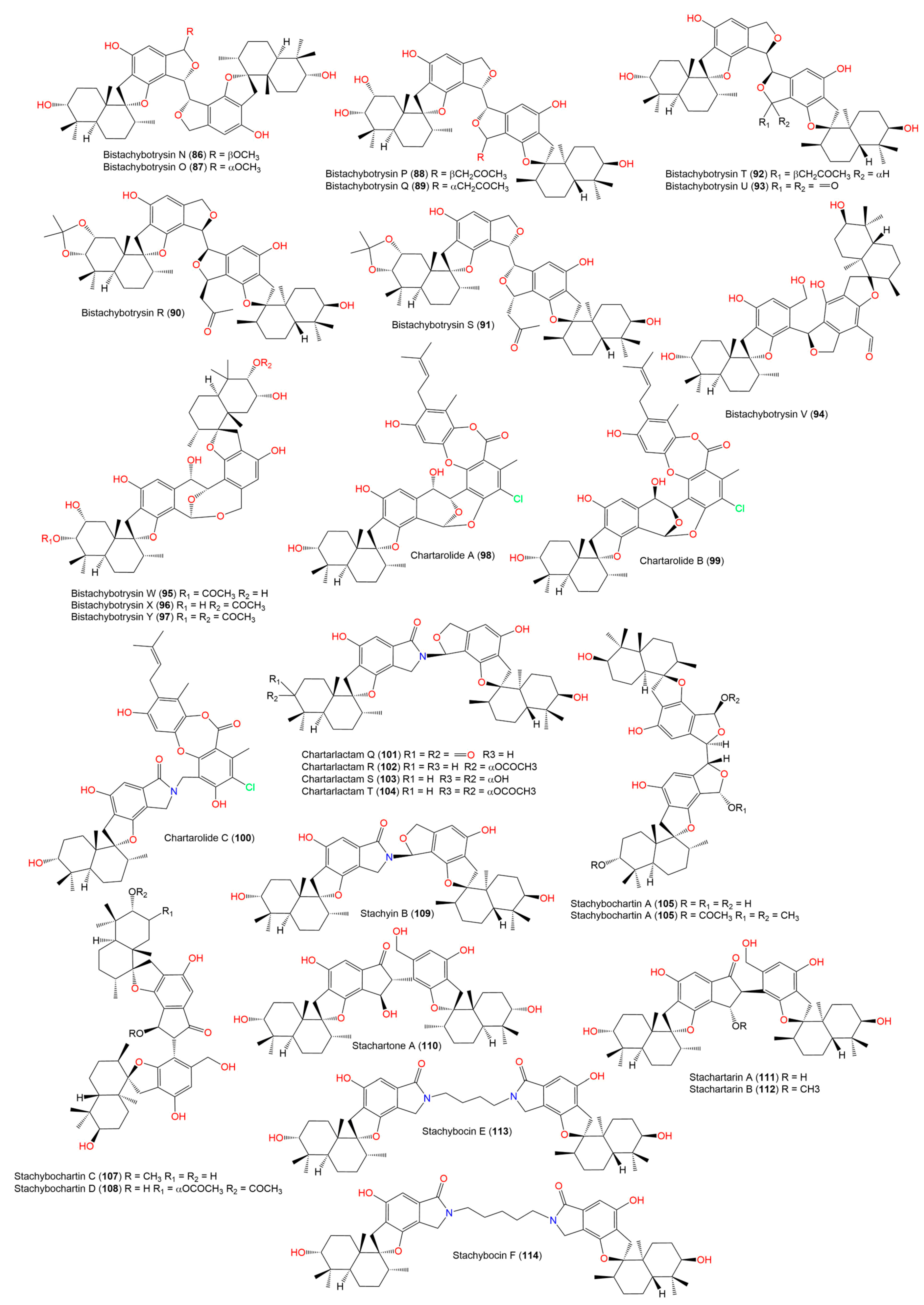
- Ibrahim, S.R.M.; Choudhry, H.; Asseri, A.H.; Elfaky, M.A.; Mohamed, S.G.A.; Mohamed, G.A. Stachybotrys chartarum—A Hidden Treasure: Secondary Metabolites, Bioactivities, and Biotechnological Relevance. J. Fungi 2022, 8, 504. [Google Scholar] [CrossRef]
- Lindemann, V.; Jagels, A.; Behrens, M.; Hübner, F.; Humpf, H.U. In Vitro Metabolism of Phenylspirodrimanes Derived from the Indoor Fungus Stachybotrys. Toxins 2022, 14, 395. [Google Scholar] [CrossRef]
- Nickel, J.; Gohlke, B.O.; Erehman, J.; Banerjee, P.; Rong, W.W.; Goede, A.; Dunkel, M.; Preissner, R. SuperPred: Update on Drug Classification and Target Prediction. Nucleic Acids Res. 2014, 42, W26–W31. [Google Scholar] [CrossRef]
- Schrödinger, LLC. Schrödinger Release 2022-3: LigPrep; Schrödinger, LLC: New York, NY, USA, 2021. [Google Scholar]
- RCSB PDB: Homepage. Available online: https://www.rcsb.org/ (accessed on 5 September 2022).
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and Ligand Preparation: Parameters, Protocols, and Influence on Virtual Screening Enrichments. J. Comput. Aided. Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Schrödinger, LLC. Schrödinger Release 2022-3: Glide; Schrödinger, LLC: New York, NY, USA, 2021. [Google Scholar]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein-Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef]
- Schrödinger, LLC. Schrödinger Release 2022-3: Maestro-Desmond Interoperability Tools; Schrödinger, LLC: New York, NY, USA, 2021. [Google Scholar]
- Schrödinger, LLC. D.E. Shaw Research. In Schrödinger Release 2022-3: Desmond Molecular Dynamics System; Schrödinger, LLC: New York, NY, USA, 2021. [Google Scholar]
- Hollingsworth, S.A.; Dror, R.O. Molecular Dynamics Simulation for All. Neuron 2018, 99, 1129–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leelananda, S.P.; Lindert, S. Computational Methods in Drug Discovery. Beilstein J. Org. Chem. 2016, 12, 2694–2718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrödinger, LLC. Schrödinger Release 2021-4: QikProp; Schrödinger, LLC: New York, NY, USA, 2021. [Google Scholar]
- Dunkel, M.; Günther, S.; Ahmed, J.; Wittig, B.; Preissner, R. SuperPred: Drug Classification and Target Prediction. Nucleic Acids Res. 2008, 36, W55–W59. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Probability * | Model Accuracy ** |
|---|---|---|
| 27 | 58% | 97% |
| 56 | 68% | 97% |
| 79 | 84% | 97% |
| 80 | 87% | 97% |
| 81 | 84% | 97% |
| 82 | 87% | 97% |
| 87 | 83% | 97% |
| 89 | 85% | 97% |
| 96 | 81% | 97% |
| 102 | 80% | 97% |
| 107 | 85% | 97% |
| 108 | 79% | 97% |
| 109 | 87% | 97% |
| 110 | 85% | 97% |
| 111 | 85% | 97% |
| 112 | 85% | 97% |
| 113 | 91% | 97% |
| 114 | 83% | 97% |
| Compound | Docking Score | Glide Gscore | Glide Emodel | XP GScore |
|---|---|---|---|---|
| E3R_6KPF | −12.12 | −12.12 | −64.537 | −12.12 |
| 56 | −11.22 | −11.222 | 31.932 | −11.222 |
| 27 | −11.106 | −11.108 | 25.961 | −11.108 |
| 23 | −10.506 | −10.507 | 37.205 | −10.507 |
| 15 | −10.193 | −10.193 | −1.513 | −10.193 |
| 55 | −10.17 | −10.171 | 36.385 | −10.171 |
| 22 | −9.957 | −9.958 | 25.109 | −9.958 |
| Molecule | Recommended Range | Stachybochartin G (15) | Stachybotrane A (22) | Stachybotrane B (23) | Stachybotrane C (27) | Chartarlactam M (55) | Chartarlactam P (56) | Reference |
|---|---|---|---|---|---|---|---|---|
| #stars | 0–5 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| #rtvFG | 0–2 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| CNS | −2 (inactive) +2 (active) | −2 | −1 | −1 | −2 | −2 | −2 | −1 |
| mol_MW | 130.0–725.0 | 388.503 | 386.487 | 428.524 | 402.486 | 385.502 | 401.502 | 421.581 |
| SASA | 300.0–1000.0 | 677.156 | 595.43 | 641.055 | 605.78 | 613.265 | 622.968 | 738.361 |
| volume | 500.0–2000.0 | 1269.411 | 1144.614 | 1260.485 | 1165.663 | 1166.122 | 1185.684 | 1374.214 |
| donorHB | 0.0–6.0 | 3 | 2 | 1 | 3 | 3 | 4 | 2 |
| accptHB | 2.0–20.0 | 6.35 | 6.2 | 6.5 | 7.9 | 5.7 | 7.4 | 5.2 |
| QPlogPw | 4.0–45.0 | 11.089 | 11.101 | 10.117 | 14.344 | 12.255 | 15.497 | 9.386 |
| QPlogPo/w | −2.0–6.5 | 3.415 | 2.834 | 3.449 | 1.887 | 2.831 | 1.866 | 5.171 |
| QPlogS | −6.5–0.5 | −4.352 | −4.597 | −5.248 | −4.125 | −4.933 | −4.428 | −6.376 |
| QPlogHERG | concern below −5 | −4.551 | −3.577 | −3.704 | −3.65 | −3.817 | −3.882 | −5.34 |
| QPPCaco | <25 poor, >500 great | 442.716 | 393.449 | 296.585 | 150.047 | 247.784 | 94.544 | 1603.15 |
| QPlogBB | −3.0–1.2 | −1.453 | −0.804 | −0.939 | −1.254 | −1.032 | −1.495 | −0.767 |
| #metab | 1–8 | 8 | 4 | 3 | 5 | 4 | 5 | 2 |
| QPlogKhsa | −1.5–1.5 | 0.287 | 0.467 | 0.709 | 0.187 | 0.537 | 0.244 | 0.939 |
| Human Oral Absorption | 1, 2, or 3 for low, medium, or high | 3 | 3 | 3 | 3 | 3 | 3 | 1 |
| Percent Human Oral Absorption | >80% is high <25% is poor | 94.3 | 89.983 | 91.385 | 76.944 | 86.374 | 73.229 | 100 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Omar, A.M.; Aljahdali, A.S.; Safo, M.K.; Mohamed, G.A.; Ibrahim, S.R.M. Docking and Molecular Dynamic Investigations of Phenylspirodrimanes as Cannabinoid Receptor-2 Agonists. Molecules 2023, 28, 44. https://doi.org/10.3390/molecules28010044
Omar AM, Aljahdali AS, Safo MK, Mohamed GA, Ibrahim SRM. Docking and Molecular Dynamic Investigations of Phenylspirodrimanes as Cannabinoid Receptor-2 Agonists. Molecules. 2023; 28(1):44. https://doi.org/10.3390/molecules28010044
Chicago/Turabian StyleOmar, Abdelsattar M., Anfal S. Aljahdali, Martin K. Safo, Gamal A. Mohamed, and Sabrin R. M. Ibrahim. 2023. "Docking and Molecular Dynamic Investigations of Phenylspirodrimanes as Cannabinoid Receptor-2 Agonists" Molecules 28, no. 1: 44. https://doi.org/10.3390/molecules28010044